

Abstract
Standard therapeutic approaches of cytotoxics and radiation in cancer are not only highly toxic, but also of limited efficacy in treatment of a significant number of cancer patients. The molecular analysis of the cancer genomes have shown a remarkable complexity and pointed to key genomic and epigenomic alterations in cancer. These discoveries are paving the way for targeted therapy approaches. However, while there are a large number of potential targets, only a few can regulate key cellular functions and intersect multiple signaling networks. The Aurora kinase family members (A, B, and C) are a collection of highly related and conserved serine/threonine kinases that fulfill these criteria, being key regulators of mitosis and multiple signaling pathways. Alterations in Aurora kinase signaling are associated with mitotic errors and have been closely linked to chromosomal aneuploidy in cancer cells. Several studies have shown amplification and/or over-expression of Aurora kinase A and B in hematologic malignancies and solid tumors. Over the past several years, Aurora kinases have become attractive targets. Several ongoing clinical trials and bench-based research are assessing the unique therapeutic potential of Aurora-based targeted therapy.
Keywords: Aurora, kinase, cancer, therapy, targets
Structure of the Aurora kinases
The ability of a cell to divide properly is a prerequisite for its normal growth and development, and this process is tightly regulated. Studies in lower organisms have shown that several serine/threonine kinases, known as mitotic kinases, include: cyclin dependent kinase 1 (CDK1: also known as p34cdc2), polo-like kinases, NIMA-related kinases, WARTS/LATS1-related kinases, and Aurora/Ip11-related kinases are playing an important role in different stages of cell division. The structure of these enzymes has been well conserved through evolution. Any aberration in the genetic pathways regulating cell growth and apoptosis leads to cell transformation and tumorigenesis. The Aurora kinase family is a collection of highly related serine/threonine kinases that are key regulators of mitosis; essential for accurate and equal segregation of genomic material from parent to daughter cells. Aurora kinases show conservation of both structure and function throughout eukaryotic organisms, members of this family have been extensively studied in a range of different model organisms (1). Invertebrates are comprised of three family members: Aurora-A, -B and -C, with one or more highly conserved orthologues being found in the yeasts, flies, worms, and other invertebrates. Saccharomyces cerevisiae cells have a single Aurora gene, IPL1 (2). The Drosophila and Caenorhabditis elegans genomes encode one member in each of the Aurora-A and -B classes (3). The homologs of Aurora-A and -B have also been found in Xenopus (4). They have a COOH-terminal catalytic domain that is highly conserved within the family and an NH2 terminal domain that is variable among organisms (5) (Figure 1). Aurora-A and-B share 71% identity in their C-terminal catalytic domain. The most conserved motif is the putative activation loop. At the amino terminal domain, three putative conserved Aurora boxes (A-boxI, A-boxII and A-boxIII) can be identified. The functional significance of these boxes is not known. Despite significant sequence homology, the localization and functions of these kinases are largely distinct from one another. The high percentage of conservation is very important in relation to the specificity of substrates and inhibitors. The mean proportion of similar amino acids estimated by pair-wise sequence comparisons is significantly higher among different families of Aurora-A, -B and -C in vertebrates (0.84+0.5) than within the same family (Aurora-A or -B) in vertebrates and invertebrates species (0.69+0.3 for both). This suggests a recent evolutionary radiation of Aurora families within vertebrates. Structural and motif based comparison suggested an early divergence of Aurora-A from Aurora-B and Aurora-C.
Figure 1.
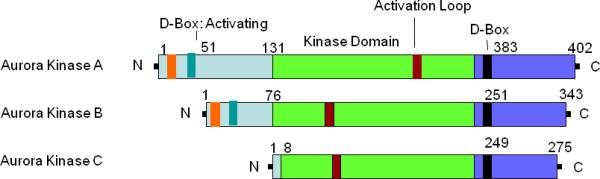
Schematic diagram of Aurora-A, -B, & -C kinase domains. N & C terminal domains contain most of the regulatory sequences. The central domain consists of catalytic kinase domain and activation loop. D-Box at the c-terminal domain is the destruction box. Brown box = Activation loop; Black box = destruction box at C terminus; Light green box = destruction box at N terminus; Light green box = Kinase domain.
Biology, function and regulations of Aurora kinases
Aurora Kinase A (AURKA)
The human AURKA gene (also known as Aurora-2/BTAK/STK15) maps to chromosome 20q13.2, and is thus far, a more extensively studied member of the aurora kinase family. AURKA is ubiquitously expressed and regulates cell cycle events occurring from late S-phase through the M phase, including: centrosome maturation, mitotic entry, centrosome separation, bipolar spindle assembly, chromosome alignment, cytokinesis, and mitotic exit (6-8). AURKA activity and protein levels both increase from late G2 through the M phase, with peak activity in pro-metaphase. The kinase activity of AURKA is tightly regulated throughout the cell cycle. It is activated through the phosphorylation of T288 (human sequence) on its activation loop. It can be inactivated through dephosphorylation of T288 by protein phosphatase 1 (PP1). Beyond phosphorylation and dephosphorylation, its activity is also regulated by its expression and degradation. AURKA binds to, and phosphorylates LIM domain containing Ajuba protein during the G2 phase and results in autophosphorylation of Aurora-A in its activating loop (7). This phosphate group is removed by protein phosphatase 1 or 2A (PP1/2A), which renders AURKA inactive. A number of co-factors including microtubule associated protein TPX2 and GTPase Ran are required for this switch to activation. Ran releases TPX2 from importins allowing TPX2 to bind to AURKA, targeting it to spindle microtubules at the pole. TPX2 activates AURKA activity by stimulating its autophosphorylation and by protecting it from the inhibitory action of PP1 (9). In the absence of TPX2 the AURKA activation segment is in an inactive conformation, with the crucial phosphothreonine exposed and accessible for deactivation. A recent report by Anderson et al (10) reported that TPX2 binding has no effect on the turnover number of AURKA and does not change its reaction mechanism. The mode of binding between TPX2 and AURKA and the conformational changes that are induced in AURKA upon binding, bear resemblance to the mode of intramolecular binding and activation of cAMP-dependent kinase. In vivo, activation of AURKA synergistically depends on (auto) phosphorylation within its activation segment (on threonine 288) and TPX2 binding, potentially in combination with microtubule binding.
Aurora Kinase B (AURKB)
AURKB maps to chromosome 17q13. It is a chromosomal passenger protein critical for accurate chromosomal segregation, cytokinesis (11) protein localization to the centrosome and kinetochore correct microtubule-kinetochore attachments, and regulation of the mitotic checkpoint. Inhibition of AURKB function results in an increase in ploidy phenotype. AURKB, mRNA and protein expression levels peak at G2/M phase, the maximum kinase activity is reached at transition during metaphase to the end of mitosis (12). AURKB is phosphorylated at multiple sites throughout the cell cycle in Xenopus (13); the upstream kinase that regulates AURKB has not been identified. AURKB functions in cooperation with its binding partners and substrates like inner centromere protein (INCEP), survivin, and borealin to ensure proper kinetochore-microtubule attachments. AURKB directly phosphorylates INCEP and this phosphorylation feeds back positively to potentiate its kinase activity in vitro (14). AURKB helps in proper chromosome bio-orientation; however, inhibition of AURKB overrides the checkpoints and drives cells through an aberrant mitosis. This phenomenon is different than inhibition of AURKA which causes arrest in mitosis. Due to this feature inhibitors of AURKB inhibitors have been referred as mitotic drivers in a recent review (15). It has been recently shown that AURKB interacts with microtubule destabilizing mitotic centrosome-associated kinesin (MCAK) to ensure proper chromosome bio-orientation (16). Some studies have reported roles of AURKB as phosphorylating histone H3 and in establishing microtubule-kinetochore associations (17).
