

Abstract
In cystic fibrosis (CF) respiratory failure caused by progressive airway obstruction and tissue damage is primarily a result of the aberrant inflammatory responses to lung infections with Pseudomonas aeruginosa. Despite considerable improvement in patient survival, conventional therapies are mainly supportive. Recent progress towards gene therapy for CF has been encouraging; however, several factors such as immune response and transduced cell turnover remain as potential limitations to CF gene therapy. As alternative gene therapy vectors for CF we examined the feasibility of using SV40-derived vectors (rSV40s) which may circumvent some of these obstacles. To accommodate the large CFTR cDNA, we removed not only SV40 Tag genes, but also all capsid genes. We therefore tested whether “gutless” rSV40s could be packaged and were able to express a functional human CFTR cDNA. Results from our in vitro analysis determined that rSV40-CFTR was able to successfully result in the expression of CFTR protein which localized to the plasma membrane and restored channel function to CFTR deficient cells. Similarly in vivo experiments delivering rSV40-CFTR to the lungs of Cftr−/− mice resulted in a reduction of the pathology associated with intra-tracheal pseudomona aeruginosa challenge. rSV40-CFTR treated mice had had less weight loss when compared to control treated mice as well as demonstrably reduced lung inflammation as evidence by histology and reduced inflammatory cytokines in the BAL. The reduction in inflammatory cytokine levels led to an evident decrease in neutrophil influx to the airways. These results indicate that further study of the application of rSV40-CFTR to CF gene therapy is warranted.
Keywords: SV40, CFTR, Cystic Fibrosis, Gene Therapy, pseudomonas, gutless viral vectors
Introduction
Cystic fibrosis (CF) is among the most common genetic diseases. Despite considerable improvement in survival recently with conventional therapies, patients with CF become increasingly debilitated and have markedly shortened life expectancies, mostly due to recurrent pulmonary infections.
The genetic defect in CF is well documented: mutations in a cell membrane chloride channel, the cystic fibrosis transmembrane conductance regulator (CFTR). The resulting defective cellular chloride transport has many consequences. CF patients produce very viscous mucous, the key consequences of which are exocrine pancreatic insufficiency and recurrent pulmonary infections. The latter leads to respiratory failure (1, 2).
Treatment to date has generally been supportive: replacing the function of the defective ion channel has not yet been possible with small molecule therapeutics. Gene therapy would seem to offer a potentially useful therapeutic approach. However, despite great effort, CFTR gene delivery to the lungs has not yet succeeded in altering the course of the disease (3–8).
CF presents many obstacles to gene delivery that explain this situation. These include uncertainty as to which are the most appropriate cellular target for gene delivery, the best route of administration, viscous, neutrophil- and bacteria-rich mucous that both degrades vectors and prevents vectors delivered through the airways from reaching target epithelial cells, antigenicity of gene delivery vehicles, and the potential need to repeat gene delivery to compensate for loss of vector DNA and turnover of transduced cells (9–11).
Recombinant SV40-derived gene delivery vehicles (rSV40s) may help overcome some of these obstacles. These vectors integrate into the cell genome to provide permanent transduction and transduce most cell types very efficiently (12, 13). Perhaps most importantly, because their mode of entry into cells bypasses the antigen processing apparatus (14, 15), and because they thereafter produce no proteins of viral origin, rSV40s do not elicit detectable immune responses neither neutralizing antibodies nor cell mediated immunity. Uniquely among gene delivery vectors, rSV40s can be administered repeatedly without loss of transduction efficiency to normal immunocompetent individuals (16).
Despite considerable effort, genetic therapy for cystic fibrosis remains elusive. Some difficulties encountered in achieving this quest reflect the inherent pathophysiology of cystic fibrosis, while others are due to limitations of gene delivery technology. Application of recombinant SV40-derived gene delivery vectors (rSV40s) to this disease may circumvent some of these obstacles, e.g., the lack of neutralizing antibodies against these vectors may facilitate a potential need for repeated administration. To accommodate the large CFTR cDNA, we had to remove not only SV40 Tag genes, but also all capsid genes. We therefore tested whether “gutless” rSV40s could be packaged, whether they could express and carry a functional coding region for human CFTR cDNA, and provide an active chloride channel to CFTR-negative cells. Our data show that such gutless rSV40 vectors are easily produced by COS-7 cells at very high titers, without helper virus or repeated cotransfection.
The current studies, undertaken in vitro, were designed to test whether SV40-derived vectors could accommodate the CFTR cDNA, deliver it to cells, and provide a functional chloride channel. These vectors deliver CFTR expression efficiently to unselected CFTR-negative target cells. Resulting CFTR protein localizes to the cell membranes and provides functional chloride channels and direct evidence of a corrective effect in vitro. Furthermore these studies led to the testing of the vector in a mouse model of CFTR related disease, where the vector was able to attenuate in some cases resolve airway inflammation associated with intra-tracheal bacterial instillation.
Results
Making rSV40-CFTR
rSV40 genome constructs reported to date lacked the 2.6 kb Tag and tag genes, but retained 2 kb containing the SV40 capsid genes, VP1, VP2, and VP3. Since wtSV40 genome is 5.25 kb, deletion of additional SV40 sequences should allow effective packaging of a vector carrying a 4.2 kb version of the human CFTR cDNA. This is shown in Fig. 1. Briefly, pSV5 contains a Tag-deleted SV40 genome with 2 tandem SV40 early promoters (SV40-EP) followed by an engineered polylinker. This modified SV40 genome was cloned as a NotI fragment into pGEM13. An additional 1.8 kb of wtSV40 DNA was excised from pSV5 as a HpaI fragment, to produce pSV5(4.3). Human CFTR cDNA was cloned into the polylinker and rSV40-CFTR was produced from this plasmid according to published procedures (17).
Figure 1. Construction of rSV40-CFTR.
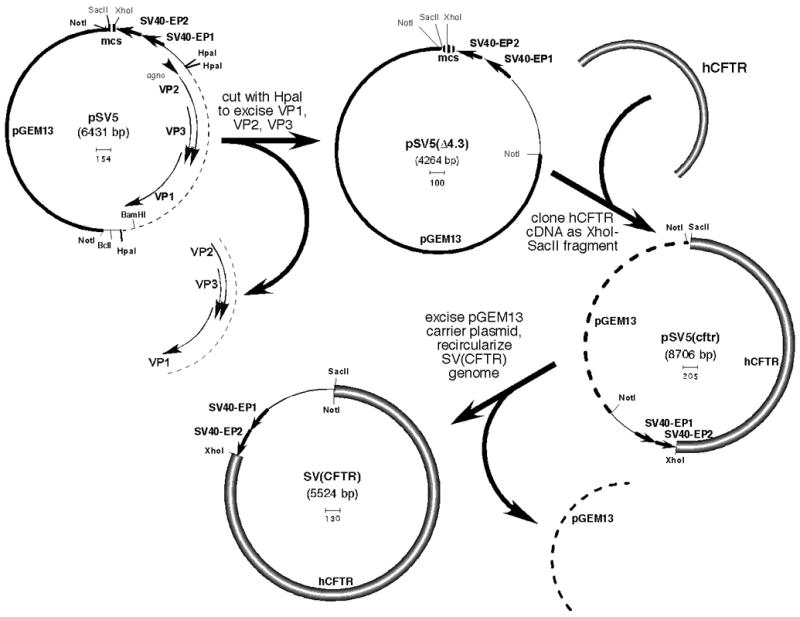
pSV5 carries an SV40 genome from which the Tag gene was deleted and replaced by a second SV40 early promoter (SV40-EP), plus a polylinker. The resulting Tag-less rSV40 genome, which is carried in pGEM13 (Promega), has been described (12).
