

Summary
Phage φC31 integrase is a recombinase that can be expressed in mammalian cells to integrate plasmids carrying an attB sequence into the genome at specific pseudo attP locations. We demonstrate by immunofluoresence that wild-type φC31 integrase is cytoplasmic and that addition of a SV40 nuclear localization signal (NLS) localizes φC31 integrase to the nucleus. Unexpectedly, the NLS depressed integration efficiency in HeLa cells and provided no benefit when used to integrate the human Factor IX (hFIX) gene into mouse liver. Since breakdown of the nuclear membrane during mitosis could allow cytoplasmic integrase access to the chromosomes, we analyzed whether cell division was required for integration into liver cells in vivo. Hepatocytes were labeled with iododeoxyuridine to mark cells that underwent DNA replication during the week following hydrodynamic injection. Hydrodynamic delivery led to DNA replication in one-third of hepatocytes. Approximately 3 out of 4 cells having φC31 integrase-mediated stable hFIX expression did not undergo replication, indicating that cell division was not required for integrase function in liver. Therefore, although the bulk of φC31 integrase protein appears to be cytoplasmic in mammalian cells, integration can still occur in the nucleus, even without cell division.
Keywords: hydrodynamic delivery, liver, non-viral gene therapy, proliferation, nuclear localization signal
Introduction
A relatively recent addition to the gene therapy field is the development of the φC31 integrase system for permanent, sequence-specific genomic integration of plasmid DNA.1 The φC31 integrase has been used extensively for gene therapy applications2-5 and for genetic engineering to create and manipulate transgenic organisms.6-10 ΦC31 integrase is a serine recombinase encoded by the Streptomyces bacteriophage φC31.11,12 In nature, the enzyme catalyzes a unidirectional reaction between the 34 bp attB bacterial attachment site and the 39 bp attP phage attachment site.13,14 For use in gene medicine, the therapeutic gene plasmid is constructed to carry the attB sequence. The integrase mediates recombination between this plasmid and sites within the genome that are similar to attP in sequence, called pseudo attP sites.15
Little is understood about how φC31 integrase functions in the context of mammalian cells. The SV40 NLS has been added to the φC31 integrase at both the N and C-termini.10,16 Using an extrachromosomal plasmid on which the attB and attP sites must be recombined to express the reporter protein, Andreas et al. determined that the φC31 integrase carrying a C-terminal NLS performed 1.7-fold better than the wild-type enzyme.16 When the same reporter plasmid was integrated into the chromosomes of NIH 3T3 cells, expression of the reporter protein was 10-fold higher on average in cells that received the φC31 integrase with the NLS compared to wild-type.16 In another study, the φC31 integrase gene was both codon-optimized for expression in mouse and simultaneously modified to carry a C-terminal NLS.10 The optimized, NLS-carrying form of φC31 integrase clearly outperformed wild-type in an intrachromosomal recombination assay in the early transgenic mouse embryo.10
However, neither of these studies confirmed cytoplasmic localization of wild-type φC31 or nuclear localization of φC31 integrase carrying the NLS. In addition, φC31 integrase with a NLS was not compared to wild-type φC31 for integration of plasmid DNA into the genome, the reaction of interest in gene therapy, in either tissue culture cells or in a gene therapy setting. The localization of φC31 integrase appeared to be cytoplasmic when an integrase-enhanced green fluorescent protein (eGFP) fusion protein was visualized.17 Despite a predominantly cytoplasmic location, wild-type φC31 integrase must obtain access to mammalian chromosomes, because genomic integration events are found in 5-10% of unmodified tissue culture cells transfected with an attB-carrying plasmid.15 The integrase could access the DNA after dissolution of the nuclear envelope during mitosis. Alternatively, a small fraction of the protein may otherwise gain entry into the nucleus, and this amount may be sufficient to achieve integration. To address these hypotheses, we designed experiments to relocalize φC31 integrase to the nucleus and to investigate if cell division was required for integration.
We investigated these properties in vivo by delivering plasmids to the mouse liver by hydrodynamic, or high-pressure, tail vein injection. This method is an effective way to transfect up to 40% of hepatocytes after one rapid, high-volume injection of plasmid DNA.18,19 It has been shown that after hydrodynamic tail-vein injection, there may be some increase in cell division in the liver.20 However, the total extent of this proliferation has not been quantified. In order to investigate whether the φC31 integrase requires cell division to integrate an attB plasmid into the genome of hepatocytes, we first determined the extent of cell division in the liver after hydrodynamic injection. We then labeled replicating DNA to mark cells that had undergone cell division and stained for long-term transgene expression to deduce if φC31 integrase had a requirement for cell division. We hoped to better define the conditions that are required for φC31-mediated integration, as well as uncover possible ways in which the enzyme might be improved.
Results
Localization of HA-tagged φC31 integrase with and without a NLS
In order to detect φC31 integrase in the cell, we constructed a modified version of the wild-type enzyme with a C-terminal HA tag for easy antibody recognition (pCSI-HA). To localize φC31 integrase, FuGene 6 was used to transfect HeLa cells with pCSI-HA. After twenty-four hours, we fixed and stained the cells using an antibody to the HA tag. As shown in Figure 1, φC31 integrase was localized to the cytoplasm. This finding agreed with another report that used an eGFP-integrase fusion protein.17 To transport integrase to the nucleus, we added the SV40 NLS to wild-type integrase before or after the HA tag at the C-terminus. Figure 1 shows that both pCSI-HA-NLS and pCSI-NLS-HA produced proteins that were recognized by the HA antibody and localized to the nucleus.
Figure 1. Localization of φC31 integrase with and without a NLS in HeLa cells.
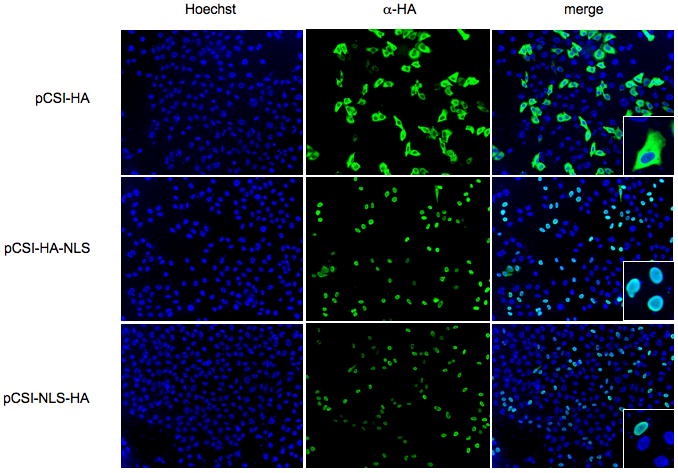
The φC31 integrase was fused to the 9 amino acid HA sequence to permit visualization, as well as an NLS before or after the HA sequence. HeLa cells grown on coverslips were transfected with the constructs indicated. The cells were fixed with 4% paraformaldehyde and stained for the HA tag and with Hoechst to label the DNA. Inset is a 9-fold amplification of the merge image. The NLS effectively relocalized the protein from the cytoplasm to the nucleus.
