

SUMMARY
Human embryonic stem cells (hESCs) provide a novel source of hematopoietic and other cell populations suitable for gene therapy applications. Preclinical studies to evaluate engraftment of hESC-derived hematopoietic cells transplanted into immunodeficient mice demonstrate only limited repopulation. Expression of a drug resistance gene, such as Tyr22-dihydrofolate reductase (Tyr22-DHFR), coupled to methotrexate (MTX) chemotherapy has the potential to selectively increase engraftment of gene-modified hESC-derived cells in mouse xenografts. Here, we describe the generation of Tyr22-DHFR – GFP expressing hESCs that maintain pluripotency, produce teratomas and can differentiate into MTXr-hemato-endothelial cells. We demonstrate that MTX administered to nonobese diabetic/severe combined immunodeficient/IL-2Rγcnull (NSG) mice after injection of Tyr22-DHFR-derived cells significantly increases human CD34+ and CD45+ cell engraftment in the bone marrow (BM) and peripheral blood of transplanted MTX-treated mice. These results demonstrate that MTX treatment supports selective, long-term engraftment of Tyr22-DHFR-cells in vivo, and provides a novel approach for combined human cell and gene therapy.
Keywords: dihydrofolate reductase, methotrexate, embryonic stem cells
INTRODUCTION
Over the past two decades, development of stem cell-based gene therapies has been challenging due to low level of gene transfer in human hematopoietic stem cells (HSCs) and difficulties with long-term stable engraftment of modified cells 1. In clinical trials for X-linked severe combined immunodeficiency (X-SCID) and adenosine deaminase deficiency (ADA-SCID), the growth advantage of gene-modified lymphocytes compensated for the low level of gene transfer to support immune reconstitution 2–5. However, in cases where the gene-of-interest does not confer a selective advantage to modified cells, drug resistance gene expression in HSCs may be clinically beneficial. For example, drug resistance gene expression coupled to chemotherapy has been shown to selectively expand gene-modified cells in vivo 6–8. In addition, drug resistance gene expression in nonmalignant chemosensitive tissues, such as the bone marrow and gastrointestinal tract, has the potential to decrease toxicity associated with high-dose chemotherapy7. In the allogeneic setting, coupling post-transplantation immunosuppressive drug treatment to drug-resistance in the allograft also has the potential to reduce the risk of graft rejection and the severity of graft-versus-host disease (GVHD).
Methotrexate is widely used in post-hematopoietic stem cell transplantation (HSCT) immunosuppressive therapy and in the treatment of some cancers 9–11. We and others have shown that gene transfer and expression of a MTX-resistant dihydrofolate reductase (Tyr22-DHFR) in hematopoietic stem cells (HSCs) confers chemoprotection to recipient animals 12–14. In these studies, MTX supported a transient drug-dependent selective expansion of gene-modified cells in the peripheral blood of treated animals. These results suggest that MTX administration may be used to regulate in vivo expansion of gene-modified cells.
Human embryonic stem cells (hESCs) are capable of self-renewal and can be induced to differentiate into diverse hematopoietic cell lineages including myeloid and lymphoid lineages of characteristic phenotypes and function 15–20. We have also found that natural killer (NK) cells derived from hESCs have potent anti-tumor activity both in vitro and in vivo 21,22. Others have characterized maturation of erythroid cells from hESCs with normal oxygen carrying capacity 23–25. However, in studies to date, only limited long-term hematopoietic potential of hESC-derived cells has been observed when transplanted into immunodeficient mice or in fetal sheep models 26–29. Several studies have focused on gene modification of hESCs, defining optimal conditions for achieving stable viral and non-viral transfer of genes encoding bioluminescent or fluorescent proteins 30–32. Other groups have demonstrated expression of antibiotic resistance genes for enrichment of drug-resistant cells 33–35, or used lineage- or stage-specific regulation of GFP to monitor differentiation of hESCs 36–39. To date, gene transfer and in vivo expression of chemotherapeutic drug resistance genes, such as Tyr22-DHFR, in hESC-derived cells has not been reported.
Clinically, Tyr22-DHFR expression by hematopoietic progenitor cells derived from either hESCs or induced pluripotent stem (iPS) cells coupled to MTX chemotherapy after HSCT could potentially protect the transplanted cells, providing a selective engraftment advantage to improve levels of long-term engraftment. Here, we describe stable expression of Tyr22-DHFR and GFP in hESCs after lentiviral transduction. These Tyr22-DHFR-GFP cells were evaluated in vitro and in vivo to demonstrate improved cell survival, maintaining hematopoietic differentiation potential while under MTX selection when transplanted into immunodeficient mice.