Efficiency of transduction by rSV40-CFTR and localization of the delivered CFTR protein at the cell membrane
CFPAC cells (CFPAC-1, ATCC) make a mutated form of CFTR that is folded incorrectly (deletion of three nucleotides resulting in a phenylalanine-508 deletion), does not reach the cell membrane, and so is more rapidly degraded than wt CFTR (18). We therefore used these cells to determine the efficiency of rSV40-CFTR transduction and subcellular localization of the delivered protein. CFPAC cells were transduced with rSV40-CFTR or rSV40-HBS, or were mock-transduced. They were then examined by fluorescence microscopy following immunostaining with antibody to CFTR (Fig. 2a). Transduction efficiency, allowing for some cell-to-cell variation in levels of transgene expression, was 85–90% in unselected cells. Higher magnification analysis (Fig. 2b) suggested localization of the immunodetectable CFTR along the cellular membrane.
Figure 2. rSV40-CFTR transduction efficiency and analysis of rSV40-CFTR delivered CFTR expression by immunostaining.

CFPAC cells, a cystic fibrosis pancreatic adenocarcinoma cell line were plated in 4 well slides at a density of 105 cells/well. The cells were transduced with rSV40-CFTR, rSV40-HBS (negative control) at an MOI of ~3 or were mock-transduced. For (a) and (b), staining was visualized using an Olympus fluorescence microscope, and digitized (Spot Image Analysis Software, Diagnostic Instruments) using Macintosh computers. (a) composite of representative immunostained fields in cells treated as noted. (b) higher magnification of rSV40-CFTR-transduced and mock-transduced cells. (c) confocal microscopic examination of rSV-CFTR-transduced and control cells (as in (a)). CFPAC cells, transduced as in (a), were examined by confocal microscopy using a Biorad MR-60 confocal microscope.
Therefore, to evaluate more definitively the subcellular localization of the rSV40-CFTR delivered protein, confocal microscopic studies were done. Again, CFPAC cells were transduced with rSV40-CFTR or rSV40-HBS, or mock-transduced, then immunostained for CFTR and viewed using a confocal microscope (Fig. 2c). These studies confirmed that the delivered CFTR localized along the cell membrane. Distribution of the protein was patchy, as has been reported by others (19).
Functional correction of CFTR chloride channel defect with rSV40-CFTR
An important measurable activity of CFTR is its function as a cell membrane chloride channel. To test whether rSV40-CFTR transduction of CFPAC cells produced a functional chloride channel, two different assays were used. In the first series of studies, mock- and rSV40-CFTR transduced cells were loaded with the fluorescent Cl-sensitive dye, N- (6-methoxyquinolyl) acetoethyl ester (MQAE, Sigma Chemical Co.) (20), and stimulated by 10 μM forskolin. Permeability to Cl- was assayed as a change in fluorescence as a function of time after adding forskolin. Compared to TC7 cells (CFTR+ normal monkey kidney cells, + control), and mock-transduced CFPAC cells (−control), rSV40-CFTR- restored Cl- channel activity in CFPAC cells to≈50% of wild type levels (Fig. 3).
Figure 3. Chloride channel activity in CFPAC cells transduced with rSV40-CFTR: MQAE assay.
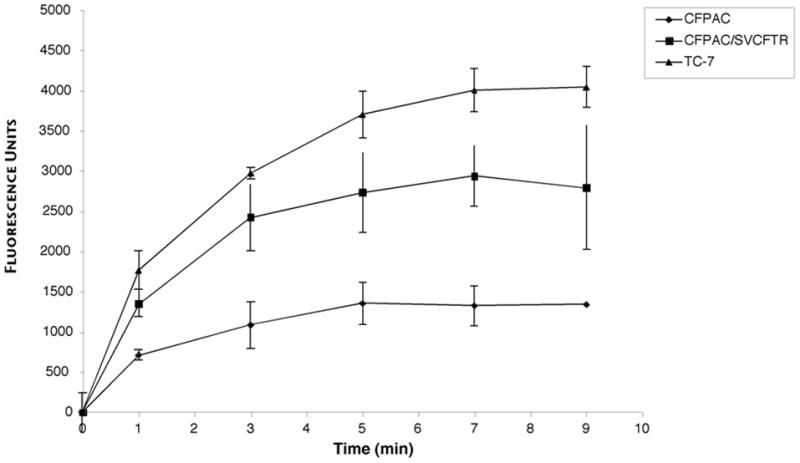
CFPAC cells were transduced with rSV-CFTR at MOI = 100, or mock transduced. Control cells were TC7 cells, which are normal simian kidney cells (courtesy, Janet Butel, Baylor College of Medicine). To measure Cl- channel activity, a microplate assay technique was used, as described by West and Molloy 22. Data are presented as Δ fluorescence (Ft−Fo), where Ft is fluorescence at time t and Fo is fluorescence at t = 0, and statistical significance (P<0.05) ascertained at time points beyond 1 minute by Wilcoxon’s signed rank test.
As an additional test of whether rSV40-CFTR could deliver a functional Cl- channel and to determine if channel activity could be restored in a different cell line, we performed 36Cl isotope tracer efflux assays on IB3–1 cells. CFTR-mutant IB3–1 cells (genotype ΔF508/W1282X) were infected with rSV40-CFTR at an MOI of 100. Mock-transduced T84 cells were the CFTR + control, mock-transduced IB3 cells were the CFTR −control. On day 10 post transduction, the cells were loaded with 5 μCi of 36Cl for 5 hours, and washed to remove excess isotope. The efflux rate was then measured at 30 second intervals for 4.0 minutes. After the final time point, the monolayer was carefully examined for integrity. Flasks with non-intact monolayers were eliminated from the experiment. Samples were counted for Cl36 activity. Transduction with rSV40-CFTR significantly increased Cl- channel activity by this assay, delivering ≈33% of wild type Cl- channel activity (P = 0.01), as compared to control T84 cells (Fig. 4)
Figure 4. Changes in36Cl efflux show CFTR channel activity in response to agonist stimulation of IB3 cells transduced with rSV40-CFTR.
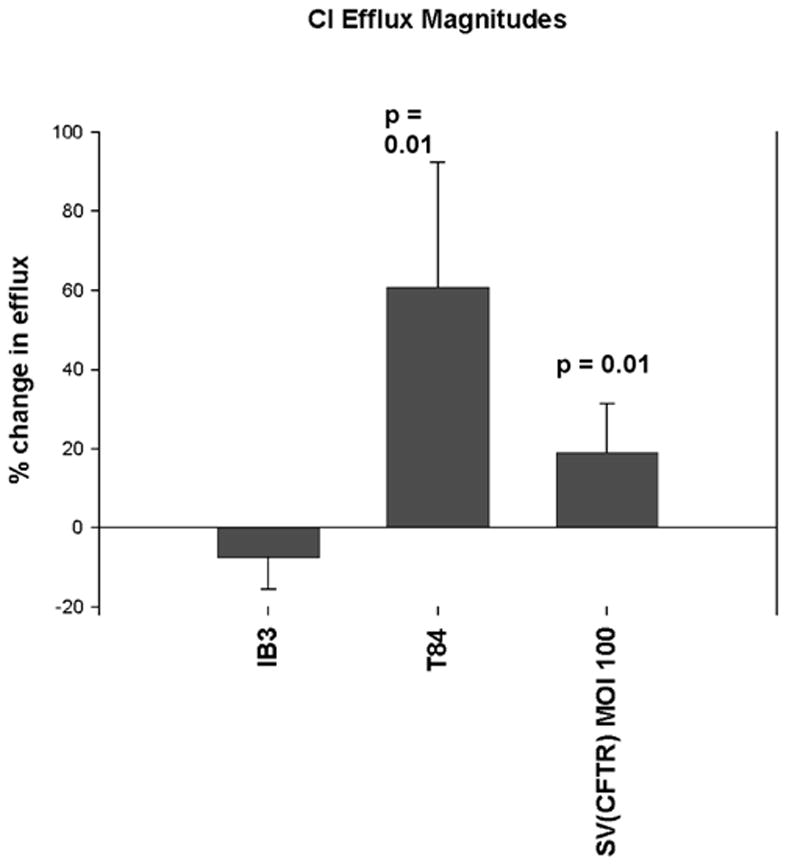
36Cl isotope tracer efflux assays – IB3 cells were plated at a density of 2×106 into T-25 flasks in LHC-8 media containing 10% FBS and 5% penicillin/streptomycin (LHC-8+) and allowed to grow overnight. The next day the media was changed with 10 ml of fresh LHC-8+ and cells were infected with rSV40-CFTR virus at an MOI of 100. Samples were mixed with CytoSafe scintillation fluid and counted for 36Cl activity. The % change from the maximal post-agonist rate (cpm) and the minimal rate (cpm) prior to that efflux event is defined as the “% change in efflux” [max post-agonist % efflux - min pre-agonist % efflux]. Cells without significant CFTR activity will not stimulate upon exposure to agonist mixture and will display a negative “% change in efflux”. Uninfected negative control IB3 cells (“IB3”), supra-physiologic CFTR positive control T-84 cells and IB3 cells infected with rSV40-CFTR are shown. t test p values are relative to IB3 neg. control cells.