To compare the efficiency of φC31 integrase with and without the NLS, we performed a colony-forming assay in vitro. HeLa cells were transfected with pDB2, a plasmid carrying an attB site for integration and a neomycin resistance gene, and a series of integrase expression plasmids. We used a 1:50 ratio between pDB2 and the integrase plasmid to decrease background levels of random integration. (Figure 2a). Few colonies were obtained with pCSmI, a negative control that expresses an integrase protein lacking the catalytic serine (green bar), while abundant colonies were seen with pCSI, which expressed the wild-type integrase (pink bar). The HA tag of pCSI-HA (orange bar) had a slightly negative effect on the level of integration. Surprisingly, the NLS decreased integration frequency by four to five-fold. Both pCSI-NLS-HA (blue) and pCSI-HA-NLS (red) produced similar, lower colony numbers. This result suggested that the NLS was detrimental to either genomic integration or cellular survival. To determine if the NLS was toxic, we stained HeLa cells transfected with pCSI-HA, pCSI-NLS-HA, and pCSI-HA for cleaved caspase-3, a marker of cell death, and did not observe increased levels of apoptosis in cells that received the NLS plasmids (data not shown). Therefore, we attributed the decreased number of colonies obtained with the nuclear-localized integrase to lower numbers of cells that underwent a genomic integration and were thus able to form colonies, reflecting decreased integration efficiency.
Figure 2. Effect of nuclear localization of integrase on transgene expression in cell culture and in liver.
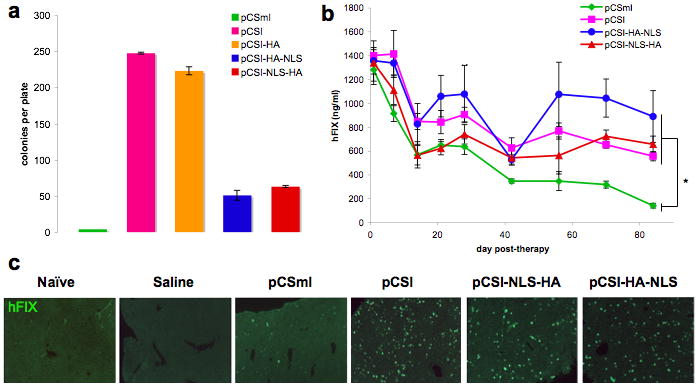
(a) HeLa cells were transfected using FuGene 6 in a 1:50 ratio of donor plasmid (pDB2) to integrase plasmid (pCSmI, inactive integrase, green; pCSI, wild-type integrase, pink; pCSI-HA, wild-type integrase with HA tag, orange; pCSI-HA-NLS, wild-type integrase with NLS after HA tag, blue; pCSI-NLS-HA, wild-type integrase with NLS before HA tag, red). Stable integrants were selected with G418, and the colonies that formed after two weeks that were greater than 1 mm in diameter were counted. p<10-7 for two-tailed t-test comparing pCSI-HA to pCSI-HA-NLS or pCSI-NLS-HA. (b) A solution containing 20 μg each of donor plasmid (pVFB, hAAT liver-specific promoter driving human Factor IX with an attB) and integrase plasmid (pCSmI, green diamond; pCSI, pink square; pCSI-HA-NLS, blue circle; pCSI-NLS-HA, red triangle) was administered to 8-10 week old female C57BL/6 mice by hydrodynamic injection. Mice were bled at the indicated time points, and ELISAs were done to determine hFIX levels in the serum. Control data were in common with another, simultaneous study, to reduce animal usage.35 (c) Staining of liver sections from mice in (b) for hFIX (green). Error bars are the standard error of the mean in (a) and (b); n ≥ 5 in (a) and (b). *p<0.005; two-tailed t-test performed on end-point data for each group, pCSI, pCSI-NLS-HA, or pCSI-HA-NLS against the pCSmI group.
To investigate the effect of the NLS on integration in an in vivo gene therapy setting, we performed hydrodynamic tail vein injection on groups of 5-7 mice with the NLS-containing integrase constructs and a human Factor IX (hFIX) attB donor plasmid (Figure 2b). Serum levels of hFIX were measured over a time course as an indication of stable integration. At the termination of the liver gene therapy experiment at 12 weeks post-injection, there was no significant difference between hFIX levels of mice in the pCSI, pCSI-NLS-HA and pCSI-HA-NLS groups, while each of these groups had significantly higher hFIX levels than the negative control, pCSmI (two-tailed t-test, p<0.005) attributed to random integration. During the period of stable hFIX expression from week 6 until week 12, the average and normalized hFIX values were 290 ng/ml (22.6% of the day 1 value) for mice given pCSmI, 651 ng/ml (46.5%) for mice given pCSI, 622 ng/ml (46.4%) for mice given pCSI-NLS-HA, and 884 ng/ml (65.2%) for mice given pCSI-HA-NLS. Although mice given pCSI-HA-NLS appeared to have higher hFIX values than those given pCSI and pCSI-NLS-HA, the levels at week 12 were not significantly different (two-tailed t-test, p=0.203 and p=0.264, respectively). To visualize hepatocyte expression of hFIX, the mice used for the study in Figure 2b were sacrificed, and sections of their livers were stained for the transgene (Figure 2c). It appeared that there was no increase in the numbers of cells staining positive for the transgene in the sections that received integrase constructs containing the NLS. Together, these data indicated that nuclear localization provided little or no benefit to φC31 integrase for liver gene therapy.