Aurora Kinase C (AURKC)
AURKC, the third member of the Aurora kinase family, is also a chromosomal passenger protein that co-localizes with AURKB and is expressed in the testis where it functions in spermatogenesis and regulation of cilia and flagella. AURKC shares a higher identity with AURKB than AURKA (83 and 71%, respectively). Expression of AURKC at both mRNA and protein levels also peaks at G2/M phase. AURKC is localized to centrosome during mitosis from anaphase to cytokinesis and plays a role(s) in centrosome function at a later stage of mitosis (18).
Aurora Kinases in Cancer
Deregulation in Aurora kinases has been linked to tumorigenesis. Out of the three family members, AURKA is consistently associated with cancers. AURKB has also recently been reported to contribute to tumorigenesis but the role of AURKC is not yet properly associated.
AURKA's role in tumor development
AURKA gene amplification and/or over-expression is a frequent finding in several malignancies including breast, colon, pancreas, ovaries, bladder, liver, and gastric cancers (19-21). AURKA over-expression can occur because of gene amplification, transcriptional induction or post-translational stabilization (22). Interest in AURKA intensified after a series of preclinical studies demonstrated the oncogenic potential of AURKA activation resulting in the in vitro and in vivo transformation of rodent fibroblast cells and the formation of multipolar mitotic spindles inducing genome instability (23) establishing AURKA as a bona fide oncogene (Figure 2). AURKA over-expression has been reported to be significantly associated with a higher grade of tumor and a poor prognosis (24). Aneuploidy is a good marker of tumor progression and prognosis caused due to chromosomal instability, the most frequent genomic damage that occurs during cancer development. In gastric carcinoma and in papillary thyroid carcinoma aneuploidy is a marker of metastasis (25, 26) and in many malignancies aneuploidy is associated with a poor outcome (27). A correlation between AURKA over-expression and aneuploidy exists in gastric cancer; clinical samples with AURKA amplification and over-expression showed aneuploidy and poor prognosis (20). AURKA plays an important role in centrosome maturation, and numerous centrosomal abnormalities are observed in AURKA-deficient cells. Centrosomal anomalies have been reported to arise at early stages of tumor formation and to expand concomitant with tumor progression (28) a process in agreement with the AURKA expression profile pattern which increases from early to late stages of tumor. Although no direct link has been found between AURKA overexpression and centrosome abnormalities in cancer, AURKA over-expression, centrosome amplification and aneuploidy are always associated. Centrosomal abnormalities lead to bipolar mitotic spindle defects, chromosomal segregation deficiency and aneuploidy. Centrosomal aberrations are found in brain (29), breast (30), lung, colon and prostrate tumors (29). Furthermore, centrosome aberrations lead to aneuploidy, suggesting that AURKA over-expression is responsible for centrosome amplification, and thus, participates in tumorigenesis.
Figure 2.
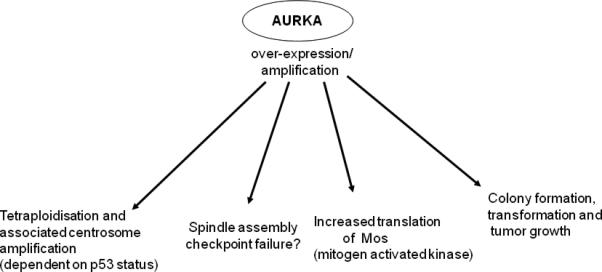
Overview of the different effects that are observed upon over-expression and/or amplification of AURKA.
AURKA binds and phosphorylates the breast cancer associated gene product, BRAC1, in vitro and in vivo to regulate its function (31). It is reported that ovarian and breast epithelial carcinomas play a role in the regulation of human telomerase reverse transcriptase mRNA levels through c-Myc (32). AURKA has also been reported to override the spindle checkpoint activated by paclitaxel (Taxol) and nocodazole (33). These defects might contribute to transformation.
AURKA interacts with the p53 pathway at multiple levels, suggesting that these proteins form a part of an integrated functional network. AURKA interferes with p53 suppressor function by at least two mechanisms: it directly phosphorylates p53 at Ser315 facilitating MDM-2 mediated degradation of p53 in cancer cells (34), and it also phosphorylates p53 at Ser215 to inactivate its transcriptional activity (35). In addition to these two mechanisms, our work suggests regulation of p53 through AKT/MDM-2 axis in gastric cancer cells (36). We also reported that AURKA over-expression suppresses TAp73 in p53 deficient cancer cells (37). TAp73, a p53 family, member has significant homology with p53 and plays an essential role in apoptosis induced by cytotoxic agents (38). The tumor suppressor proteins p53 and p73 can activate genetic programs that halt cell proliferation transiently (G1 and G2 cell cycle arrest) or permanently (senescence) or eliminate the cell altogether (apoptosis). Regulation of p53 and p73 by AURKA over-expression can lead to suppression of apoptosis of tumor cells. AKT is a major pro-proliferative serine/threonine kinase that promotes cell survival in a variety of cell types and prevents apoptosis induced by various apoptotic stimuli (39). We (36), and others (40), have reported that AURKA up-regulates AKT phosphorylation at Ser473. We reported the regulation of GSK-3ß and ß-catenin (41) by AURKA over-expression in gastric cancer cells. A schematic overview of possible AURKA interaction is shown in Figure 3.
Figure 3.
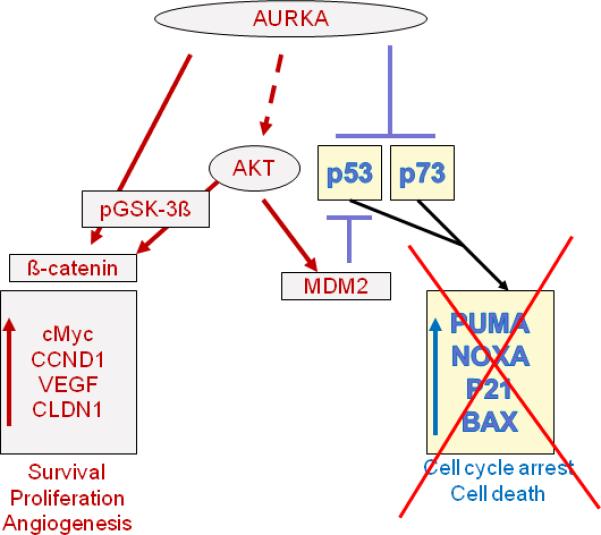
Schematic representation of AURKA interactions. AURKA over-expression inhibits p53 family members and suppresses apoptosis and cell cycle arrest. AURKA interacts directly with p53 by phosphorylating it at Ser215 (34) and 315 (35) causing its degradation through MDM2 or inactivating it at transcription level, respectively. AURKA regulates p73 and its downstream targets (37). It also up-regulates the PI3 kinase pathway that enhances cell survival and proliferation either directly interacting with GSK-3β (41) or by regulating AKT (36, 75).
AURKB
AURKB regulates kinetochore-microtubule attachment and ensures faithful chromosome segregation (42). It is over-expressed in various human tumors like breast, colorectal, kidney, lung, and prostrate (43). An increased level of AURKB correlates with advanced stages of colorectal cancer (44). Its over-expression results in multi-nucleation and polyploidy in human cells (43); however, this phenotype is exacerbated in absence of p53 (45). It has also been reported that AURKB over-expression induces chromosomes lagging in metaphase, chromosome segregation error and errors in cytokinesis, and thus, playing a role in carcinogenesis (46). AURKB does not transform cells alone but is reported to induce H-Ras mediated transformation (47). AURKB over-expression is reported to correlate with the level of genomic instability within a tumor indicating that it contributes to the acquisition of genetic alterations critical for neoplastic transformation.