In vivo correction of the mouse CF lung phenotype with rSV40-CFTR
Having determined the functional expression of the CFTR channel in vitro, it was necessary to determine if the delivery of a functional chloride channel with rSV40 vectors would also have beneficial effects on a CF lung disease phenotype. Therefore, we used our in vitro characterized vectors to deliver the CFTR gene to Cftr−/− mice that were used to recapitulate a well-characterized Pseudomonas aeruginosa (PA01 strain) agarose bead airway challenge model (21).
Two sets of independent experiments were performed with this model. In the initial Ps-bead challenge experiment matched groups of 5 Cftr−/− mice were treated with 4.0×107 particles of either rSV40-CFTR vector or the irrelevant rSV40-BUGT negative control via intra-tracheal injection and housed in SPF conditions. Due to the highly stable expression of rSV40 vectors it was possible to allow the mice to completely heal form the rSV40 intra-tracheal injection before performing the second intra-tracheal injection with the Pseudomonas bead slurry. Thus, eight weeks later the mice were challenged with 30 ul of a 50 % Pseudomonas aeruginosa (PA01) agarose bead slurry (as determined by 30 minute gravity sedimentation), as previously described (21, 22). This initial bead preparation had a bacterial count of 3.8×106 CFU/ml and resulted in the previously characterized excessive weight loss in Cftr−/− mice. Interestingly, in our studies, mice rescued with rSV40-CFTR had statistically significant less weight loss on the first three days after the bacterial challenge (Fig. 5) when compared to rSV40-BUGT treated mice. On the last and fourth day before sacrifice rSV40-CFTR rescued mice also demonstrated a trend toward less weight loss (p=0.06). The hallmark endpoint of this model has been the excessive weight loss seen in Cftr−/− mice observed in previous studies (21). In our experiments the day 3 weight loss difference between rSV40-BUGT treated mice (~26 %) and rSV40-CFTR rescued mice (~19 %) was ~7 % (Fig. 5).
Figure 5. Amelioration of weight loss by rs40-CFTR gene therapy in Pseudomonas-infected cystic fibrosis (CF) mice.
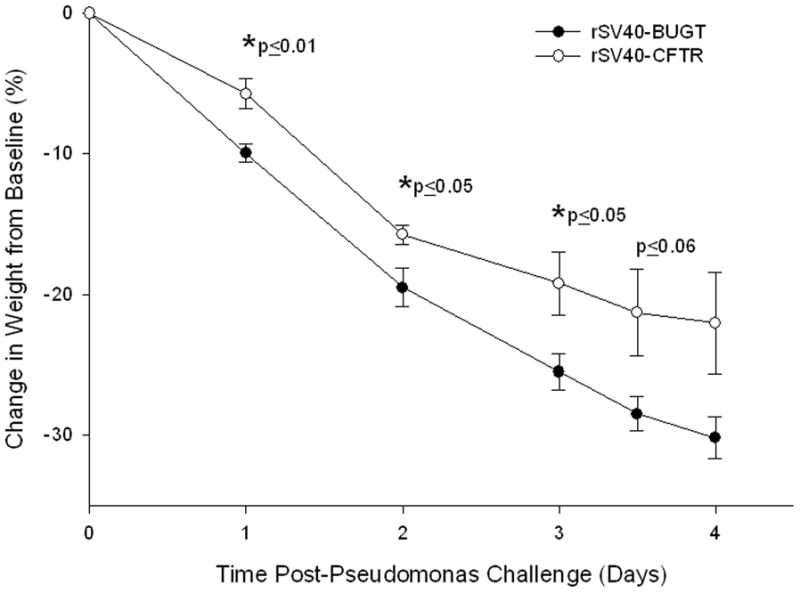
Shown are the means ± standard deviations of the percent changes in weight after Pseudomonas-agarose bead intra-tracheal challenge for the CFTR−/− mice in the rS40-CFTR group and in rS40-BUGT-treated control group. N=4 for each timepoint. *p≤0.05 difference between the two groups.
To determine if these differences in weight lose were consistent with CFTR expression; RT-PCR assays to detect the SV40 driven human CFTR transcript were performed on lung tissue collected 4 days after bacterial challenge. As shown in figure 6, human CFTR expression was detected in mice receiving the rSV40-CFTR, while none was detectable by RT-PCR in mice receiving the BUGT vector. RT-PCR using a no reverse transcriptase control to rule out the possibility of rSV40-CFTR DNA amplification was negative (data not shown). It should be noted that this expression is representative of mice ~ 9 weeks post rSV40-CFTR intra-tracheal delivery and after they have undergone pseudomonas agar bead challenges, thus highlighting the stability of transgene expression achieved by rSV40 gene delivery. This stability is further evidence by a separate experiment in which IHC analysis of lung tissues from mice receiving an AU1 epitope tagged rSV40 vector sacrificed at 2, 4 and 9 weeks post delivery showed no apparent decrease in the expression of the epitope tag at the various time points (data not shown). Further analysis of the mice 9 weeks post delivery revealed that rSV40 transduction as determined by AU1 epitope expression was evident throughout the airways including in mucous producing cells, as determined by overlay of AUI expressing cells and the lysozyme expressing mucous producing cells (figure 7). It has been previously reported that lysozyme is produced by sub-mucosal glands and goblet cells(23, 24).
Figure 6. Stable expression of CFTR in mouse lungs after intra-tracheal delivery of rSV40-CFTR.
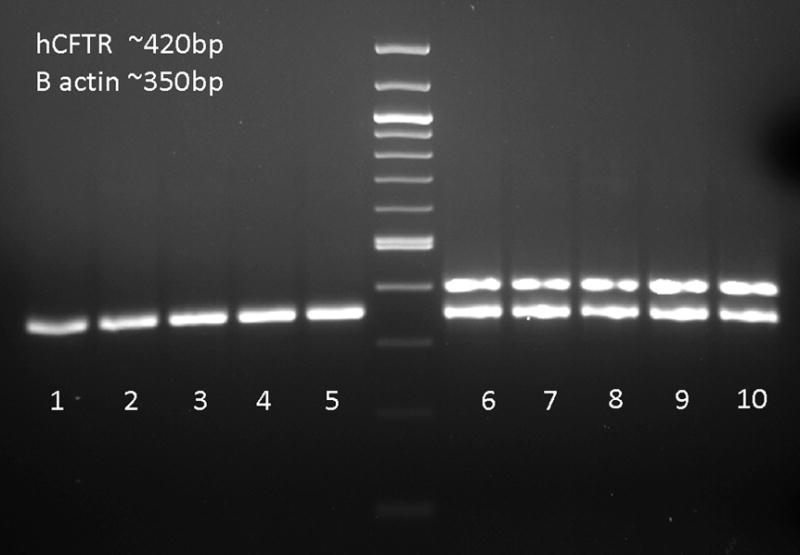
RT-PCR for Human CFTR mRNA was assayed from lungs 4 days after pseudomonas challenge. Lungs were homogenized and RNA was extracted to run RT-PCR reactions with human CFTR and B-actin primers; lanes 1–5 rSV40-CFTR, lanes 6–10 rSV40-BUGT. The ‘no reverse transcriptase’ controls were negative for both the CFTR and B-actin bands (data not shown)
Figure 7. Transduction of Mucous Producing Cells in the Airways with rSV40.