We also performed an experiment to examine the levels of the different forms of φC31 integrase. All constructs expressed φC31 integrase from the constitutive cytomegalovirus (CMV) promoter, so variations in protein levels might be due to differential degrees of solubility, stability, and degradation of the protein, rather than differential rates of synthesis. Although the same procedure for protein purification from HeLa cells was used, Int-NLS-HA and Int-HA-NLS were found to have different solubilities. The soluble and insoluble protein fractions were visualized by Western blot (Figure 3). While Int-NLS-HA was similar in solubility to Int-HA, a higher proportion of the Int-HA-NLS protein remained in the insoluble pellet after cell lysis. Furthermore, there was less Int-NLS-HA present than Int-HA. These differences in solubility and amount of the different forms of integrase could have effects on integrase activity.
Figure 3. The levels of soluble and insoluble φC31 integrase protein in HeLa cells.
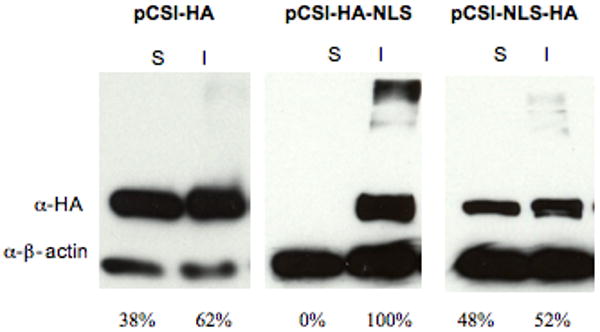
HeLa cells were transfected with 1 μg of pCSI-HA, pCSI-HA-NLS or pCSI-NLS-HA using FuGene 6. The soluble (S) and insoluble (I) protein extracts were subjected to immunoblotting for either the HA tag or β-actin. Protein amounts were normalized between samples transfected with pCSI-HA, pCSI-HA-NLS, or pCSI-NLS-HA by analyzing the soluble protein fraction by Bradford assay. For each sample, the same volume of soluble and insoluble protein was loaded onto the gel. Below each immunoblot is the percentage of the total protein in each pool of transfected cells that was either soluble or insoluble, as quantified by densitometry and normalized to the β-actin loading control.
Hydrodynamic DNA delivery induces hepatocyte proliferation
Since the NLS did not aid integration significantly, we hypothesized that integration via wild-type φC31 might occur preferentially during mitosis, when the nuclear envelope was dissolved. While the liver is normally quiescent, hepatocytes might be stimulated to divide by the hydrodynamic injection procedure. To test the extent of hepatocyte division following the gene therapy, liver cells were labeled with a nucleoside analogue during defined windows of time spanning two weeks following high-pressure tail vein injection.
Mice were given a hydrodynamic injection of 20 μg of an eGFP-expressing donor plasmid to verify that the DNA delivery procedure was successful. Animals were given IdU in their drinking water to label cells that were going through S-phase during the time period indicated (Figure 4a). Immediately following IdU exposure, mice were sacrificed and their sectioned livers were fixed and stained with DAPI and antibodies recognizing IdU and eGFP. Both the total number of nuclei and the number of IdU positive nuclei were counted (Figure 4b). The data showed that the period during which the most proliferation occurred after hydrodynamic delivery was between day one and two; 22.1% of nuclei were IdU positive. In mice that were given IdU for two weeks, 38.0% of nuclei were IdU positive, or about one-third of nuclei if the percentage observed after two weeks of IdU administration to a naïve mouse (6.2%) was subtracted. Similarly, the additive percentage of IdU positive nuclei over the first week was 34.7%. Because the number of dividing cells over the first week was similar to the number of dividing cells over the first two weeks, we concluded that the majority of cell division was completed by one week after hydrodynamic injection.
Figure 4. Hydrodynamic tail vein injection leads to the proliferation of hepatocytes.
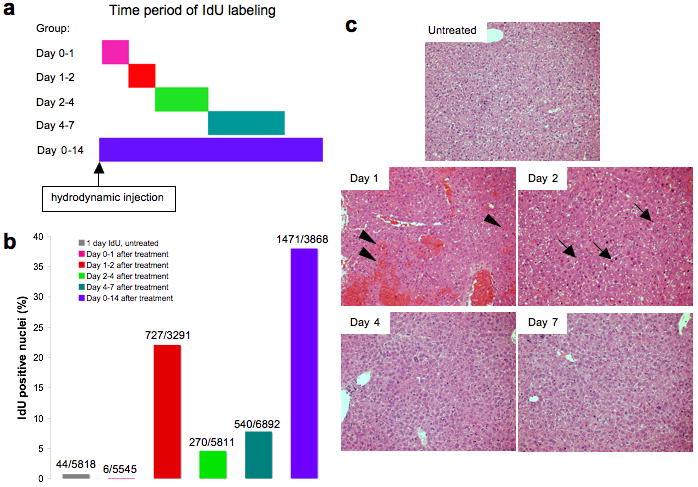
(a) Experimental design. Adult BALB/C mice received 20 μg of pDB2 via hydrodynamic injection and were given IdU in their drinking water for the time period indicated by the bar. Mice were then sacrificed and their livers were fixed in paraformaldehyde and subjected to sectioning. (b) Sectioned livers were stained for eGFP to confirm a successful injection and IdU to detect cells that replicated their DNA. IdU positive and all DAPI-stained negative nuclei were then counted. The exact numbers of cells counted are given above each bar (IdU+/all nuclei). The colors correspond to those in (a): grey, no injection and IdU labeled for one day (n=1); pink, IdU labeled between day 0-1 (n=2); red, day 1-2 (n=2); green, day 2-4 (n=2); turquoise, day 4-7 (n=2); purple, day 0-14 (n=1). (c) Hematoxylin and eosin stained representative sections of the livers from this experiment. Note the damage at day 1 indicated by erythrocytes among the hepatocytes (arrowheads), the large numbers of mitotic bodies at day 2 (condensed DNA; three are indicated by arrows), and apparently normal histology at days 4 and 7.
Serial sections stained with hematoxylin and eosin indicated that after hydrodynamic tail-vein injection, there was liver damage that was rapidly repaired. Figure 4c shows that at one day post-injection, there were large numbers of erythrocytes within the liver (arrowheads), indicating damage to the vasculature, as has been observed by others.21 A period of proliferation and repair took place at two days post-injection, during which condensed DNA, indicative of mitotic cells, was widespread (thin arrows). By 4 days post-injection, the liver was histologically indistinguishable from normal. Similarly, DNA replication subsided and was at background levels by 7 days post-injection. These histological were consistent with the IdU studies and indicated that a substantial level of cell division occurred, presumably in response to liver damage.