RESULTS
Stable expression of DHFR and GFP in undifferentiated hESCs
In order to determine the vector design that would best support long-term DHFR and GFP expression in hESCs, the human H9 cell line was transduced with three different bifunctional self-inactivating (SIN)-lentivirus vectors, each containing the human EF1-α promoter regulating transcription of the murine Tyr22-DHFR variant (Figure 1a). Other genetic features include a posttranscriptional regulatory element (pre), rev response element (rre) and a central polypurine tract. In the EFDIG lentivirus vector, the DHFR and GFP coding sequences are separated by an internal ribosomal entry site (IRES) for translation of both proteins from one transcribed message. We also engineered a vector (DL2G) expressing a DHFR-GFP fusion protein with the intent that GFP expression would more accurately reflect drug resistance in transduced cells. Our previous transduction studies in mouse HSCs showed that EFDIG conferred a higher level of chemoprotection in vivo, compared to DL2G 12. We also evaluated a two-promoter configuration (REDPeG), because our recent studies in canine HSCs have shown that this vector supports a higher level of GFP expression compared to EFDIG and DL2G, thus providing easier detection of gene-modified cells in vivo (unpublished observation). Also included in all studies is a GFP-only control (CSIIEG), which has also been previously described 40. All studies here were done with the H9 hESC line that has been commonly used for studies to derive hematopoietic and other cell lineages. This line is quite amenable to stable genetic modification while retaining a normal karyotype and pluripotency.31
Figure 1. GFP expression in DHFR-GFP transduced hESCs.

(a) DHFR-GFP lentivirus vector schematics. The human EF1-α promoter regulates transcription of Tyr22-DHFR and/or GFP, along with an internal ribosomal entry site (ires) between the two coding sequences (EFDIG) or as a genetic fusion (DL2G). In a two-promoter configuration (REDPeG), EF1-α regulates Tyr22-DHFR and the human PGK promoter regulates GFP expression. Other features (described in Materials and Methods) include the HIV-1 R, U5, self-inactivating U3, and rev response element (rre), and the post-transcriptional regulatory element from the woodchuck hepatitis virus ([w2]wpre). (b) GFP expression was evaluated by fluorescence microscopy of the lentivirus transduced hES cell line H9 4 days after transduction. All images (10–20× objective) were captured at the same exposure time and modified identically in Photoshop CS2 version 9 (Brightness altered to −60, contrast +35 then applied smart sharpen). (c) Flow cytometric evaluation of GFP expression at 4 weeks. Transduced cells were stained with PE-conjugated anti-SSEA-4 to assess co-expression of GFP and surface expression of SSEA-4 as a marker for undifferentiated hESCs.
Four days after transduction (as detailed in Materials and Methods), characteristic colonies emerged, some of which were GFP+ when evaluated by fluorescence microscopy. The GFP+ colonies transduced with the vectors described above retained characteristic hESC morphology, and GFP expression was distributed evenly among the cells in each colony (Figure 1b). One month after transduction, the cells maintained the morphology of undifferentiated hESCs and 40 to 80% of the transduced populations were positive for both GFP and stage specific embryonic antigen-4 (SSEA-4), indicating stable transgene expression in undifferentiated cells (Figure 1c). All gene-modified cell populations retained a normal karyotype (Supplemental Figure. 1). While fluorescence intensity was highest for CSIIEG- and DL2G-hESCs and lower for EFDIG- and REDPeG-hESCs, all gene-modified populations contained 1–2 vector copies per genome equivalent as determined by quantitative PCR. Since GFP expression was similar in EFDIG and DL2G cell populations (~60% GFP+ cells), we also determined MTXr-DHFR enzyme activities in cell extracts. In the absence of MTX, there was no significant difference in DHFR activities among the different transduced cell populations (Table 1). However, in the presence of MTX, DHFR activity was significantly higher in extracts from EFDIG-H9 (p<0.005) and DL2G-H9 (p<0.01) populations compared to CSIIEG-HP (GFP only control) extracts, in which MTXrDHFR activity was undetectable. Interestingly, DHFR activity was not significantly increased by Tyr22-DHFR expression in the absence of MTX. Given the higher level of enzyme activity associated with EFDIG-hESCs and our previous studies showing that EFDIG confers a higher level of chemoprotection in mice compared to DL2G 12, we selected the EFDIG-modified hESCs for subsequent in vivo studies.
Table 1.
MTXr-Tyr22DHFR Enzyme Activity in DHFR-GFP hESC Extracts
|
−MTX U/mga |
−MTX p valueb |
+MTX U/mga |
+MTX p valueb |
|
|---|---|---|---|---|
| EFDIG-H9 | 15 ± 3 | 0.151 | 15 ±4 | 0.001 |
| DL2G-H9 | 12 ± 8 | 0.988 | 13 ±7 | 0.007 |
| CSIIEG-H9 | 12 ± 2 | 0 |
Cell lysates from transduced or untransduced H9 cells were assayed for DHFR activity in the presence and absence of 0.25 µM MTX. One unit of enzyme activity is defined as 1 nmol/min. Results shown are the mean of three assays ±S.D.
Enzyme activity in extracts from EFDIG-H9 cells and DL2G-H9 cells were compared to CSIIEG-H9 cells using an unpaired, two-tailed student’s t-test.