Mice received 1.5×107 particles of a rSV40 vector with and AU1 epitope tag and were sacrificed 9 weeks post delivery. The far left frame is an H&E stain of the lung tissue, with black arrows depicting goblet cells. On the right are three fluorescence micrographs, the upper one using an antibody directed at lysozyme, to delineate the mucous-producing cells, the middle using an antibody against the AU1 tag, and the lower panels shows a merge of the two stains where the white arrows show representative doubly positive cells.
Further analysis of the lung compartment of these mice revealed that rSV40 mediated correction of the Cftr−/− mice, not only attenuated the weight loss phenotype, but it had also had an effect on the inflammatory cytokine profiles recovered from the broncho-alveolar lavages (BALs). While the cytokine levels in rSV40-CFTR rescued mice all trended lower, IL-1beta was statistically significantly lower (Fig 8). Also importantly for CF related lung disease there was a strong trend (p≤0.1) towards lower KC levels in treated mice. KC is considered the mouse analogue to IL-8 and is mainly responsible for the recruitment of neutrophils into the lung.
Figure 8. Attenuation of inflammatory cytokine profiles in the lung compartment of CFTR corrected mice.
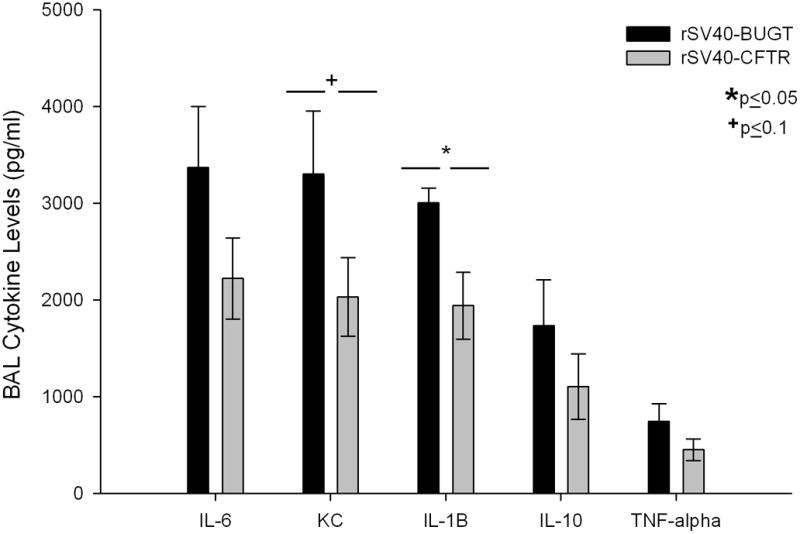
Cytokine levels in BALs of Pseudomonas challenge on the cytokine levels in the BALs of rS40-CFTR and rS40-BUGT-treated Cftr −/− Mice. Results are expressed as means ± S.E.M (N=4).
In order to determine if this attenuated inflammatory profile in the BALs of rSV40-CFTR recued mice was accompanied by a reduction in the influx of neutrophils and lung pathology, a second set of aged matched mice were IT injected with either rSV40-CFTR or rSV40-BUGT. As in the first experiment described above, 8 weeks after rSV40 delivery, mice were challenged with Ps-bead slurry, albeit of a slightly lower CFU count due to preparation variations (3.0×106CFU/ml). Histopathological analysis of lung sections from this second cohort showed a dramatic reduction in the infiltration and inflammation of the lung tissue (Figure 9, 10). Mice treated with the control vector showed severe inflammatory infiltration of the airways with focal disease in the parenchyma, often accompanied with epithelial disintegration and sloughing. This was accompanied by a prominent presence of neutrophils and type II pneumocyte hyperplasia. These findings were highly attenuated in rSV40-CFTR treated animals; the statistically significant magnitude of these changes can be appreciated in the inflammatory scores obtained from an independent blinded pathologist (table 1). As evidenced in the first set of experiments, KC and other inflammatory cytokine levels in the BAL were lower in CFTR corrected mice and the effect of these attenuated cytokine responses can be appreciated in the significant reduction of neutrophils recovered form the BALs in the second set of experiments (Figure 10).
Figure 9. Reduction and Prevention of lung inflammation by rS40-CFTR gene therapy in Pseudomonas-infected cystic fibrosis (CF) mice.
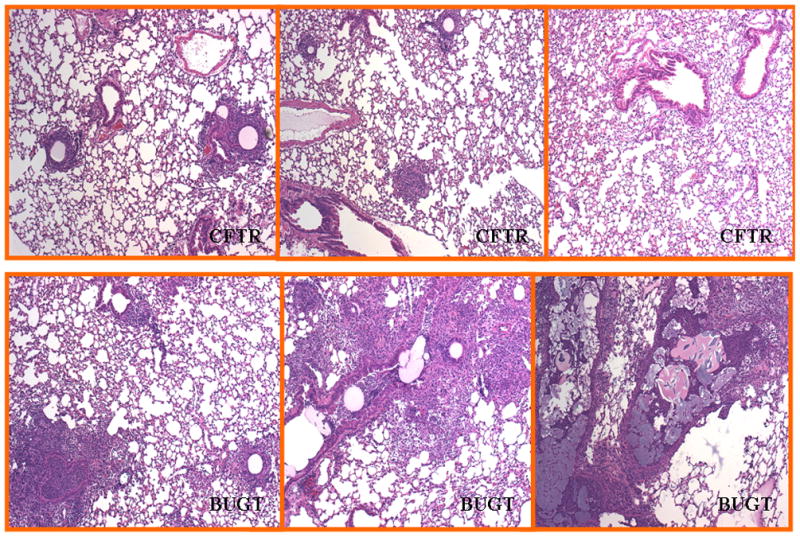
The top panels show randomly selected, representative fields of hematoxylin-eosin stained lung sections from rS40-CFTR-treated CFTR−/− mice 4 days after Pseudomonas challenge. The bottom panels show similar fields from animals pretreated with rS40-BUGT. There were 5–6 animals in each group in this second experiment.
Figure 10. Reduction in the recruitment of neutrophils into the airways of rSV40-CFTR corrected mice.
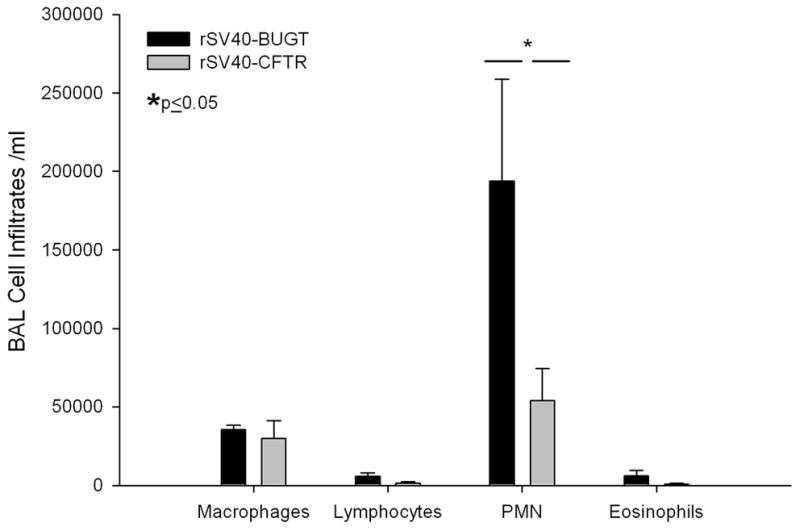
BAL Cell Counts after Pseudomonas exposure in rS40-CFTR and rS40-BUGT-treated CFTR−/− mice. Results are expressed as means ± S.E.M (N=5).