Cell division is not required for long-term transgene expression mediated by φC31 integrase
Since we determined that a significant degree of proliferation occurred in the liver following the hydrodynamic procedure, we were in a position to ask whether φC31 integrase had a direct requirement for cell division. We administered various integrase constructs along with the attB-hFIX donor plasmid pVFB by hydrodynamic injection, followed by a week-long dose of IdU drinking water. Livers were harvested at 13 weeks. At this time point, we have shown that the levels of unintegrated pVFB plasmid DNA was are very low and unlikely to make a major contribution to the abundant hFIX expression (Chavez, C.L., A.K., L.E.W., R.T.H., J. Chu, and M.P.C., manuscript in preparation).4 For example, very few kanamycin-resistant pVFB colonies formed when microgram amounts of total liver DNA were transformed into electrocompetent E. coli. At the same time, a PCR band representing the junction between the attB site on pVFB and an integration hotspot in the mouse genome appeared by three hours after injection and persisted at high intensity throughout the six-month time course of the experiment. Indeed, we have shown that a substantial portion of the extrachromosomal attB plasmid in liver becomes integrated when integrase is co-transfected.4 We therefore assumed that the majority of the hFIX positive cells at this time-point had undergone φC31 integrase-mediated genomic integration events, and thus that the enzyme had been active in those cells after hydrodynamic delivery. If cell division were required for integration, we would have expected a high fraction of the hFIX positive hepatocytes that received wild-type φC31 integrase to have IdU positive nuclei.
Liver sections from mice that were given no treatment, saline-only injection, or a hydrodynamic injection of both the hFIX-expressing donor plasmid (pVFB) and the integrase expression vector (pCSI, pCSI-HA-NLS, or pCSI-NLS-HA) were stained with antibodies for hFIX (green) and IdU (red), as well as DAPI to label the nuclei (blue). The merged image is shown for no injection, saline-only injection, and pVFB and pCSI injection (Figure 5a). 71% of hFIX-positive cells had IdU-negative nuclei in the animals that received integrase. This large fraction of hFIX-positive, non-proliferated hepatocytes was evidence that, although cell division was common in the post-hydrodynamic liver, φC31 integrase did not require cell division for integration to occur.
Figure 5. Cell division is not a requirement for integration and is slowed by integrase protein expression.
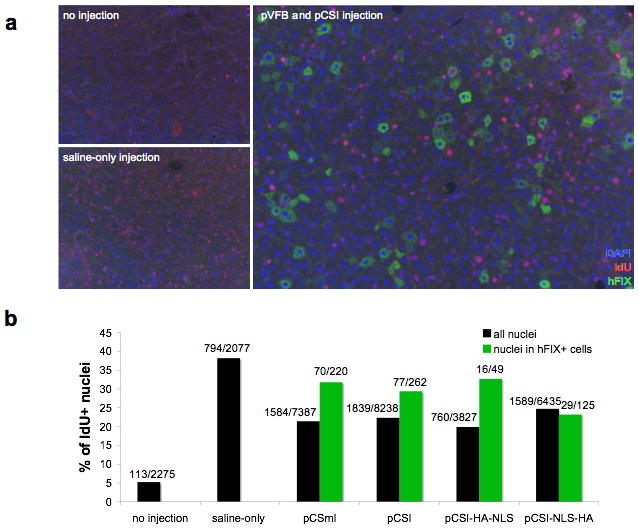
C57BL/6 mice were given 20 μg of pVFB and 20 μg of integrase expression vector (pCSmI, pCSI, pCSI-HA-NLS, or pCSI-NLS-HA) via hydrodynamic injection. Mice were provided with IdU-laced drinking water for one week following hydrodynamic injection. 13 weeks after DNA delivery, livers were fixed and stained for IdU to label cycling cells and hFIX to label cells that were likely to have received integration events. (a) Representative sections are shown for mice given no injection, saline-only injection, or injection of pVFB and pCSI. In many of the hFIX-positive cells, the nuclei do not stain for IdU, indicating that DNA replication did not occur during the week following the procedure. (b) Multiple sections from the injected mice were quantified by counting the number of IdU positive cells within all nuclei (black bars) or the number of IdU positive nuclei within cells that stained positive for hFIX (green bars). The sections chosen deliberately focused on areas that had a high number of hFIX-positive cells, so that a maximum number of hFIX-positive cells could be counted for each group. Consequently, the percentage of cells that were hFIX-positive in Fig. 5b was not representative of the total percentage of hFIX positive cells in the sample. The percentage of IdU positive cells was graphed and the raw data used to calculate that percentage is shown over each bar. All mice used for quantification had the expected serum hFIX level the day following the injection, indicating that the procedure was successful. Hydrodynamic injection without DNA significantly increased proliferation in the liver (Chi-squared test, p<0.0001). There was a decrease in proliferation relative to saline-only injected mice when hFIX and any form of integrase protein were expressed (p<0.0001). The number of IdU positive cells in the population staining positive for hFIX was significantly higher than the overall population for pCSmI (n=5; p<0.0046) and pCSI (n=4; p<0.0369) but not for pCSI-NLS-HA (n=3) or pCSI-HA-NLS (n=2). In cells that were positive for hFIX (green bars), 71% were not replicating their DNA after the hydrodynamic procedure, indicating that φC31 integrase did not require cell division for integration into hepatocyte genomic DNA.
For every group, we scored thousands of cells, including dozens of hFIX-positive cells, for their co-staining for IdU (Figure 5b). To maximize the number of hFIX-positive cells counted, sections were chosen that had the greatest numbers of such cells. Therefore, note that while the sections shown in Fig 2c were chosen to be representative of the percentage of hepatocytes that express hFIX in each group, the numbers of hFIX positive cells counted for Fig 5b do not reflect the total percentage of hFIX positive cells in the sample. For example, while the number of hFIX cells that were scored in Fig. 5b was similar for pCSmI and pCSI, the overall number of hFIX positive cells was significantly lower for pCSmI, as indicated in Fig. 2c.