AURKC
AURKC is a chromosome messenger protein expressed in the testis and not in somatic cells. However, it is reported to be highly expressed in cancer cells such as HepG2, HuH7, MDA-MD-453, and HeLa cells (18). Very little information is available regarding the role of AURKC in tumors; further functional analysis is required to understand its role in molecular pathways in cancer.
Targeting Aurora kinases
Aurora Kinase family members generated great interest after their over-expression and amplification was reported in a number of tumors. Their over-expression and association with genetic instability in tumors have made them the focus of drug discovery. Due to their involvement in a wide range of cell cycle events (e.g. centrosome function, mitotic entry, kinetochore function, spindle assembly, chromosome segregation, microtubule dynamics, spindle checkpoint function, and cytokinesis), they attracted a lot of attention from pharmaceutical companies to develop potential inhibitors against them. The design, approach and development of Aurora kinase inhibitors have been discussed in the review by Pollard et al (48). A growing number of inhibitors of Aurora Kinases have been developed, either at preclinical or clinical stages like Hesperidin (11), ZM-447439 (49), VX-680 (50), MLN8054 and MLN8237 (51) (Table 1). However, these drugs differ in specificities for different family members.
Table 1.
Aurora kinase inhibitors in clinical trials*
| Inhibitor | AURKA inhibition | AURKB inhibition | AURKC inhibition | Manufacturer | Clinical Status |
|---|---|---|---|---|---|
| AZD1152 | + | + | − | Astra Zeneca | Phase I |
| VX-680 | + | + | + | Vertex/Merck | Discontinued |
| MLN8054 | + | + | − | Millinnium | Discontinued |
| MLN8237 | + | + | − | Millinnium | Phase II |
| PHA-680632 | + | + | Nerviano | Preclinical | |
| PHA-739358 | + | + | + | Nerviano | Phase II |
| Hesperidin | − | + | − | Boehringer-Ingelhiem | Preclinical |
| ZM447439 | + | + | − | Astra Zeneca | Phase I |
| JNJ-770621 | + | + | − | Johnson & Johnson | Preclinical |
| SU6668 | + | + | − | Pfizer | Discontinued |
| CCT129202 | + | + | + | Chroma Therapeutics Ltd. | Preclinical |
| AT9283 | + | + | − | Astrex Therapeutics | Phase I |
| MP529 | + | + | − | Supregen | Preclinical |
| SNS314 | + | + | + | Sunesis Pharmaceuticals | Phase I |
| R763 | − | + | − | Rigel Pharmaceuticals | Phase I |
| ENMD2076 | + | + | − | EntreMed | Phase I |
| XL228 | + | + | + | Exelixis | Phase I |
| TTP607 | + | + | + | TransTech Pharma | Phase I |
| PF-03814735 | + | + | − | Pfizer | Phase I |
| CYC116 | + | + | + | Cyclacel | Phase I |
The source of data is http://www.clinicaltrial.gov
AZD1152
AZD1152 (AstraZeneca) is a novel acetanilide-substituted pyrazole-aminoquinazoline drug that is converted rapidly to the active drug AZD1152 hydroxy-QPA (AZD1152-HQPA) in human plasma (52). AZD1152-HQPA is a specific inhibitor of the enzymatic activity of Aurora kinases, with selectivity for AURKB (IC50 of 0.37nM versus 1368nM for AURKB and AURKA kinases, respectively, Table 1) and had even less activity against a panel of greater than 50 other serine-threonine and tyrosine kinases including FLT3, JAK2 and Abl (52). AZD1152-HQPA in vitro induces chromosome misalignment, prevents cell division; and consequently, reduces cell viability and induces apoptosis (52). AZD1152 blocks phosphorylation of histone H3 and increases the population of cells with 4N/8N DNA content (53). Preclinical efficacy of AZD1152 in human leukemia cells was also recently demonstrated (53). It inhibited the proliferation of acute myeloid cell lines (HL-60, NB4, and MOML13), acute lymphocytic leukemia cell line (PALL-2), biphenotypic leukemia (MV4-11), acute eosinophilic leukemia (EOL-1), and the blast crisis of chronic myeloid leukemia K562 cells with an AC50 ranging from 3nM to 40nM, as measured by thymidine uptake on the day of culture. AZD1152 synergistically increased the antiproliferative effect of vincristine and daunorubicin (53). Recently, in a phase I clinical trial in solid tumor patients AZD1152 was reported to be tolerated up to 300mg when administered intravenously with significant disease stabilization reported in five of eight patients (54). AZD1152 was given as a weekly 2 hr infusion to patients with advanced pretreated solid tumors. Dose limiting toxicity was neutropenia with little non-hematologic toxicity. Despite the preclinical data suggesting a potent suppression of lymphocyte or platelet function by AZD1152, no lymphopenia or thrombocytopenia occurred because of exposure to the drug.
VX-680
VX-680 (Vertex/Merck) inhibits all three family members (Ki's of 0.6, 18 and 4.6nmol/L for Aurora-A, -B and -C, respectively Table 1). VX-680 causes accumulation of cells with 4N DNA content and inhibits the proliferation of a variety of tumor cells (50). VX-680 treatment results in cells with high levels of cyclin B1 and 4N DNA content 8 to 12 hrs after release from a G1-S block, indicating that cells can enter mitosis. VX-680 induces the accumulation of cells arrested in a pseudo-G1 state with 4N DNA content or the accumulation of cells with >4N DNA content, the latter population representing cells that exit mitosis and subsequently proceed through S-phase in the absence of cell division (50). VX-680 caused endoreduplication in absence of p53 function that was accompanied by loss of viability (55). However, in the presence of p53 function suppression of endoreduplication correlated with the induction of p21Waf1/Cip1. Recently, VX-680 was shown to be effective against multiple myeloma, especially in patients with (receptor for hyaluronan-mediated motility) RHAMM over-expression (50). More interestingly, VX-680 demonstrated potent anticancer activity in chronic myeloid leukemia (CML) harboring imatinib-resistant T351I and dasatinib-resistant V299L Bcr-Abl mutations (56). Recently, it was reported that VX-680 induced apoptosis preferentially in the leukemic blasts with high AURKA expression, but not in normal bone marrow mononuclear cells (BMMCs) or AURKA low acute myeloid leukemia (AML) cells, suggesting a potential pharmacologic window for VX-680 therapeutic response in AURKA-high AMLs (57). Moreover, Haung et al (57) reported reduction of phosphorylated AKT-1, activation of cellular caspases, and an increase in the Bax/Bcl-2 ratio, a known favorable survival factor in AML, by VX-680 treatment and synergistic enhancement in the cytotoxic effect of VP16 with VX-680 in AML cells. VX-680 inhibits phosphorylation of histone H3 on Ser 10, causing a marked reduction in tumor size in human AML (HL-60) xenograft model treated with 75mg/Kg twice a day for 13 days. In preclinical models, VX-680 blocked tumor xenograft growth and induced tumor regressions (50). In its first phase I clinical trial, VX-680 was given as a continuous i.v. infusion over several days to patients with previously treated solid tumors. The principal dose-limiting toxicity (DLT) was grade 3 neutropenia, accompanied by some nonspecific side effects, including; low-grade nausea and fatigue. Disease stabilization was observed in one patient with lung cancer and in one patient with pancreatic cancer. This inhibitor entered in Phase II clinical trial on patients with chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia (Vertex Pharmaceuticals Reports 2007 Pipeline Progress). It has to be mentioned, however, that Merck has recently suspended the enrollment in clinical trials of the Aurora kinase inhibitor, VX-680, pending a full analysis of all safety data for the drug. The decision was based on preliminary safety data, in which a QTc prolongation was observed in one patient. Patients currently enrolled in these trials may continue to be treated with VX680 with additional monitoring for QTc prolongation.