Table 1.
Gross pathology scores in Cftr−/− mice 4 days after inoculation with pseudomonas-laden agarose beads
| Vector Treatment | Lung Inflammation Score and Observations | |
|---|---|---|
| rSV40-CFTR | 0 | |
| rSV40-CFTR | 2 | multifocal, small, dilated airways surrounded by layer of PMNs and macrophages, exudate in lumens, minigranulomas |
| rSV40-CFTR | 0 | |
| rSV40-CFTR | 0 | |
| rSV40-CFTR | 2.5 | multifocal; bronchioles are surrounded by infiltrates but rarely invaded |
| Average Score rSV40-CFTRM | *0.9 | |
| rSV40-BUGT | 3.5 | major bronchioles invaded and disintegrating epithelium and alveolar cells, granulomas, bulli |
| rSV40-BUGT | 1.5 | multifocal mild infiltrates and epithelial damage |
| rSV40-BUGT | 2 | multifocal; bronchioles are surrounded by infiltrates but rarely invaded. |
| rSV40-BUGT | 3.5 | multifocal infiltrates- severe, |
| rSV40-BUGT | 3.5 | multifocal infiltrates- severe, mixed populations, (“casts”; airway epithelium disintegration with sloughting epithelium into the lumen; exudate in airway “bulli |
| rSV40-BUGT | 2 | two foci infiltrates- mixed with PMN in lumen, numerous macrophages accumulating at point of infiltrate. |
| Average Score rSV40-BUGT | *2.7 | |
p≤0.05 as determined by a Bonferonni corrected Kruskal-Wallis Test
Discussion
These studies were designed to test whether “gutless” rSV40 vectors carrying human CFTR cDNA could be effectively packaged and deliver CFTR expression after transduction, and whether the CFTR so provided constituted a functional chloride channel. It was important to ascertain that “gutless” rSV40 vectors carrying the 4.2 kb CFTR cDNA insert could be packaged by COS-7 cells. rSV40-CFTR was packaged by COS-7 cells at yields comparable to those obtained for other rSV40s that carry capsid genes: approximately 1011 IU/ml. Thus, the size of the vector genome, 5.5 kb, was within the packaging limits of this system. Some other vector systems require simultaneous cotransfection of packaging cells with multiple plasmids, or coinfection with helper viruses, in order to produce gutless vectors, but the effective packaging of a rSV40 lacking capsid genes in COS-7 cells indicates that the packaged rSV40 genomes need not carry SV40 capsid genes. The latter are expressed adequately, driven by their own promoter, by the COS-7 cells: neither helper virus nor cotransfection is involved. Preliminary studies show that SV40 capsid genes, under the control of the SV40 late promoter, are not expressed constitutively in COS-7 cells, but that the presence of a replicating rSV40 genome that includes the late promoter is sufficient to activate COS-7 transcription of the capsid genes in trans (M. Mariere and D.S. Strayer, unpublished data).
A possible explanation for this phenomenon is that replicating SV40 genomes titrate out a cellular repressor that inhibits transcription of integrated SV40 late genes (25). In our system, then, the rSV40 genome replicating in COS-7 cells would bind the cellular repressor and, in so doing, trans-activate the SV40 promoter to drive expression of the capsid genes. Replicating rSV40 genomes would then de-repress expression of SV40 capsid genes from the integrated copy of the wt SV40 genome in COS-7 cells. Thus, recombinant SV40-derived vectors do not need to carry SV40 capsid genes and gutless rSV40 vectors are made as efficiently as standard rSV40s.
CFPAC cells are a pancreatic adenocarcinoma line that carries the most common known loss of function mutation in the CFTR gene: ΔF508 (18). The mutation causes an in-frame loss of a phenylalanine at position #508. The resulting abnormal protein can actually form a functional Cl- channel (26), but loss of F508 causes the protein to be misfolded and consequently shunted to the cellular proteosome for degradation, so it does not reach the cell membrane to serve as an ion channel.
Transduction with rSV40-CFTR delivered detectable CFTR protein by immunostaining in vitro and detectable transcription as determined by RT-PCR in vivo. The in vitro studies also demonstrated that rSV40 gene delivery transduced the great majority of cells in these cultures, without selection. This finding is consistent with previous data demonstrating that at rSV40s can deliver transgene expression efficiently and do not require the use of a selective agent (13). It is useful to point out that expression of proteins delivered by rSV40s, while permanent, tends to be at lower levels than are seen using other gene delivery vector systems.
Most importantly, treatment with rSV40-CFTR provided a functional chloride channel activity that was lacking in both CFPAC and IB3 cells. Furthermore, channel function was demonstrated by two different assays of Cl- channel activity, both of which demonstrated similar functionality in the ion channel delivered by rSV40-CFTR. The level of Cl- channel activity in transduced cells was 1/3 to 1/2 of that seen in normal cells. Estimates of several investigators suggest that this level of Cl- channel activity is sufficient to avoid the most harmful consequences of mutation in CFTR (27, 28). Thus, if the level of activity seen here could be achieved and maintained in vivo, it would be within the therapeutic range.
In vivo studies carried out in the bacterial agarose bead model lend further evidence of the functional and corrective nature of the rSV40 delivered CFTR gene. These results indicate that further study of the application of rSV40-CFTR to CF gene therapy is warranted. Indeed, these studies, conducted by instillation of rSV40-CFTR into airways of CFTR-knockout mice, suggest that this vector may be able to provide significant functional correction in vivo as well as in vitro. Our studies report a statistically significant difference in weight loss between our two groups ~26% for the BUGT treated mice and ~19% for the CFTR rescued mice at day 3. In comparison, the original study using the pseudomonas bead model by Van-Heeckeren et al. reports a weight loss of ~18% in Cftr−/− mice versus ~13% in wild type mice (21). In our experiments the weight loss was steeper and more prominent; this is probably due to the typical variability encountered in the preparations of the pseudomonas agar beads. However, one can appreciate the difference in weight loss between groups was proportional in the two separate studies. More importantly for CF related pathology rSV40-CFTR rescue resulted in the reduction of KC and IL-1B which correlated with the dramatic reduction of neutrophils influx to the lung and with a significantly improved overall inflammatory state of the lung as judged by histology.
CF gene therapy has stumbled at the transition between effective in vitro and in vivo gene delivery for many reasons (4, 8, 29, 30). Applying rSV40 vectors to the treatment of CF may help address some of these issues: impermanence of transgene expression (rSV40s integrate and express their transgenes indefinitely (12), cytolytic inflammatory responses against transduced cells (cells transduced with rSV40s do not elicit cytotoxic responses (31), and need for repeat dosing (rSV40s, alone of all viral gene delivery vectors, do not elicit neutralizing antibodies (16). Other questions, such as the best route of administration (32), e.g., the most appropriate target cell (33) are dilemmas that have stimulated considerable thought and experimentation, but that remain to be resolved (10).
Many hurdles remain before rSV40 gene delivery in CF can reach clinical fruition. Many issues, including vector safety and appropriate production procedures, all need to be considered. The unique ability of rSV40s to avoid eliciting neutralizing antibody has been demonstrated in rodents (12, 13), but needs to be confirmed in higher animals. Finally, the route of delivery remains a major concern, since neither aerosolization nor instillation of other vectors through the airways has not yet yielded promising results. Because SV40 is small (≈40 nm in diameter), it may be delivered more efficiently to the lungs in CF patients via the vascular system. We have administered rSV40 vectors carrying marker genes intravenously and detected abundant transgene expression in the pulmonary airway epithelium (34).
Beyond questions of effectiveness, issues relating to vector safety are important. To date, our preparations of rSV40 vectors have been free of Tag+ revertant wtSV40s. Animal recipients of these vectors have not shown evidence of adverse effects or toxicity (13).