The percentage of cells in S-phase in mice that received no treatment was about 5%, whereas in the mice that received a saline-only injection, 38% of cells experienced S-phase (Figure 5b). This number agreed well with the data reported in Figure 4b in mice that received a plasmid DNA injection of an eGFP-expressing plasmid. This concordance both confirmed that about one-third of hepatocytes synthesized new DNA at some point during the week after hydrodynamic delivery and indicated that injection of DNA to produce a marker protein had no effect on hepatocyte proliferation. Unexpectedly, when the pVFB donor plasmid expressing hFIX and any form of the φC31 integrase protein were present, including the inactive enzyme, the number of IdU positive cells throughout the liver was significantly reduced (Fig. 5b; Chi-squared test comparing each group to saline-only control, p<0.0001 in each case). This decrease in proliferation appeared less pronounced in the cells that expressed hFIX long-term for pCSmI, pCSI, and pCSI-HA-NLS. Statistical analysis indicated that among hFIX positive cells, the fraction of cells that were dividing was significantly higher for pCSmI (Chi-squared test, p=0.0046) and pCSI (p=0.0369), but not for pCSI-NLS-HA (p=0.084) or pCSI-HA-NLS (p=0.7643). This result suggested that cell division was somewhat favorable for integration without the NLS, but most integration still occurred in non-dividing cells.
Discussion
Our data reveal new information about the interaction of φC31 integrase with mammalian cells. First, we demonstrated that while the majority of φC31 integrase protein does not enter the nucleus of mammalian cells, nuclear localization could be achieved by the addition of the SV40 NLS. However, nuclear localization did not aid integrase efficiency either in cell culture or in vivo at the task of inserting an attB-containing plasmid into genomic pseudo attP sites. Second, we showed that hepatocyte proliferation was widespread after hydrodynamic tail vein injection. Even so, the majority of integration events occurred in non-dividing cells, indicating that the integrase did not require cell division to integrate a transgene permanently into the genome of mouse liver cells.
Unless φC31 integrase contained a nuclear localization signal, the 69 kDa protein was expected to be localized to the cytoplasm, because nuclear pore complexes inhibit the nuclear entry of proteins over about 40 kDa.22 We demonstrated such cytoplasmic localization, making it somewhat surprising that this phage integrase has generally been successful at mediating recombination with mammalian genomic DNA residing in the nucleus. We attempted to increase the likelihood of the integrase encountering the genomic DNA by adding a NLS to the protein, but instead discovered that nuclear localization failed to improve integration in cell culture or in a mouse model of liver gene therapy. Retroviral integrases follow a clear pattern of either possessing nuclear localization sequences of their own to allow nuclear entry in non-dividing cells or else functioning only in mitotic populations.23 Interestingly, the residues that permit DNA binding are often the same amino acids that allow nuclear entry.24 It is possible that the DNA-binding domains of φC31 integrase also permit a small fraction of integrase protein to enter the nucleus. In addition, cytoplasmic and nuclear φC31 integrase could be degraded by different mechanisms.
Nuclear localization may be more detrimental in tissue culture than in vivo due to key differences between these tests of integration efficiency. It is possible that the aggregated, insoluble fraction of φC31 integrase was more easily sorted and degraded in the cytoplasm, resulting in cells that were able to divide faster and therefore form a higher number of colonies, explaining why the cytoplasmic form provided higher integration efficiency in HeLa cells. In contrast, in liver, cells were not required to divide continuously for two weeks to form a colony, but rather simply to produce hFIX protein from the integrated transgene, in which case the bulk of nuclear integrase may have been less of an impediment. Additionally, the length of expression of φC31 integrase from the CMV promoter 28,29 is shorter in hepatocytes than in cultured cells (our unpublished data). Perhaps because of these differences between the assays, the nuclear form was less disadvantaged in vivo than it was in vitro.
We concluded that the SV40 NLS provided no significant, reproducible benefit to integration with φC31 integrase when used in conjunction with hydrodynamic liver gene therapy. Whether this result translates directly to other gene therapy approaches is unknown. For use of φC31 integrase in other applications, performance of integrase with and without the NLS should be evaluated, because the NLS may actually decrease the number of integrants in some instances, as evidenced by our cell culture data (Figure 2a). Since our integrase-NLS constructs showed differences in solubility and protein level from wild-type integrase, it is possible that other nuclear localization signals or different placement of these additional amino acids within the protein could result in a positive effect on integration efficiency. Additionally, the HA tag may have interfered somehow with the function of the protein in conjunction with the NLS. In further experiments, other methods of nuclear localization than the one described here should be tested. Reducing the amount of nuclear integrase protein may be beneficial. We are currently investigating way to decrease and possibly regulate the overall amount of integrase in the cell, while increasing the amount of soluble, functioning integrase.
Our IdU labeling studies indicated that large numbers of hepatocytes that entered the cell cycle after high-pressure tail vein injection, suggesting that the procedure is approximately as damaging to the liver as a one-third partial hepatectomy, in the amount of regeneration needed.25 This damage is reflected by the transient elevation of several liver enzymes after hydrodynamic DNA delivery.26 Fortunately, the liver is a highly regenerative organ that can recover from damage. Relevant to translation of the hydrodynamic procedure to humans, the level of liver damage incurred during a hydrodynamic clinical protocol, which was similar to procedures already performed in pig,27-30 appeared to be acceptable.27 The clinical procedure targeted only one lobe of the liver rather than the entire area,27 and thus perhaps only 5% of the entire liver was damaged, then regenerated. Use of the hydrodynamic procedure may need to be limited to serious disease to prevent mitotic entry and thus tumor promotion in cases when tumor initiators have already been active,31 such as in patients with cirrhosis. In any case, work in rats has already indicated that the procedure may not be effective in fibrotic liver tissue such as is present in patients with liver cirrhosis.32
Transfection with φC31 integrase and hFIX plasmids appeared to slow hepatocyte proliferation, as shown in Figure 5b. Because the same effect was observed regardless of integrase activity, the effect cannot be attributed to the catalytic activity of the integrase and may be due to the presence of proteins encoded by genes on the plasmids. One possibility is that if synthesis of φC31 integrase and/or hFIX saturated the resources available to the cell, decreased proliferation could result. If this were the case, one would expect transfected cells to be less likely to proliferate, but we observed the opposite. Cells that still expressed hFIX at 13 weeks post-injection were more likely to enter S-phase during the week following the procedure. Rather, it appears that the transfected cells may have produced cytokines or other factors that were capable of acting in trans to modulate the proliferative capacity of the entire liver.
Our results indicate that φC31 integrase activity does not depend upon cell division in mouse liver. We have found that the bulk of unintegrated pVFB plasmid DNA is integrated or lost from the liver by 48 hours after hydrodynamic injection (Chavez, C.L., A.K., L.E.W., R.T.H., and M.P.C., manuscript in preparation), so the vast majority of hFIX-expressing cells at 3 months after hydrodynamic injection were likely to carry an integrated hFIX transgene. Most of the cells that expressed the hFIX transgene at that time-point did not replicate their DNA during the regenerative phase following the procedure. The flexibility of the enzyme with regard to proliferative state could be specific for hepatocytes or a more generalized phenomenon. We do not know if the DNA delivery method and/or differences in the cellular environment of other tissues will affect this feature of the φC31 integrase. In the future, studies will be undertaken to determine the requirement for cell division in other tissues and in cell culture. Preliminarily, the ability of φC31 integrase to act without cell division indicates that it may provide a method for gene addition in tissues that are largely quiescent.