MLN8054
MLN8054 (Millennium) is a recently discovered ATP-competitive Aurora Kinase family inhibitor; it is highly specific to AURKA but at a higher concentration can inactivate AURKB (Table 1). MLN8054 is >40-fold more selective for AURKA than AURKB, it does not degrade or down-regulate AURKA but inhibits its phosphorylation (T288) (51). MLN8054, at higher concentrations, inhibits histone H3 phosphorylation; an indication for AURKB inhibition. It induces abnormal mitotic spindles, G2/M accumulation, cell death through apoptosis, and phenotypes consistent with AURKA inhibition (58). Cells treated with MLN8054 develop an abnormal DNA content (58). These abnormalities with MLN8054 treatment become more pronounced with time. In contrast to various pan-Aurora kinases, MLN8054 is more AURKA specific due to its ability to inhibit T288 phosphorylation, increasing in the mitotic cells in vivo (51). We recently reported (37) induction of TAp73 at protein level along with various pro-apoptotic genes, PUMA, NOXA and p21 by MLN8054 in different p53 deficient tumor cells. p53 deficient cells are resistant to chemotherapy. This observation whereby MLN8054 induced TAp73 could prove to be beneficial in targeting tumors lacking p53.
MLN8237
MLN8237 (Millennium) is a second-generation AURKA inhibitor and has recently entered phase I/II clinical trials (Table 1). It inhibits Aurora-A with an IC50 of 1nM in biochemical assays and has 200-fold selectivity for AURKA over AURKAB in cell assays. A broad screen of receptors and ion channels showed no significant cross-reactivity. The compound blocks the growth of multiple tumor cell lines with GI50 values as low as 16nM. Growth inhibition is associated with mitotic spindle abnormalities, accumulation of cells in mitosis, polyploidy, and apoptosis. It is orally available and rapidly absorbed. At effective doses a transient inhibition of histone H3 phosphorylation is observed (consistent with Aurora-B inhibition being dominant) followed by marked elevation of histone H3 phosphorylation (consistent with Aurora-A inhibition being dominant). Maximum in vivo efficacy, in multiple xenografts, has been achieved with oral doses of 20mg/kg given twice a day for 21 consecutive days, although other regimens are also effective. MLN8237 in combination Rituximab was found to reduce tumor burden in an additive and/or synergistic mechanism in multiple Diffuse Large B-cell Lymphoma tumor models (59).
PHA-680632
PHA-680632 (Nerviano) is a potent inhibitor of Aurora kinase family members with IC50s of 27, 135 and 120nmol/L for Aurora-A, -B and -C, respectively; and shows the strongest cross reactivity for FGFR1 (60). PHA-608632 is reported to have a potent antiproliferative activity in a wide range of cancer cell lines (60). PHA-680632 inhibits AURKA autophosphorylation at T288 and AURKB mediated phosphorylation of histone H3 (60, 61) phenotypes, which are consistent with the inhibition of AURKA and AURKB. Inhibition of AURKA by PHA-680632 in p53-/- HCT116 cells followed by radiation treatment enhanced response in apoptosis (61). This additive effect of PHA-680632 and IR radiation delayed tumor growth in xenografts model (61), inhibiting colony formation and induced polyploidy. PHA680632 brought about additive interaction with radiation in terms of induced cell death in p53 non-functional cells. Such additivity may be beneficial in chemo-radiotherapeutic combinations. PHA680632 and radiotherapy might be used concomitantly or in close temporal proximity, potentially without acute or late healthy tissue complications.
PHA-739358
PHA-739358 (Nerviano) is more potent than its predecessor PHA-680632 and inhibits all three Aurora Kinases A, B and C with IC50s of 13, 79 and 61nmol/L, respectively (62). It has a high cross-reactivity for other kinases mutated or over-expressed in cancers like Ret, Trk-A and Abl. It inhibits phosphorylation of AURKA on T288 and reduces histone H3 phosphorylation indicating AURKB inhibition (62). Recently, PHA-739358 has been reported to show strong antiproliferative action in chronic myeloid leukemia (CML) cells and is effective against Imatinib-resistant Bcr-Abl mutations including T3151 (63) that could lead to its use as a therapeutic target for myeloid leukemia patients, especially those who developed resistance to Gleevec. PHA-739358 is currently being evaluated in a phase II clinical trial in CML, including patients with T315I mutation. PHA-739358 has significant antitumor activity in transgenic tumor models with a favorable preclinical safety profile; principal target organs of PHA-739358 are the hemolymphopoietic system, gastrointestinal tract, male reproductive organs and kidneys. Renal effects, however, are only seen at high drug exposure.
Hesperidin
Hesperidin (Boehringer-Ingelheim) is specific for AURKB as indicated by the reduction of histone H3 phosphorylation and exhibiting the similar phenotype to AURKB knockdown (11). It has cross reactivity for six other kinases (AMPK, Lck, MKK1, MAPKAP-K1, CHK1, and PHK) (11) and proved useful to understand the biology of AURKB function. Hesperidin impairs the localization of checkpoint proteins such as BUB1 and BUBR1 to kinetochore, and induces cytokinesis and polyploidy. Hesperidin was instrumental in understanding the role of AURKB in syntelic orientation of chromosomes and spindle assemble checkpoint.
ZM447439
ZM447439 (AstraZeneca) inhibits Aurora-A and -B with IC50 values of 110 and 130nM resulting in the reduction of phosphorylation of histone H3 (49). ZM447439 treatment causes defects in chromosome alignment, segregation, and cytokinesis; most likely by interfering with the spindle integrity checkpoint (64). Cells treated with ZM447439 pass through S-phase, fail to divide and then enter a second S-phase due to failure in chromosome alignment and segregation. In p53 deficient cells ZM447439 enhanced endoreduplication, compared to p53 proficient cells, suggesting that p53-independent mechanisms may also affect ZM447439-induced tetraploidization. The effects mediated by ZM447439 (reduction in H3 phosphorylation, inhibition of kinetochore localization of BUBR1, Mad2, and Cenp-E) are characteristic to AURKB inhibition rather than AURKA (49). ZM447439 treatment on xenopus eggs exhibited no detectable effects on frequency or amplitude of oscillations in cdc2, cdc25, and MAPK activities (64). ZM447439 induces apoptosis in a concentration- and time-dependent manner, following polyploidization. Moreover, apoptosis induced by inhibition of Aurora kinases occurs via the mitochondrial pathways, depending on both Bak and Bax. Apoptosis as a secondary event in response to Aurora kinase inhibitors, depends not only on polyploidization, but also on the intracellular apoptotic signaling of treated cells. Thus, therapeutic options that stimulate apoptosis may act synergistically with Aurora kinase inhibitors to potentiate their anti-tumoral effects.