Conventional therapies have improved survival for patients with CF dramatically. Interesting new approaches continue to offer benefits, or potential benefits, to some such individuals (35). Some thoughtful approaches have recently been reported that may circumvent or decrease the barrier created by the airway inflammation and viscous mucous, so as to facilitate gene delivery (36). However, gene therapy remains tantalizing, precisely because of its promise that CF treatment could one day actually give afflicted patients something approaching normal lives.
Whether rSV40 vectors make a contribution here remains to be seen. But our data suggest that they may provide an additional gene delivery option in treating cystic fibrosis.
Material and Methods
pSV40(CFTR)
This vector utilizes a 4.2 kb (full length) human CFTR cDNA paired to 2 tandem SV40 early promoters. rSV40 genome constructs reported to date lacked the 2.6 kb Tag and tag genes, but still carried the 2 kb of wtSV40 capsid protein genes, VP1, VP2, and VP3. Since wtSV40 genome is 5.25 kb, deletion of additional SV40 sequences should allow effective packaging of a vector carrying a 4.2 kb version of the human CFTR cDNA. pSV5 contains a Tag-deleted SV40 genome with 2 tandem SV40 early promoters (SV40-EP) followed by an engineered polylinker. This modified SV40 genome was cloned as a NotI fragment in pGEM13. An additional 1.8 kb of wtSV40 DNA was excised from pSV5 as a HpaI fragment, to produce pSV5(Δ4.3). Human CFTR cDNA was cloned into the polylinker and SV(CFTR) was produced from this plasmid.
pSV(BUGT) and pSV(HBS)
This vector utilizes a human bilirubin uridine diphosphate glucuronoside transferase (BUGT) cDNA paired to 2 tandem SV40 early promoters and was used as a control vector for in vivo studies. pSV(HBS) was used as a control vector for in vitro studies.
Simian Virus 40 Vectors
The resulting Tag-less rSV40 genome, which is carried in pGEM13 (Promega), has been described (12). pSV5(Δ4.3) was made by removing a 1.8 kb HpaI fragment from pSV5. pSVCFTR was made cloning a PCR product from hCFTR into pGEM-TEasy (Promega). A 5′ primer containing an XhoI site (GGCTCGAGGGACCCCAGCGCCCGAGAGA) and a 3′ primer containing a SacII site (CACGTTCTATGTTCCGAAATTGGCGCCGG) were used to make a 4.5 kb PCR product containing the full length CFTR cDNA. This fragment was then cloned into pGEM-TEasy, amplified, and the CFTR cDNA was cut out using SacII and XhoI. This fragment was then cloned into the SacII and XhoI sites in pSV5(Δ4.3), to make pSV5(CFTR). The procedures for making gene delivery vectors from cloned rSV40 genomes have been described in detail elsewhere (17). Briefly, the SV(CFTR) viral genome was excised from this plasmid using NotI, gel purified, recircularized and transfected into COS-7 cells. COS-7 cells, a simian kidney cell line immortalized by stable transfection with a wild type (wt) SV40 genome that is deficient at the origin of replication (16), were maintained in DMEM supplemented with 10% newborn calf serum (NCS, Hyclone). Virus was allowed to grow for 7 days after which lysates were harvested. Lysates were used to amplify SV(CFTR) by infecting COS-7 cells(ATCC, Washington, DC, USA), without helper virus and without additional transfections. Virus was purified by density centrifugation and virus stocks were titered by in situ PCR, as described previously (17). In order to measure the infectivity of replication-deficient SV-40, the viral stocks were titrated by in situ PCR. Infectivity of our rSV40 viral vector stocks was generally approximately 109 infectious units (IU)/ml.
CFTR Immuno-staining
CFPAC cells, a cystic fibrosis pancreatic adenocarcinoma cell line (ATCC, #CRL-1918) (18), were maintained in Iscove’s media supplemented with 10% NCS. Both media were supplemented with 2 mM L-glutamine and 1% penicillin-streptomycin and all cells were grown in 5% CO2 and 100% humidity. The cells were plated in 4 well slides at a density of 105 cells/well. The cells were transduced with rSV40-CFTR, rSV40-HBS (negative control) or were mock-transduced. Cells were fixed in acetone-methanol (50:50) for 5 minutes. Blocking was performed by 1 hour incubation with 10% goat serum. Cells were incubated for one hour at room temperature with the primary antibody, mouse monoclonal anti-CFTR, C-terminus (Research Diagnostics, Inc., NJ) at a 1:100 dilution, then washed 3 times with PBS, and incubated with 1:100 diluted FITC-conjugated rabbit anti-mouse IgG (Dako, Carpinteria, CA). Mounting medium containing DAPI (Vector Laboratories, Burlingame, CA) was used to visualize the nuclei.
Chloride Ion Channel Activity Mircoplate Assay
CFPAC cells were transduced with rSV40-CFTR at MOI = 100, or mock transduced. Control cells were TC7 cells, which are normal simian kidney cells (courtesy, Janet Butel, Baylor College of Medicine). To measure Cl- channel activity, a microplate assay technique was used, as described by West and Molloy 22. Specifically, 6d after transduction with rSV40-CFTR, CFPAC cells were plated in 96 well culture plates at a density of 104 cells/well. 48 hours after plating, cells were loaded overnight with 10 mM N-(6-methoxyquinolyl) acetoethyl ester (MQAE). The following day cells were washed with a chloride-containing buffer ( 2.4 mM K2HPO4; 0.6 mM KH2PO4; 10 mM HEPES; 10 mM dextrose; 1 mM MgSO4; 130 mM NaCl). This buffer was then replaced with a chloride-free buffer (2.4 mM K2HPO4; 0.6 mM KH2PO4; 10 mM HEPES; 10 mM dextrose; 1 mM MgSO4; 130 mM NaNO3), containing 10 μM forskolin. Repetitive fluorescence measurements were performed immediately using a Millipore Cytoflour 3050 plate reader (excitation; 360 nm, emission; 460).
36Cl isotope tracer efflux assay
IB3–1 cells were seeded at a density of 2×106 cells/T25 flask in LHC-8+ and left overnight in a 37°C humidified CO2 incubator. The next day monolayers had achieved subjectively between 60–70% confluence. T84 cells were also seeded as a supra-physiologic CFTR positive control and allowed to grow to ~70% at this time. The wells containing IB3–1 cells were assigned into either negative control plates that received PBS treatment or CFTR test plates that received either infected with viral rSV40-CFTR at an MOI of 100 (particles/cell). The virus containing media (LHC-8+) was left on the cells until day 5 and then was changed with 10 ml of fresh LHC-8+ medium. The LHC-8+ medium was replaced again on day 8.
On day 10 cells were loaded with 5mCi of 36Cl for 5 hours at 37°C. Each flask was then transferred into a modified 37°C dry heat incubator and placed upon a metal block heat sink. Each flask was washed 5 times with lactated Ringers solution (LRS) for 30 seconds/wash to remove excess isotope and the efflux rate was then measured at 30 second intervals for 4.0 minutes. All washes and the first efflux collection were obtained by removing the entire fluid volume (1.5 ml) from the flask with a disposable fine tipped transfer pipette, and replacing an equal amount of 37°C lactated Ringers solution (LRS). The replacement solution for second through eighth samples contained a combination of 2.5 mM forskolin, 250 mM 8-Br-cAMP, and 250 mM CPT-cAMP, added to increase intracellular levels of cAMP. At the conclusion of the final time point, the monolayer was carefully examined for integrity. Flasks with non-intact monolayers were eliminated from the experiment. Samples were mixed with CytoSafe scintillation fluid and counted for 36Cl activity. Statistical analyses were conducted by a two tailed t test, P(T≤t). The % change from the maximal post-agonist rate (cpm) and the minimal rate (cpm) prior to that efflux event is defined as the “% change in efflux”. Cells without significant CFTR activity will not stimulate upon exposure to agonist mixture and will display a negative “% change in efflux”. Uninfected negative control IB3 cells (“IB3”), supra-physiologic CFTR positive control T-84 cells, and IB3 cells infected with rSV40-CFTR are shown. t test p values are relative to IB3 neg. control cells.