Materials and Methods
Plasmids
The attB-carrying pDB2,33 pNBL2,34 and pVFB35 donor plasmids have been described. Briefly, pDB2 carries eGFP gene driven by the CMV promoter and pVFB carries the hFIX gene driven by a liver-specific promoter composed of the human alpha-1 anti-trypsin promoter with an apolipoprotein E enhancer. pDB2 also carries the neomycin resistance gene driven by an SV40 promoter.
pCSI (or pCMVInt),15 pCSmI,36 and pCSI-HA35 have been described previously. pCSI-NLS-HA and pCSI-HA-NLS were cloned in parallel by the same method. All NLS and HA tags were placed onto the carboxy-terminal region of the protein. PCR was performed with the forward primer 5′-CTGAGGTGATCTACAAGAA-3′ and the reverse primer 5′-ATTCCGGGATCCAGTCTAAGCGTAGTCTGGGACGTCGTATGGGTAGCCAACCTTCCTCTTCTTCTTAGGGCCCGCCGCTACGTCTTCCGTG-3′ for pCSI-NLS-HA or 5′-ATTCCGGGATCCAGTCTAAACCTTCCTCTTCTTCTTAGGGCCAGCGTAGTCTGGGACGTCGTATGGGTAGCCCGCCGCTACGTCTTCCGTG-3′ to make pCSI-HA-NLS. The resulting PCR products were sequentially BamHI and BstEII digested to produce 950 bp fragments, which were substituted for the wild-type sequence in pCSI.
Tissue Culture
Human cervical cancer HeLa cells (CCL-2; ATCC, Manassas, VA, USA) were grown in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 9% fetal bovine serum and 1% penicillin/streptomycin (Invitrogen). Plates of cells were transfected according to manufacturer's instructions using FuGene 6 (Roche, Palo Alto, CA, USA) with DNA prepared by Maxiprep kit (Qiagen, Valencia, CA, USA) at a 3:1 reagent (μl) to DNA (μg) ratio.
To perform colony assays to determine integration efficiency, one well from a 6-well plate was split 1:7 onto a 10 cm plate, and neomycin resistant cells were selected with G418 supplemented media. Five plates were counted and averaged for each group in the experiment. Colonies that formed after two weeks were stained with methylene blue and counted.
To obtain protein, the cells were washed with PBS, lysed by adding 300 μl of fresh protein lysis buffer [50mM Tris pH 7.5-7.8, 100 mM NaCl, 5 mM EDTA, 0.4% Triton X-100, 0.04 M β-glycerophosphate, 0.05 M NaF, 0.002M Na orthovanadate, 0.001 M PMSF, 0.01% Aprotonin (Sigma, St. Louis, MO, USA)] for 7 minutes on ice, then centrifuged at 4°C, 12k RPM for 7 minutes. The supernatant was transferred to a new Eppendorf tube, kept on ice, flash frozen in liquid nitrogen, and stored at -80°C until use. To analyze insoluble protein, the pellet obtained after centrifugation was resuspended in a volume of additional lysis buffer equal to that of the supernatant.
Mice
8-10 week old BALB/c or C57BL/6 mice were ordered from Charles Rivers (Wilmington, MA, USA) or Jackson Laboratories (Bar Harbor, ME, USA). Mice were kept in the Stanford Research Animal Facility and given water and food ad libitum. For the IdU experiments, a 0.8 or 1 mg/mL solution of IdU in water, protected from light and refreshed several times per week, was given for the time period indicated. The Stanford Research Animal Committee approved all animal studies.
Hydrodynamic Tail-Vein Injection
A DNA solution was prepared that diluted 20 μg of each indicated plasmid in 1.8 mL of buffer. All DNA was prepared by endotoxin-free Maxiprep (Qiagen) according to the manufacturer's protocol, with the exception that the DNA pellets were reconstituted in either high-quality water (Qiagen) or HBSS (Invitrogen). For the initial set of proliferation studies using pDB2, BALB/c mice were anesthetized with isoflourane. Heat pads were placed on the tail to dilate the vein, and mice were injected with 20 μg of pDB2 diluted in 1.8 mL of calcium and magnesium-free PBS (Invitrogen). Alternatively, in all other studies, the animals were placed under a heat lamp until their tail veins were clearly dilated, at which time they were placed into a mouse restrainer (Braintree Scientific, Braintree, MA, USA) and injected with a DNA solution diluted in calcium and magnesium-free HBSS (Invitrogen). All mice were injected with 1.8 mL of DNA solution as quickly as possible (∼3-6 seconds) using a 3 mL syringe (BD Biosciences) and a 27-gauge regular or butterfly needle (BD Biosciences).
Immunoflourescence and Live Cell Imaging
To stain HeLa cells, the cells were seeded onto 6-well plates containing 18 mm diameter glass coverslips (VWR, Westchester, PA, USA) and transfected with FuGene 6 (Roche). 24 hours later, the cells were fixed with 4% paraformaldehyde (PFA) in PBS for 20 minutes at room temperature, followed by five PBS washes. Coverslips were then removed from wells and placed in a dark humidified chamber. Cells were permeabilized with PBS + 0.1% Triton X and blocked in block solution (PBS + 10% goat serum, 5% BSA, and 0.1% Triton-X 100). A FITC-conjugated anti-HA antibody (F-7, Santa Cruz Biotechnology, Santa Cruz, CA, USA) was diluted 1:500 in block solution and incubated on the cells for one hour, followed by 5 washes in PBS + 0.05% Tween 20. Then, the dye Hoechst (Invitrogen) was added to stain the DNA, followed by five more washes. Cells were mounted onto microscope slides with Aqua-Mount (Thermo Fisher Scientific, Pittsburgh, PA, USA) and sealed with clear nail polish.