JNJ-770621
JNJ-770621 (Johnson and Johnson) is a potent cell cycle inhibitor targeting cyclin dependent kinases (CDK) and Aurora Kinases. JNJ-770621 has specificity for AURKA and AURKB in addition to CDK1, CDK2, CDK4, and CDK6 (65). The phenotypes exhibited by JNJ-770621 treatment are similar to AURKB inhibition, for example; decrease in the phosphorylation of histone H3, compromised spindle checkpoint function, and endoreduplication. JNJ-770621 was reported to be a substrate of ATP-binding cassette transporter family member (ABCG2) in HeLa cells selected for resistance to JNJ-770621 (66). JNJ-7706621 shows potent antiproliferative activity in cancer cells regardless of p53, retinoblastoma status, or P-glycoprotein expression level, and is several fold less potent at inhibiting normal cell growth. The principal effects of this compound on cells stem from its ability to delay transit through the cell cycle and induce a G2-M arrest.
SU6668
SU6668 (Pfizer) was basically characterized as an ATP-competitive inhibitor of PDGFR, VEGFR2 and FGFR1 RTKs in vitro (67); however, it has been recently shown to inhibit Aurora kinases (68). SU6668 inhibits AURKA and AURKB, as evidenced by destabilizing the microtubule organization and suppression in the phosphorylation of histone H3, respectively (68). SU6668 induces defects in centrosome organization, spindle assembly and histone modification; and as a consequence, leads to an arrest in cell cycle progression (68). SU6668 was reported as an Aurora kinase inhibitor only in a single study, its development was discontinued in favor of a more potent inhibitor of VEGF receptors, sunitinib, which makes its use unlikely on a clinical level.
CCT129202
CCT129202 is an ATP-competitive pan-Aurora Kinase inhibitor inhibiting all three family members Aurora-A, -B, and -C with IC50 values as 0.042, 0.198 and 0.227, respectively. It does not affect protein levels of Aurora-A and -B at IC50, but at higher concentrations (>5x IC50). CCT129202 caused G2-M accumulation and induces formation of abnormal mitotic spindles with various degrees of chromosome alignment defects (69). The molecular mechanism of the action of CCT129202 is consistent with the inhibition of Aurora -A and -B as evidenced by the reduction in the phosphorylation of histone H3 and p53 stabilization, respectively. CCT129202 has been reported to affect the p21/Rb/E2F pathway and down-regulate thymidine kinase 1 (TK1) (69). Antitumor activity has also been reported in human tumor xenografts. Taken into account that TK1 is required for [18F] FLT uptake in vivo (70), Chan et al (69) have effectively shown that [18F] FLT-PET can be used to monitor the biological effects of CCT129202 in vivo and reported reduction in tumor [18F] FLT retention using noninvasive PET imaging.
AT9283
AT9283 (Astrex Therapeutics), a multitargeted kinase inhibitor, inhibits several closely related tyrosine and serine/threonine kinases with an IC50 of <10nM including Aurora-A and -B, JAK and ABL. Exposure of solid tumor cell lines to AT9283 in vitro induces an “aurora inhibitory” phenotype. Cell survival decreases with increased duration of exposure. A phase I dose escalation study has been reported using a 72 hr continuous i.v. infusion schedule repeated three times weekly according to a standard “3+3” design (71). Thirty-three patients with a median age of 61 (range 33 to 76 years) had been treated in this study. The maximum tolerated dose (MTD) was 9mg/m2/day. Treatment was well tolerated with febrile neutropenia the only dose limiting toxicity. Other adverse events considered possibly related to AT9283 were reversible and included gastrointestinal disturbance and fatigue. Biological evidence of Aurora-B inhibition manifest as a reduction in histone H3 phosphorylation in skin biopsies during the infusion was observed at all dose levels (71). A plateau steady state plasma concentration of AT9283 was reported to be achieved within 24 hrs of initiating drug infusion at all dose levels and exposure increased linearly with dose. Seven patients received an initial oral dose of AT9283 as an aqueous solution in a fasting state at a dose of 0.9mg mg/m2 one week prior to starting i.v. treatment. Interim pharmacokinetic analysis indicated that the median oral bioavailability was 27% (range 17-45%) The best response to treatment was a partial response in one patient with NSCLC (ongoing). An additional four patients received at least six cycles of therapy (squamous cell carcinoma of the lung, adenocarcinoma of the esophagus and colorectal carcinoma) with a best response of stable disease. The MTD of AT9283 when administered as a 72 hr continuous i.v. infusion was 9mg/m2/day.
SNS314
SNS314 (Sunesis Pharmaceuticals) is a pan-Aurora inhibitor with good affinity against all three isoforms (72) (IC50 for Aurora-A, -B, and -C of 9, 31, and 3nM, respectively) and has selectivity over the majority of kinases (199 out of 219 kinases had IC50 values greater than 1 µM). In keeping with other pan-Aurora inhibitors, SNS314 potently blocks proliferation in a diverse panel of human cancer cell-lines (72) (IC50 values in the range 1.8-24nM) and leads to accumulation of cells with >4N DNA content. In xenograft models the compound blocks tumor growth at doses of 50-170mg/kg administered i.p. twice a week for 3 weeks. Apoptosis of tumor tissue along with inhibition of histone H3 phosphorylation (Ser10) in tumor, skin, and bone marrow is observed SNS314 is currently being assessed in a dose-escalating phase I study in advanced solid tumors as an i.v. infusion given once a week for 3 weeks.
CYC116
CYC116 (Cyclacel) is a pan-Aurora kinase and VEGFR2 inhibitor (IC50 for Aurora-A,-B and -C of 44, 19 and 65nM, respectively) (73). It inhibits the spindle checkpoint and cytokinesis, resulting in polyploidy and induction of apoptosis (73). It has antitumor activity in various human solid tumors and leukemia xenograft models. CYC116 is presently in phase 1 clinical trail in advanced solid tumors and is orally bioavailable.
PF-03814735
PF-03814735 (Pfizer) is a novel oral ATP-competitive, reversible inhibitor of Aurora-A and -B kinases with a broad spectrum of preclinical activity (74) (IC50 for Aurora-A and -B of 5nM and 0.8nM, respectively). In a study, 20 patients have received a median of 2 cycles (1-4) across 7 dose levels from 5-100mg/day for five days (74). Tumor types included in the study were colorectal {5}, breast {3}, NSCLC {4}, SCLC {2}, bladder, melanoma, ovarian, renal, head and neck, and cancer of unknown primary (1 each). The dose was doubled in single patient cohorts until treatment-related grade 2 diarrhea occurred in one patient at 40 mg/day. Afterwards, cohorts included 3-7 patients with 20-50% dose increments per cohort. In the first 16 patients, the most common treatment-related adverse events were mild to moderate diarrhea (50%), vomiting (25%), anorexia, fatigue, and nausea (19% each). Dose-limiting febrile neutropenia was observed in 2/7 patients treated at 100mg/day. The maximum tolerated dose was defined as 80mg/day for five days. This dose level is currently being expanded to obtain proof-of-mechanism data at the recommended phase II dose.