Pseudomonas agarose-bead Challenges
In the challenge experiments, CFTRtm1Unc-TgN(FABPCFTR)#Jaw mice (gut-corrected CFTR knockout mice) (CFTR−/− )mice were treated intra-trachealy with rSV40-CFTR vector or rSV40-BUGT vector or phosphate-buffered saline (PBS), housed in specific pathogen-free (SPF) conditions for 8 weeks, and then challenged with 30 ul of an approximately 50% Pseudomonas agarose-bead mixture (as determined by 30-min gravity sedimentation) of OD600 2.0 containing ~ 3.0×106 cfu/ml. Mice were randomized with regards to injection order to avoid any unrecognized systematic error. Weights were recorded at time of challenge and at time of sacrifice on day 4. Histopathologic analysis of hematoxylin-eosin stained sections of formalin-fixed, paraffin embedded lung tissue samples taken at the time of sacrifice was performed by a pathologist blinded to the assignment of animals to the two groups.
RT-PCR for Detection of hCFTR Expression
To ensure the expression of hCFTR by the lungs, following intra-tracheal injections of rSV40-CFTR, lungs were collected from all the CFTR−/− mice. A 100mg of tissue sample was used to extract mRNA using the Qiagen (Valencia, CA) total RNA columns. For Reverse transcriptase first strand synthesis the Invitrogen Superscript III kit was used with the oligo(dt) primers and the optional DNase digestion according to the manufacture’s protocol. This was followed by a PCR using Eppendorf Taq polymerase master mix and gene specific primers for hCFTR (hCFTRfor_5′ aaacttctaatggtgatgacag, hCFTRrev_5′agaaattcttgctcgttgac) or B-actin (B-actin1_5′gctcgtcgtcgacaacggtc, Bactin2_5′caaacatgatctgggtcatcttctc). The no RT controls were subjected to the same PCR conditions and primers but in the absence of reverse transcriptase in the initial cDNA synthesis.
Immunodetection of rSV-40 Transduction
Mice were intra-trachealy injected with an rSV40 vector expressing an AU1 epitope tagged gene, 9 weeks post delivery mice where sacrificed and their lungs were inflated with a 50/50 mixture of PBS and OCT and subsequently frozen in OCT medium. For immunofluorescence, 10 um cryostat sections were processed for indirect immunofluorescence. Blocking was performed by 60 minutes incubation with 10% goat serum in 0.10 M PBS (pH 7.4). Then, cryostat sections were incubated with mouse FITC-labeled anti-AU1 (IgG2a; 1: 100) (Covance, Emeryville, CA) FITC and rabbit anti-human lysozyme (Accurate Chemical and Scientific Corp., Westbury, NY). Incubation with primary antibody was performed for 1 h and followed by incubation for 1 h with secondary antibody TRITC-conjugated goat anti-mouse IgG (Sigma, Saint-Louis, MO), diluted 1: 100. Incubations were at room temperature. Double immunofluorescence was performed as previously described (37). Incubation was followed by extensive washing with PBS. Specimens were finally examined under a Leica DMRBE microscope (Leica Microsystems, Wetzlar, Germany). For negative controls slides were pre-incubated with PBS and 0.01% BSA, non-immune isotype-matches control antibody was substituted for primary antibody, and/or the primary antibody was omitted.
Lung Broncho-alveolar Lavage for Cell Differentials and Cytokine Profiles
The BAL fluid was retrieved from each animal via cannulation of the exposed trachea and gentle flushing of the lungs with two separate 1 ml aliquots of PBS. Aliquots were pooled for individual animals preceding centrifugation and separation of pelleted cells and supernatant. Cytospin cell preparations made with 200 μls of BAL were stained using Hema 3 (Biochemical Sciences, Swedesboro, NJ) differential stain and relative cell populations were determined using standard morphological criteria. Total cell numbers were calculated by counting the cells on the cytospin slide obtained from the 200 ul BAL aliquot. Assessments of cytokine profiles from the BAL were performed using a commercially available multiplexed kit(Biorad Mouse Multi-Cytokine Detection System; BioRad Laboratories) and the Bioplex Suspension Array System. Simultaneous measurement of several cytokines was performed. All assays were performedaccording to the manufacturer’s protocols. Cytokine concentrations were determinedutilizing Bioplex software with four-parameter data analysis. The sensitivity of the assay is less than 10pg/ml and has a range from 0.2–32,000 pg/ml with an inter and intra-assay CV of less than 10%.
Acknowledgments
This work was supported by grants AI48244, RR13156, AI41399 and R01HL69877 from the National Institutes of Health as well as by a fellowship from the Parker B Francis Foundation and the Diabetes and Endocrinology Research Center of the University of Massachusetts Medical School (supported by Grant P30 DK32520). The authors are grateful to Miss Maria Lamothe and Mr. Charles Ko for technical assistance. Dr. Janet S. Butel, Baylor College of Medicine, generously provided us with the original SV40 genomic constructs from which these vectors were derived. We are grateful to Dr. John Engelhardt for the human CFTR cDNA used in these studies.
References
- 1.Schwiebert EM, Benos DJ, Fuller CM. Cystic fibrosis: a multiple exocrinopathy caused by dysfunctions in a multifunctional transport protein. Am J Med. 1998 Jun;104(6):576–90. doi: 10.1016/s0002-9343(98)00119-3. [DOI] [PubMed] [Google Scholar]
- 2.Southern KW. delta F508 in cystic fibrosis: willing but not able. Arch Dis Child. 1997 Mar;76(3):278–82. doi: 10.1136/adc.76.3.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flotte TR. Gene therapy progress and prospects: recombinant adeno-associated virus (rAAV) vectors. Gene Ther. 2004 May;11(10):805–10. doi: 10.1038/sj.gt.3302233. [DOI] [PubMed] [Google Scholar]
- 4.Ferrari S, Geddes DM, Alton EW. Barriers to and new approaches for gene therapy and gene delivery in cystic fibrosis. Adv Drug Deliv Rev. 2002 Dec 5;54(11):1373–93. doi: 10.1016/S0169-409X(02)00145-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Driskell RA, Engelhardt JF. Current status of gene therapy for inherited lung diseases. Annu Rev Physiol. 2003;65:585–612. doi: 10.1146/annurev.physiol.65.092101.142426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griesenbach U, Ferrari S, Geddes DM, Alton EW. Gene therapy progress and prospects: cystic fibrosis. Gene Ther. 2002 Oct;9(20):1344–50. doi: 10.1038/sj.gt.3301791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flotte TR. Recombinant adeno-associated virus vectors for cystic fibrosis gene therapy. Curr Opin Mol Ther. 2001 Oct;3(5):497–502. [PubMed] [Google Scholar]
- 8.Mueller C, Flotte TR. Gene Therapy for Cystic Fibrosis. Clin Rev Allergy Immunol. 2008 Jul 4; doi: 10.1007/s12016-008-8080-3. [DOI] [PubMed] [Google Scholar]