For immunostaining, liver tissues were paraffin-embedded and sectioned by Histo-tec Laboratory (Hayward, CA, USA) or the Stanford University histology core. The staining procedure has been described previously,35 with the following modifications. If the slides needed to be double-stained, the tissues were fixed after the first secondary antibody washes were complete with 4% PFA in PBS for 15 minutes, washed, blocked again, and incubated with the second primary antibody overnight at 4°C followed by the second secondary antibody for one hour. Slides were then either incubated with DAPI diluted 1:1000 for 5 minutes and washed again, then mounted with Mowiol mounting media (for eGFP staining), or mounted with ProLong Gold Antifade Mounting Media with DAPI (all other staining; Invitrogen). The antibodies used were: rabbit α-GFP (A6455; Invitrogen), mouse α-BrdU (for IdU) (BD Biosciences), goat α-hFIX (GAFIX-AP; Affinity Biologicals, Ancaster, ON, Canada), Alexafluor goat α-rabbit-488 (Invitrogen), Alexafluor goat α-mouse-594 (Invitrogen), Alexafluor rabbit α-goat-488 (Invitrogen), Alexafluor rabbit α-mouse-594 (Invitrogen). All were used at a 1:500 dilution in serum buffer. The serum buffer used contained PBS + 0.1% Tween 20 and either 10% goat serum + 1% BSA (for staining with the eGFP antibody) or 10% rabbit serum (for staining with the hFIX antibody). The antibody to be stained with a 488-conjugated secondary was stained for first when two antigens were visualized together. Images of stained cells were taken on an Axioskop 2 plus microscope with a FluoArc power supply with an AxioCam MRc camera (Zeiss, Thornwood, NY, USA).
Immunoblotting
Frozen protein samples were analyzed by Bradford assay (BioRad, Hercules, CA) for protein content. Controls, protein ladders, and time point samples (15 μg of protein) were run on 7.5% Tris HCl Ready Gels (BioRad) and transferred to nitrocellulose TransBlot Transfer Medium (BioRad). Membranes were blocked with 5% non-fat dry milk in TN buffer (0.03 M Tris pH 7.6, 0.15 M NaCl). The membranes were cut at the 50 kDa marker into two halves. The upper half was incubated in rabbit α-HA (H6908, Sigma) diluted 1:1000 in milk buffer overnight, then washed five times for seven minutes in TTBS (TN + 0.05% Tween 20), followed by the goat α-rabbit-HRP secondary antibody (Calbiochem, Darmstadt, Germany) diluted 1:10,000 in milk buffer. The lower half was incubated in mouse α-β-actin (NB 600-501, Novus Biologicals, Littleton, CO, USA) diluted 1:1000 in milk buffer overnight, then washed five times for seven minutes in TTBS followed by the goat α-mouse-HRP secondary antibody (Calbiochem) diluted 1:10,000 in milk buffer. After incubation with the secondary antibodies, the blots were washed five times in TTBS and once in TN, then incubated with SuperSignal West Pico Substrate (Thermo Scientific, Waltham, MA, USA) and imaged with BioMax film (Kodak, Rochester, NY, USA). Densitometry was performed using ImageJ software.
Acknowledgments
Many thanks to Julien Sage and members of his laboratory at Stanford for assistance with the liver staining experiments. Thanks to Anne Brunet and members of her lab for assistance with their microscope. We appreciate the contributions of Christopher L. Chavez to the conclusions made in this paper. PHS Grant Number CA09302, awarded by the National Cancer Institute, DHHS, supported LEW. RTH was supported by a medical research training fellowship from the Howard Hughes Medical Institute. This work was supported by NIH grant HL068112 to MPC.
References
- 1.Calos MP. The phiC31 integrase system for gene therapy. Curr Gene Ther. 2006;6:633–645. doi: 10.2174/156652306779010642. [DOI] [PubMed] [Google Scholar]
- 2.Chalberg TW, Genise HL, Vollrath D, Calos MP. phiC31 integrase confers genomic integration and long-term transgene expression in rat retina. Invest Ophthalmol Vis Sci. 2005;46:2140–2146. doi: 10.1167/iovs.04-1252. [DOI] [PubMed] [Google Scholar]
- 3.Ortiz-Urda S, Thyagarajan B, Keene DR, Lin Q, Calos MP, Khavari PA. PhiC31 integrase-mediated nonviral genetic correction of junctional epidermolysis bullosa. Hum Gene Ther. 2003;14:923–928. doi: 10.1089/104303403765701204. [DOI] [PubMed] [Google Scholar]
- 4.Olivares EC, Hollis RP, Chalberg TW, Meuse L, Kay MA, Calos MP. Site-specific genomic integration produces therapeutic Factor IX levels in mice. Nat Biotechnol. 2002;20:1124–1128. doi: 10.1038/nbt753. [DOI] [PubMed] [Google Scholar]
- 5.Bertoni C, Jarrahian S, Wheeler TM, Li Y, Oliveres EC, Calos MP, et al. Enhancement of plasmid-mediated gene therapy for muscular dystrophy by directed plasmid integration. Proc Natl Acad Sci U S A. 2006;103:419–424. doi: 10.1073/pnas.0504505102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belteki G, Gertsenstein M, Ow DW, Nagy A. Site-specific cassette exchange and germline transmission with mouse ES cells expressing phiC31 integrase. Nat Biotechnol. 2003;21:321–324. doi: 10.1038/nbt787. [DOI] [PubMed] [Google Scholar]
- 7.Allen BG, Weeks DL. Transgenic Xenopus laevis embryos can be generated using phiC31 integrase. Nat Methods. 2005;2:975–979. doi: 10.1038/nmeth814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khan MS, Khalid AM, Malik KA. Phage phiC31 integrase: a new tool in plastid genome engineering. Trends Plant Sci. 2005;10:1–3. doi: 10.1016/j.tplants.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Bateman JR, Lee AM, Wu CT. Site-specific transformation of Drosophila via phiC31 integrase-mediated cassette exchange. Genetics. 2006;173:769–777. doi: 10.1534/genetics.106.056945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raymond CS, Soriano P. High-efficiency FLP and PhiC31 site-specific recombination in mammalian cells. PLoS ONE. 2007;2:e162. doi: 10.1371/journal.pone.0000162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuhstoss S, Rao RN. Analysis of the integration function of the streptomycete bacteriophage phi C31. J Mol Biol. 1991;222:897–908. doi: 10.1016/0022-2836(91)90584-s. [DOI] [PubMed] [Google Scholar]