Concluding Remarks
The principal goal in the development of Aurora kinase inhibitors is to assess whether or not the administration of these small molecules to patients will yield a clinical benefit. For this reason, it is essential to answer several different questions, such as those regarding the effect of these inhibitors on other kinase proteins, the effect of the same drugs on the three different members of the Aurora kinase family, and the protein involved in Aurora kinase inhibition. For example, the interaction between Aurora kinase and p53 might select a patient for inclusion in the study according to the p53 status. On the other hand, recent studies indicate that AURKA inhibitors can activate p73-dependent apoptosis raising the possibility that these inhibitors may function irrespective of the p53 status. Furthermore, it will be important to identify a safe dose for target inhibition in humans, tumor types that most likely respond to these drugs, reversibility of the effect on normal cells, and the dependence on this dose and duration of exposure. Neutropenia being the primary dose limiting phase I toxicity in several studies suggest that these agents have collateral anti-proliferation toxicity on the bone marrow. Aurora kinase inhibitors induce polyploidy in normal mammary epithelial cell cultures (49), thus raising the issue of long-term clinical effects. Clinical tolerability has generally been good, however, and no severe mucositis, peripheral neuropathy, diarrhea, or alopecia has been observed. Additional parameters include the toxicity effects observed in patients, effect of these drugs on disease-free and overall survival, and the effect of these drugs when used with other chemotherapy agents. These drugs may be particularly effective in combination with drugs that depend on the spindle checkpoint such as taxanes and others. However, the dose-limiting cytopenias seen with AURKA inhibitors so far mandate careful phase I studies to assess the safest combinations of these drugs with potentially less overlapping toxicity. One question for the future will therefore be: are there tumors that are exceptionally sensitive to such compounds, enabling delivery of minimally toxic doses that have significant antitumor effects?. It is clear that we are entering a new era in anti-mitotic therapy with the identification and now clinical translation of new targets in mitosis beyond tubulin, but many questions remain with regard to Aurora function. The answers will be of great interest, not only to basic researchers but to clinicians and patients as well.
Both pharmaceutical companies as well as clinicians presently consider Aurora kinases “hot property.” Pharmaceutical companies are investing in the development of different inhibitors to target Aurora kinases. Correlation of AURKA with tumor progression, interaction with tumor suppressors such as p53, BRCA1, p73, GSK3B, and lats2 is a clear indication of a real connection to oncogenesis. For a clinician, the fact that small molecule Aurora kinase inhibitors could be effective at killing cancer cells has shed more light on these kinases; however, it seems appropriate to voice a cautionary note as to the overall efficacy of such inhibitors in cancer treatment. Although aurora inhibitors may trigger apoptosis in a proportion of cells and lead to the arrest of tumor growth in model systems, it is notable that these treatments induce a modest increase in the proportion of apoptotic cells. Nothing is known about how the inhibitors cause cell death (e.g. is it the result of polyploidization), to what extent this happens in vivo and whether the long-term outcome of their inhibition is favorable for maintaining long-term remission. At face value, inhibition of any kinase required for stable chromosome inheritance is dangerous because of a greater probability of genetic heterogeneity, hence the potential for tumor evolution. Undoubtedly, massive chromosome loss does, in the majority of cells, lead to cell death, but at what point does increased chromosome instability trigger cell death pathways? In addition, AURKB is required for cytokinesis. Its inhibition leads to polyploidization – a condition that may result in the survival of a severely aneuploidy cancerous cell. Very little is understood of how this is sensed in the cell. There is no doubt that studies are required to ascertain the long-term effects of Aurora kinase inhibitors administration in a suitable model organism. Never the less, the frequent over-expression of Aurora kinases in solid tumors and their contribution to biological processes and signaling pathways, critical for cancer cells, highlight them as the rising stars in targeted therapy and the future of personalized therapy in cancer.
Acknowledgments
This work was supported in part by the Vanderbilt CTSA grant UL1 RR024975 from NCCR/NIH and by the National Cancer Institute Grant CA131225 (WER). The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute or Vanderbilt University Medical Center.
References
- 1.Adams RR, Carmena M, Earnshaw WC. Chromosomal passengers and the (aurora) ABCs of mitosis. Trends Cell Biol. 2001;11:49–54. doi: 10.1016/s0962-8924(00)01880-8. [DOI] [PubMed] [Google Scholar]
- 2.Francisco L, Wang W, Chan CS. Type 1 protein phosphatase acts in opposition to IpL1 protein kinase in regulating yeast chromosome segregation. Mol Cell Biol. 1994;14:4731–40. doi: 10.1128/mcb.14.7.4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meraldi P, Honda R, Nigg EA. Aurora kinases link chromosome segregation and cell division to cancer susceptibility. Curr Opin Genet Dev. 2004;14:29–36. doi: 10.1016/j.gde.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 4.Mendez R, Hake LE, Andresson T, Littlepage LE, Ruderman JV, Richter JD. Phosphorylation of CPE binding factor by Eg2 regulates translation of c-mos mRNA. Nature. 2000;404:302–7. doi: 10.1038/35005126. [DOI] [PubMed] [Google Scholar]
- 5.Giet R, Prigent C. Aurora/Ipl1p-related kinases, a new oncogenic family of mitotic serine-threonine kinases. J Cell Sci. 1999;112(Pt 21):3591–601. doi: 10.1242/jcs.112.21.3591. [DOI] [PubMed] [Google Scholar]
- 6.Berdnik D, Knoblich JA. Drosophila Aurora-A is required for centrosome maturation and actin-dependent asymmetric protein localization during mitosis. Curr Biol. 2002;12:640–7. doi: 10.1016/s0960-9822(02)00766-2. [DOI] [PubMed] [Google Scholar]
- 7.Hirota T, Kunitoku N, Sasayama T, et al. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114:585–98. doi: 10.1016/s0092-8674(03)00642-1. [DOI] [PubMed] [Google Scholar]
- 8.Marumoto T, Honda S, Hara T, et al. Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem. 2003;278:51786–95. doi: 10.1074/jbc.M306275200. [DOI] [PubMed] [Google Scholar]
- 9.Eyers PA, Erikson E, Chen LG, Maller JL. A novel mechanism for activation of the protein kinase Aurora A. Curr Biol. 2003;13:691–7. doi: 10.1016/s0960-9822(03)00166-0. [DOI] [PubMed] [Google Scholar]
- 10.Anderson K, Yang J, Koretke K, et al. Binding of TPX2 to Aurora A alters substrate and inhibitor interactions. Biochemistry. 2007;46:10287–95. doi: 10.1021/bi7011355. [DOI] [PubMed] [Google Scholar]