- 9.Harvey BG, Leopold PL, Hackett NR, Grasso TM, Williams PM, Tucker AL, et al. Airway epithelial CFTR mRNA expression in cystic fibrosis patients after repetitive administration of a recombinant adenovirus. J Clin Invest. 1999 Nov;104(9):1245–55. doi: 10.1172/JCI7935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Virella-Lowell I, Poirier A, Chesnut KA, Brantly M, Flotte TR. Inhibition of recombinant adeno-associated virus (rAAV) transduction by bronchial secretions from cystic fibrosis patients. Gene Therapy. 2000;7:1783–9. doi: 10.1038/sj.gt.3301268. [DOI] [PubMed] [Google Scholar]
- 11.Zeitlin PL. Cystic fibrosis gene therapy trials and tribulations. Mol Ther. 2000 Jan;1(1):5–6. doi: 10.1006/mthe.1999.0016. [DOI] [PubMed] [Google Scholar]
- 12.Strayer D, Branco F, Zern MA, Yam P, Calarota SA, Nichols CN, et al. Durability of transgene expression and vector integration: recombinant SV40-derived gene therapy vectors. Mol Ther. 2002 Aug;6(2):227–37. doi: 10.1006/mthe.2002.0657. [DOI] [PubMed] [Google Scholar]
- 13.Strayer DS, Zern MA, Chowdhury JR. What can SV40-derived vectors do for gene therapy? Curr Opin Mol Ther. 2002 Aug;4(4):313–23. [PubMed] [Google Scholar]
- 14.Norkin LC. Simian virus 40 infection via MHC class I molecules and caveolae. Immunol Rev. 1999 Apr;168:13–22. doi: 10.1111/j.1600-065x.1999.tb01279.x. [DOI] [PubMed] [Google Scholar]
- 15.Pelkmans L, Puntener D, Helenius A. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science. 2002 Apr 19;296(5567):535–9. doi: 10.1126/science.1069784. [DOI] [PubMed] [Google Scholar]
- 16.Kondo R, Feitelson MA, Strayer DS. Use of SV40 to immunize against hepatitis B surface antigen: implications for the use of SV40 for gene transduction and its use as an immunizing agent. Gene Ther. 1998 May;5(5):575–82. doi: 10.1038/sj.gt.3300623. [DOI] [PubMed] [Google Scholar]
- 17.Strayer DS, Lamothe M, Wei D, Milano J, Kondo R. Generation of recombinant SV40 vectors for gene transfer. Methods Mol Biol. 2001;165:103–17. doi: 10.1385/1-59259-117-5:103. [DOI] [PubMed] [Google Scholar]
- 18.Schoumacher RA, Ram J, Iannuzzi MC, Bradbury NA, Wallace RW, Hon CT, et al. A cystic fibrosis pancreatic adenocarcinoma cell line. Proc Natl Acad Sci U S A. 1990 May;87(10):4012–6. doi: 10.1073/pnas.87.10.4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Egan ME, Glockner-Pagel J, Ambrose C, Cahill PA, Pappoe L, Balamuth N, et al. Calcium-pump inhibitors induce functional surface expression of Delta F508-CFTR protein in cystic fibrosis epithelial cells. Nat Med. 2002 May;8(5):485–92. doi: 10.1038/nm0502-485. [DOI] [PubMed] [Google Scholar]
- 20.West MR, Molloy CR. A microplate assay measuring chloride ion channel activity. Anal Biochem. 1996 Oct 1;241(1):51–8. doi: 10.1006/abio.1996.0377. [DOI] [PubMed] [Google Scholar]
- 21.Heeckeren A, Walenga R, Konstan MW, Bonfield T, Davis PB, Ferkol T. Excessive inflammatory response of cystic fibrosis mice to bronchopulmonary infection with Pseudomonas aeruginosa. J Clin Invest. 1997;100(11):2810–5. doi: 10.1172/JCI119828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sirninger J, Muller C, Braag S, Tang Q, Yue H, Detrisac C, et al. Functional characterization of a recombinant adeno-associated virus 5-pseudotyped cystic fibrosis transmembrane conductance regulator vector. Human Gene Therapy. 2004 Sep;15:832–41. doi: 10.1089/hum.2004.15.832. [DOI] [PubMed] [Google Scholar]
- 23.Dajani R, Zhang Y, Taft PJ, Travis SM, Starner TD, Olsen A, et al. Lysozyme secretion by submucosal glands protects the airway from bacterial infection. Am J Respir Cell Mol Biol. 2005 Jun;32(6):548–52. doi: 10.1165/rcmb.2005-0059OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akinbi HT, Epaud R, Bhatt H, Weaver TE. Bacterial killing is enhanced by expression of lysozyme in the lungs of transgenic mice. J Immunol. 2000 Nov 15;165(10):5760–6. doi: 10.4049/jimmunol.165.10.5760. [DOI] [PubMed] [Google Scholar]
- 25.Wiley SR, Kraus RJ, Zuo F, Murray EE, Loritz K, Mertz JE. SV40 early-to-late switch involves titration of cellular transcriptional repressors. Genes Dev. 1993 Nov;7(11):2206–19. doi: 10.1101/gad.7.11.2206. [DOI] [PubMed] [Google Scholar]
- 26.Pasyk EA, Foskett JK. Mutant (delta F508) cystic fibrosis transmembrane conductance regulator Cl- channel is functional when retained in endoplasmic reticulum of mammalian cells. J Biol Chem. 1995 May 26;270(21):12347–50. doi: 10.1074/jbc.270.21.12347. [DOI] [PubMed] [Google Scholar]
- 27.Johnson LG, Olsen JC, Sarkadi B, Moore KL, Swanstrom R, Boucher RC. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat Genet. 1992 Sep;2(1):21–5. doi: 10.1038/ng0992-21. [DOI] [PubMed] [Google Scholar]
- 28.Dorin JR, Farley R, Webb S, Smith SN, Farini E, Delaney SJ, et al. A demonstration using mouse models that successful gene therapy for cystic fibrosis requires only partial gene correction. Gene Ther. 1996 Sep;3(9):797–801. [PubMed] [Google Scholar]
- 29.Flotte TR. Recombinant adeno-associated virus gene therapy for cystic fibrosis and alpha(1)-antitrypsin deficiency. Chest. 2002;121(3 Suppl):98S–102S. doi: 10.1378/chest.121.3_suppl.98s. [DOI] [PubMed] [Google Scholar]
- 30.Mueller C, Flotte TR. Clinical gene therapy using recombinant adeno-associated virus vectors. Gene Ther. 2008 Jun;15(11):858–63. doi: 10.1038/gt.2008.68. [DOI] [PubMed] [Google Scholar]
- 31.Sauter BV, Parashar B, Chowdhury NR, Kadakol A, Ilan Y, Singh H, et al. A replication-deficient rSV40 mediates liver-directed gene transfer and a long-term amelioration of jaundice in gunn rats. Gastroenterology. 2000 Nov;119(5):1348–57. doi: 10.1053/gast.2000.19577. [DOI] [PubMed] [Google Scholar]
- 32.Middleton PG, Alton EW. Gene therapy for cystic fibrosis: which postman, which box? [see comments] Thorax. 1998;53(3):197–9. doi: 10.1136/thx.53.3.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferkol T, Perales JC, Eckman E, Kaetzel CS, Hanson RW, Davis PB. Gene transfer into the airway epithelium of animals by targeting the polymeric immunoglobulin receptor. J Clin Invest. 1995;95(2):493–502. doi: 10.1172/JCI117690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strayer DS, Milano J. SV40 mediates stable gene transfer in vivo. Gene Ther. 1996 Jul;3(7):581–7. [PubMed] [Google Scholar]
- 35.Ruiz FE, Clancy JP, Perricone MA, Bebok Z, Hong JS, Cheng SH, et al. A clinical inflammatory syndrome attributable to aerosolized lipid-DNA administration in cystic fibrosis. Hum Gene Ther. 2001 May 1;12(7):751–61. doi: 10.1089/104303401750148667. [DOI] [PubMed] [Google Scholar]
- 36.Zahm JM, Debordeaux C, Maurer C, Hubert D, Dusser D, Bonnet N, et al. Improved activity of an actin-resistant DNase I variant on the cystic fibrosis airway secretions. Am J Respir Crit Care Med. 2001 Apr;163(5):1153–7. doi: 10.1164/ajrccm.163.5.2005002. [DOI] [PubMed] [Google Scholar]
- 37.Louboutin JP, Liu B, Chekmasova AA, Reyes BA, van Bockstaele EJ, Strayer DS. Delivering genes to the organ-localized immune system: long-term results of direct intramarrow transduction. J Gene Med. 2007 Oct;9(10):843–51. doi: 10.1002/jgm.1084. [DOI] [PubMed] [Google Scholar]