- 12.Rausch H, Lehmann M. Structural analysis of the actinophage phi C31 attachment site. Nucleic Acids Res. 1991;19:5187–5189. doi: 10.1093/nar/19.19.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groth AC, Olivares EC, Thyagarajan B, Calos MP. A phage integrase directs efficient site-specific integration in human cells. Proc Natl Acad Sci U S A. 2000;97:5995–6000. doi: 10.1073/pnas.090527097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thorpe HM, Smith MC. In vitro site-specific integration of bacteriophage DNA catalyzed by a recombinase of the resolvase/invertase family. Proc Natl Acad Sci U S A. 1998;95:5505–5510. doi: 10.1073/pnas.95.10.5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thyagarajan B, Oliveres EC, Hollis RP, Ginsburg DS, Calos MP. Site-specific genomic integration in mammalian cells mediated by phage phiC31 integrase. Mol Cell Biol. 2001;21:3926–3934. doi: 10.1128/MCB.21.12.3926-3934.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andreas S, Schwenk F, Küter-Luks B, Faust N, Kühn R. Enhanced efficiency through nuclear localization signal fusion on phage PhiC31-integrase: activity comparison with Cre and FLPe recombinase in mammalian cells. Nucleic Acids Res. 2002;30:2299–2306. doi: 10.1093/nar/30.11.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen JZ, Ji CN, Xu GL, Pang RY, Yao JH, Zhu HZ, et al. DAXX interacts with phage PhiC31 integrase and inhibits recombination. Nucleic Acids Res. 2006;34:6298–6304. doi: 10.1093/nar/gkl890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–1266. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 19.Zhang G, Budker V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum Gene Ther. 1999;10:1735–1737. doi: 10.1089/10430349950017734. [DOI] [PubMed] [Google Scholar]
- 20.Rossmanith W, Chabicovsky M, Herkner K, Schulte-Hermann R. Cellular gene dose and kinetics of gene expression in mouse livers transfected by high-volume tail-vein injection of naked DNA. DNA Cell Biol. 2002;21:847–853. doi: 10.1089/104454902320908496. [DOI] [PubMed] [Google Scholar]
- 21.Suda T, Gao X, Stolz DB, Liu D. Structural impact of hydrodynamic injection on mouse liver. Gene Ther. 2007;14:129–137. doi: 10.1038/sj.gt.3302865. [DOI] [PubMed] [Google Scholar]
- 22.Bonner WM. Proximity and accessibility studies of histones in nuclei and free nucleosomes. Nucleic Acids Res. 1978;5:71–85. doi: 10.1093/nar/5.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whittaker GR, Helenius A. Nuclear import and export of viruses and virus genomes. Virology. 1998;246:1–23. doi: 10.1006/viro.1998.9165. [DOI] [PubMed] [Google Scholar]
- 24.Gallay P, Hope T, Chin D, Trono D. HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway. Proc Natl Acad Sci U S A. 1997;94:9825–9830. doi: 10.1073/pnas.94.18.9825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lambotte L, Saliez A, Triest S, Tagliaferri EM, Barker AP, Baranski AG. Control of rate and extent of the proliferative response after partial hepatectomy. Am J Physiol. 1997;273:G905–912. doi: 10.1152/ajpgi.1997.273.4.G905. [DOI] [PubMed] [Google Scholar]
- 26.Budker VG, Subbotin VM, Budker T, Sebestyén MG, Zhang G, Wolff JA. Mechanism of plasmid delivery by hydrodynamic tail vein injection. II Morphological studies. J Gene Med. 2006;8:874–888. doi: 10.1002/jgm.920. [DOI] [PubMed] [Google Scholar]
- 27.Khorsandi SE, Bachellier P, Weber JC, Greget M, Jaeck D, Zacharoulis D, et al. Minimally invasive and selective hydrodynamic gene therapy of liver segments in the pig and human. Cancer Gene Ther. 2008;15:225–230. doi: 10.1038/sj.cgt.7701119. [DOI] [PubMed] [Google Scholar]
- 28.Yoshino H, Hashizume K, Kobayashi E. Naked plasmid DNA transfer to the porcine liver using rapid injection with large volume. Gene Ther. 2006;13:1696–1702. doi: 10.1038/sj.gt.3302833. [DOI] [PubMed] [Google Scholar]
- 29.Aliño SF, Herrero MJ, Noguera I, Dasí F, Sánchez M. Pig liver gene therapy by noninvasive interventionist catheterism. Gene Ther. 2007;14:334–343. doi: 10.1038/sj.gt.3302873. [DOI] [PubMed] [Google Scholar]
- 30.Fabre JW, Grehan A, Whitehorne M, Sawyer GJ, Dong X, Salehi S, et al. Hydrodynamic gene delivery to the pig liver via an isolated segment of the inferior vena cava. Gene Ther. 2008;15:452–462. doi: 10.1038/sj.gt.3303079. [DOI] [PubMed] [Google Scholar]
- 31.Schulte-Hermann R. Initiation and promotion in hepatocarcinogenesis. Arch Toxicol. 1987;60:179–181. doi: 10.1007/BF00296976. [DOI] [PubMed] [Google Scholar]
- 32.Yeikilis R, Gal S, Kopeiko N, Paizi M, Pines M, Braet F, et al. Hydrodynamics based transfection in normal and fibrotic rats. World J Gastroenterol. 2006;12:6149–6155. doi: 10.3748/wjg.v12.i38.6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keravala A, Portlock JL, Nash JA, Vitrant DG, Robbins PD, Calos MP. PhiC31 integrase mediates integration in cultured synovial cells and enhances gene expression in rabbit joints. J Gene Med. 2006;8:1008–1017. doi: 10.1002/jgm.928. [DOI] [PubMed] [Google Scholar]
- 34.Thyagarajan B, Calos MP. Site-specific integration for high-level protein production in mammalian cells. Methods Mol Biol. 2005;308:99–106. doi: 10.1385/1-59259-922-2:099. [DOI] [PubMed] [Google Scholar]
- 35.Keravala A, Lee S, Thyagarajan B, Olivares EC, Gabrovsky VE, Woodard LE, et al. Mutational derivatives of PhiC31 integrase with increased efficiency and specificity. Mol Ther. 2009;17:112–120. doi: 10.1038/mt.2008.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Portlock JL, Keravala A, Bertoni C, Lee S, Rando TA, Calos MP. Long-term increase in mVEGF164 in mouse hindlimb muscle mediated by phage phiC31 integrase after nonviral DNA delivery. Hum Gene Ther. 2006;17:871–876. doi: 10.1089/hum.2006.17.871. [DOI] [PubMed] [Google Scholar]