- 11.Hauf S, Cole RW, LaTerra S, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281–94. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terada Y, Tatsuka M, Suzuki F, Yasuda Y, Fujita S, Otsu M. AIM-1: a mammalian midbody-associated protein required for cytokinesis. Embo J. 1998;17:667–76. doi: 10.1093/emboj/17.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolton MA, Lan W, Powers SE, McCleland ML, Kuang J, Stukenberg PT. Aurora B kinase exists in a complex with survivin and INCENP and its kinase activity is stimulated by survivin binding and phosphorylation. Mol Biol Cell. 2002;13:3064–77. doi: 10.1091/mbc.E02-02-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bishop JD, Schumacher JM. Phosphorylation of the carboxyl terminus of inner centromere protein (INCENP) by the Aurora B Kinase stimulates Aurora B kinase activity. J Biol Chem. 2002;277:27577–80. doi: 10.1074/jbc.C200307200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keen N, Taylor S. Mitotic drivers--inhibitors of the Aurora B Kinase. Cancer Metastasis Rev. 2009;28:185–95. doi: 10.1007/s10555-009-9184-9. [DOI] [PubMed] [Google Scholar]
- 16.Bourhis E, Hymowitz SG, Cochran AG. The mitotic regulator Survivin binds as a monomer to its functional interactor Borealin. J Biol Chem. 2007;282:35018–23. doi: 10.1074/jbc.M706233200. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka TU, Rachidi N, Janke C, et al. Evidence that the Ipl1-Sli15 (Aurora kinase-INCENP) complex promotes chromosome bi-orientation by altering kinetochore-spindle pole connections. Cell. 2002;108:317–29. doi: 10.1016/s0092-8674(02)00633-5. [DOI] [PubMed] [Google Scholar]
- 18.Kimura M, Matsuda Y, Yoshioka T, Okano Y. Cell cycle-dependent expression and centrosome localization of a third human aurora/Ipl1-related protein kinase, AIK3. J Biol Chem. 1999;274:7334–40. doi: 10.1074/jbc.274.11.7334. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka T, Kimura M, Matsunaga K, Fukada D, Mori H, Okano Y. Centrosomal kinase AIK1 is overexpressed in invasive ductal carcinoma of the breast. Cancer Res. 1999;59:2041–4. [PubMed] [Google Scholar]
- 20.Sakakura C, Hagiwara A, Yasuoka R, et al. Tumour-amplified kinase BTAK is amplified and overexpressed in gastric cancers with possible involvement in aneuploid formation. Br J Cancer. 2001;84:824–31. doi: 10.1054/bjoc.2000.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goepfert TM, Adigun YE, Zhong L, Gay J, Medina D, Brinkley WR. Centrosome amplification and overexpression of aurora A are early events in rat mammary carcinogenesis. Cancer Res. 2002;62:4115–22. [PubMed] [Google Scholar]
- 22.Farruggio DC, Townsley FM, Ruderman JV. Cdc20 associates with the kinase aurora2/Aik. Proc Natl Acad Sci U S A. 1999;96:7306–11. doi: 10.1073/pnas.96.13.7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou H, Kuang J, Zhong L, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20:189–93. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]
- 24.Jeng YM, Peng SY, Lin CY, Hsu HC. Overexpression and amplification of Aurora-A in hepatocellular carcinoma. Clin Cancer Res. 2004;10:2065–71. doi: 10.1158/1078-0432.ccr-1057-03. [DOI] [PubMed] [Google Scholar]
- 25.Sasaki O, Kido K, Nagahama S. DNA ploidy, Ki-67 and p53 as indicators of lymph node metastasis in early gastric carcinoma. Anal Quant Cytol Histol. 1999;21:85–8. [PubMed] [Google Scholar]
- 26.Sturgis CD, Caraway NP, Johnston DA, Sherman SI, Kidd L, Katz RL. Image analysis of papillary thyroid carcinoma fine-needle aspirates: significant association between aneuploidy and death from disease. Cancer. 1999;87:155–60. doi: 10.1002/(sici)1097-0142(19990625)87:3<155::aid-cncr9>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 27.Abad M, Ciudad J, Rincon MR, et al. DNA aneuploidy by flow cytometry is an independent prognostic factor in gastric cancer. Anal Cell Pathol. 1998;16:223–31. doi: 10.1155/1998/158243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pihan GA, Purohit A, Wallace J, Malhotra R, Liotta L, Doxsey SJ. Centrosome defects can account for cellular and genetic changes that characterize prostate cancer progression. Cancer Res. 2001;61:2212–9. [PubMed] [Google Scholar]
- 29.Pihan GA, Purohit A, Wallace J, et al. Centrosome defects and genetic instability in malignant tumors. Cancer Res. 1998;58:3974–85. [PubMed] [Google Scholar]
- 30.Lingle WL, Salisbury JL. Altered centrosome structure is associated with abnormal mitoses in human breast tumors. Am J Pathol. 1999;155:1941–51. doi: 10.1016/S0002-9440(10)65513-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ouchi M, Fujiuchi N, Sasai K, et al. BRCA1 phosphorylation by Aurora-A in the regulation of G2 to M transition. J Biol Chem. 2004;279:19643–8. doi: 10.1074/jbc.M311780200. [DOI] [PubMed] [Google Scholar]
- 32.Yang H, Ou CC, Feldman RI, Nicosia SV, Kruk PA, Cheng JQ. Aurora-A kinase regulates telomerase activity through c-Myc in human ovarian and breast epithelial cells. Cancer Res. 2004;64:463–7. doi: 10.1158/0008-5472.can-03-2907. [DOI] [PubMed] [Google Scholar]
- 33.Jiang Y, Zhang Y, Lees E, Seghezzi W. AuroraA overexpression overrides the mitotic spindle checkpoint triggered by nocodazole, a microtubule destabilizer. Oncogene. 2003;22:8293–301. doi: 10.1038/sj.onc.1206873. [DOI] [PubMed] [Google Scholar]
- 34.Katayama H, Sasai K, Kawai H, et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. 2004;36:55–62. doi: 10.1038/ng1279. [DOI] [PubMed] [Google Scholar]
- 35.Liu Q, Kaneko S, Yang L, et al. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J Biol Chem. 2004;279:52175–82. doi: 10.1074/jbc.M406802200. [DOI] [PubMed] [Google Scholar]
- 36.Dar AA, Zaika A, Piazuelo MB, et al. Frequent overexpression of Aurora Kinase A in upper gastrointestinal adenocarcinomas correlates with potent antiapoptotic functions. Cancer. 2008;112:1688–98. doi: 10.1002/cncr.23371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dar AA, Belkhiri A, Ecsedy J, Zaika A, El-Rifai W. Aurora kinase A inhibition leads to p73-dependent apoptosis in p53-deficient cancer cells. Cancer Res. 2008;68:8998–9004. doi: 10.1158/0008-5472.CAN-08-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thottassery JV, Westbrook L, Someya H, Parker WB. c-Abl-independent p73 stabilization during gemcitabine- or 4'-thio-beta-D-arabinofuranosylcytosine-induced apoptosis in wild-type and p53-null colorectal cancer cells. Mol Cancer Ther. 2006;5:400–10. doi: 10.1158/1535-7163.MCT-05-0409. [DOI] [PubMed] [Google Scholar]
- 39.Khwaja A. Akt is more than just a Bad kinase. Nature. 1999;401:33–4. doi: 10.1038/43354. [DOI] [PubMed] [Google Scholar]
- 40.Guan Z, Wang XR, Zhu XF, et al. Aurora-A, a negative prognostic marker, increases migration and decreases radiosensitivity in cancer cells. Cancer Res. 2007;67:10436–44. doi: 10.1158/0008-5472.CAN-07-1379. [DOI] [PubMed] [Google Scholar]
- 41.Dar AA, Belkhiri A, El-Rifai W. The aurora kinase A regulates GSK-3beta in gastric cancer cells. Oncogene. 2009;28:866–75. doi: 10.1038/onc.2008.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biggins S, Murray AW. The budding yeast protein kinase Ipl1/Aurora allows the absence of tension to activate the spindle checkpoint. Genes Dev. 2001;15:3118–29. doi: 10.1101/gad.934801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tatsuka M, Katayama H, Ota T, et al. Multinuclearity and increased ploidy caused by overexpression of the aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Cancer Res. 1998;58:4811–6. [PubMed] [Google Scholar]
- 44.Katayama H, Ota T, Jisaki F, et al. Mitotic kinase expression and colorectal cancer progression. J Natl Cancer Inst. 1999;91:1160–2. doi: 10.1093/jnci/91.13.1160. [DOI] [PubMed] [Google Scholar]
- 45.Meraldi P, Honda R, Nigg EA. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. Embo J. 2002;21:483–92. doi: 10.1093/emboj/21.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ota T, Suto S, Katayama H, et al. Increased mitotic phosphorylation of histone H3 attributable to AIM-1/Aurora-B overexpression contributes to chromosome number instability. Cancer Res. 2002;62:5168–77. [PubMed] [Google Scholar]
- 47.Kanda A, Kawai H, Suto S, et al. Aurora-B/AIM-1 kinase activity is involved in Ras-mediated cell transformation. Oncogene. 2005;24:7266–72. doi: 10.1038/sj.onc.1208884. [DOI] [PubMed] [Google Scholar]
- 48.Pollard JR, Mortimore M. Discovery and development of aurora kinase inhibitors as anticancer agents. J Med Chem. 2009;52:2629–51. doi: 10.1021/jm8012129. [DOI] [PubMed] [Google Scholar]
- 49.Ditchfield C, Johnson VL, Tighe A, et al. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–80. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harrington EA, Bebbington D, Moore J, et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10:262–7. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- 51.Manfredi MG, Ecsedy JA, Meetze KA, et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci U S A. 2007;104:4106–11. doi: 10.1073/pnas.0608798104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilkinson RW, Odedra R, Heaton SP, et al. AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin Cancer Res. 2007;13:3682–8. doi: 10.1158/1078-0432.CCR-06-2979. [DOI] [PubMed] [Google Scholar]
- 53.Yang J, Ikezoe T, Nishioka C, et al. AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo. Blood. 2007;110:2034–40. doi: 10.1182/blood-2007-02-073700. [DOI] [PubMed] [Google Scholar]
- 54.Schellens JH, Boss D, Witteveen PO, et al. Phase I and pharmacological study of the novel aurora kinase inhibitor AZD1152. Journal of Clinical Oncology. 2006;24 abstr 3008. [Google Scholar]
- 55.Gizatullin F, Yao Y, Kung V, Harding MW, Loda M, Shapiro GI. The Aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Res. 2006;66:7668–77. doi: 10.1158/0008-5472.CAN-05-3353. [DOI] [PubMed] [Google Scholar]
- 56.Shah NP, Skaggs BJ, Branford S, et al. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J Clin Invest. 2007;117:2562–9. doi: 10.1172/JCI30890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang XF, Luo SK, Xu J, et al. Aurora kinase inhibitory VX-680 increases Bax/Bcl-2 ratio and induces apoptosis in Aurora-A-high acute myeloid leukemia. Blood. 2008;111:2854–65. doi: 10.1182/blood-2007-07-099325. [DOI] [PubMed] [Google Scholar]
- 58.Hoar K, Chakravarty A, Rabino C, et al. MLN8054, a small-molecule inhibitor of Aurora A, causes spindle pole and chromosome congression defects leading to aneuploidy. Mol Cell Biol. 2007;27:4513–25. doi: 10.1128/MCB.02364-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang M, Huck J, Hyer M, Ecsedy J, Manfredi M. Effect of Aurora A kinase inhibitor MLN8237 combined with rituximab on antitumor activity in preclinical B-cell non-Hodgkin's lymphoma models. Journal of Clinical Oncology. 2009;27 abstr 8553. [Google Scholar]
- 60.Soncini C, Carpinelli P, Gianellini L, et al. PHA-680632, a novel Aurora kinase inhibitor with potent antitumoral activity. Clin Cancer Res. 2006;12:4080–9. doi: 10.1158/1078-0432.CCR-05-1964. [DOI] [PubMed] [Google Scholar]
- 61.Tao Y, Zhang P, Frascogna V, et al. Enhancement of radiation response by inhibition of Aurora-A kinase using siRNA or a selective Aurora kinase inhibitor PHA680632 in p53-deficient cancer cells. Br J Cancer. 2007;97:1664–72. doi: 10.1038/sj.bjc.6604083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carpinelli P, Ceruti R, Giorgini ML, et al. PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol Cancer Ther. 2007;6:3158–68. doi: 10.1158/1535-7163.MCT-07-0444. [DOI] [PubMed] [Google Scholar]
- 63.Gontarewicz A, Balabanov S, Keller G, et al. Simultaneous targeting of Aurora kinases and Bcr-Abl kinase by the small molecule inhibitor PHA-739358 is effective against Imatinib-resistant BCR-ABL mutations including T315I. Blood. 2008 doi: 10.1182/blood-2007-09-113175. [DOI] [PubMed] [Google Scholar]
- 64.Gadea BB, Ruderman JV. Aurora kinase inhibitor ZM447439 blocks chromosome-induced spindle assembly, the completion of chromosome condensation, and the establishment of the spindle integrity checkpoint in Xenopus egg extracts. Mol Biol Cell. 2005;16:1305–18. doi: 10.1091/mbc.E04-10-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Emanuel S, Rugg CA, Gruninger RH, et al. The in vitro and in vivo effects of JNJ-7706621: a dual inhibitor of cyclin-dependent kinases and aurora kinases. Cancer Res. 2005;65:9038–46. doi: 10.1158/0008-5472.CAN-05-0882. [DOI] [PubMed] [Google Scholar]
- 66.Seamon JA, Rugg CA, Emanuel S, et al. Role of the ABCG2 drug transporter in the resistance and oral bioavailability of a potent cyclin-dependent kinase/Aurora kinase inhibitor. Mol Cancer Ther. 2006;5:2459–67. doi: 10.1158/1535-7163.MCT-06-0339. [DOI] [PubMed] [Google Scholar]
- 67.Laird AD, Vajkoczy P, Shawver LK, et al. SU6668 is a potent antiangiogenic and antitumor agent that induces regression of established tumors. Cancer Res. 2000;60:4152–60. [PubMed] [Google Scholar]
- 68.Godl K, Gruss OJ, Eickhoff J, et al. Proteomic characterization of the angiogenesis inhibitor SU6668 reveals multiple impacts on cellular kinase signaling. Cancer Res. 2005;65:6919–26. doi: 10.1158/0008-5472.CAN-05-0574. [DOI] [PubMed] [Google Scholar]
- 69.Chan F, Sun C, Perumal M, et al. Mechanism of action of the Aurora kinase inhibitor CCT129202 and in vivo quantification of biological activity. Mol Cancer Ther. 2007;6:3147–57. doi: 10.1158/1535-7163.MCT-07-2156. [DOI] [PubMed] [Google Scholar]
- 70.Barthel H, Perumal M, Latigo J, et al. The uptake of 3'-deoxy-3'-[18F]fluorothymidine into L5178Y tumours in vivo is dependent on thymidine kinase 1 protein levels. Eur J Nucl Med Mol Imaging. 2005;32:257–63. doi: 10.1007/s00259-004-1611-0. [DOI] [PubMed] [Google Scholar]
- 71.Kristeleit R, Calvert H, Arkenau H, et al. A phase I study of AT9283, an aurora kinase inhibitor, in patients with refractory solid tumors. J Clin Oncol. 2009;27:2566. [Google Scholar]
- 72.Arbitrario JP, Belmont BJ, Evanchik MJ, et al. SNS-314, a pan-Aurora kinase inhibitor, shows potent anti-tumor activity and dosing flexibility in vivo. Cancer Chemother Pharmacol. 2009 doi: 10.1007/s00280-009-1076-8. [DOI] [PubMed] [Google Scholar]
- 73.Griffiths G, Scaerou F, Midgley C, et al. Anti-tumor activity of CYC116, a novel small molecule inhibitor of Aurora kinases and VEGFR2. AACR Meeting Abstracts. 2008:5644. [Google Scholar]
- 74.Jones SF, Burris HA, III, Dumez H, et al. Phase I accelerated dose-escalation, pharmacokinetic (PK) and pharmacodynamic study of PF-03814735, an oral aurora kinase inhibitor, in patients with advanced solid tumors: Preliminary results. Journal of Clinical Oncolgy. 2008;26:2517. [Google Scholar]
- 75.Yang H, He L, Kruk P, Nicosia SV, Cheng JQ. Aurora-A induces cell survival and chemoresistance by activation of Akt through a p53-dependent manner in ovarian cancer cells. Int J Cancer. 2006;119:2304–12. doi: 10.1002/ijc.22154. [DOI] [PubMed] [Google Scholar]