MTX dose-escalation and teratoma formation in immunodeficient mice
Previous studies in immunocompetent mice showed that transplantation with marrow that expresses MTXr-DHFR (MTXr-DHFR marrow) protects recipients from chemotoxicity 12. In preparation for long-term MTX treatment of immunodeficient mice, we first transplanted NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice with MTXr-DHFR or GFP lentivirus-transduced bone marrow as described in Materials and Methods. MTX was administered i.p. daily, starting at 0.25 mg/kg day. The dose was increased if both animal weight and hematocrit were stable among the mice in a particular cohort (Supplemental Figure 2). The goal of this dose escalation experiment was to determine the maximum MTX dose that could be tolerated by DHFR-transduced BMT NSG mice without causing anemia. We found that mice transplanted with GFP-transduced marrow could tolerate up to 0.5 mg/kg/day, while the DHFR-transduced BMT mice tolerated up to 2 mg/kg/day while maintaining stable health and hematocrits (Supplemental Figure 2).
Using these mice engrafted with MTXr marrow, we next evaluated the ability of EFDIG-hESCs to give rise to advanced derivatives of all three germ layers in teratomas that were allowed to develop in the presence and absence of MTX. In this assay, EFDIG and CSIIEG undifferentiated hESCs were injected into the left and right hind limbs of DHFR-chemoprotected NSG mice, respectively. One month after hESC injection, animals were treated daily with either PBS or MTX for one month and tumor growth monitored. Both gene-modified hESC populations gave rise to teratomas. There was a trend toward growth inhibition of CSIIEG teratomas (with no DHFR expression) compared to the growth of EFDIG teratomas (that express DHFR), as would be expected by expression of DHFR, although the difference between these groups was not statistically significant (Figure 2a). Notably, in this study, MTX treatment and tumor growth was terminated when tumor size was 1.5 cm, approximately 1 month after the start of MTX treatment. Therefore, this difference in teratoma growth between the treatment groups would likely have been greater if tumors were allowed to grow beyond 1.5 cm (as limited by our animal protocol). QPCR analysis showed that lentiviral integrants per genome equivalent were maintained or increased slightly during differentiation, compared to GFP marking by qPCR in the input undifferentiated ESC populations (Figure 2a). Derivatives of all three germ layers and GFP+ cells were detected in the teratomas, with no discernible developmental differences among treatment groups (Figure 2b,c). These data indicate that the gene-modified ESCs maintained their multipotency and gene expression during MTX administration.
Figure 2. MTXr-teratoma formation during methotrexate administration.

Four million undifferentiated EFDIG- and CSIIEG-hESCs were injected into the left and right hind limbs of DHFR-BM transplanted NSG mice (n=8), respectively. One month after hESC injection, mice were treated 5–7 days/wk with PBS or MTX for one month and euthanized at day 60 post hESC-transplantation. (a) Endpoint teratoma diameter in PBS and MTX treated mice. CSIIEG teratomas trended toward growth inhibition in the presence of MTX. Teratomas were also evaluated for GFP marking by qPCR (bottom). The fold change in GFP copy number was calculated by dividing the mean number of copies per genome equivalent in the teratomas divided by the copies per genome equivalent in the input undifferentiated cell populations (b) Representative teratoma sections were stained with hematoxylin and eosin (H&E) to show the presence of germ layer cell derivatives including: Mixed (bone, pigmented epithelium, smooth muscle), Ectoderm, Mesoderm (cartilage), and Endoderm (gut-like structure surrounded by pigmented epithelium). Frozen teratoma sections were stained with DAPI and anti-GFP to detect transgene expression after multi-lineage differentiation. Left to Right: PBS/CSIIEG, PBS/EFDIG, MTX/CSIIEG, MTX/EFDIG, all stained with rabbit anti-GFP primary and FITC conjugated donkey anti-rabbit secondary antibodies.
DHFR and GFP expression during hESC differentiation on M210 stromal cells
We next compared GFP expression and hematopoietic differentiation in gene-modified hESCs. Here, the four hESC lines transduced with CSIIEG, EFDIG, DL2G, or REDPeG were allowed to differentiate by co-culture with M2-10B4 stromal cells as previously described 17,41. The level of GFP expression was maintained in CSIIEG cells during this process, but was progressively reduced in DHFR-GFP populations during the 5-week time course (Figure 3a). Peak development of CD34+ cells ranged between 10 and 33% (Figure 3b), similar to what we have previously found for unmodified hESCs 42. Subpopulations of all transduced differentiated cells also expressed endothelial progenitor markers (ACE, CD31, Flk1)(Fig. 3c and data not shown), also as previously shown for unmodified hESCs 42,43. By day 21, subpopulations emerged that co-expressed CD34 and the pan-hematopoietic marker CD45 (Fig. 3d). Peak expression of CD45 ranged from 1% (EFDIG) to 6% (REDPeG). Importantly, subpopulations of CD34+ were also GFP+ (Figure 3e). Of the DHFR-GFP transduced cell populations evaluated, co-expression of CD34 and GFP was the highest for EFDIG transduced cells.
Figure 3. Hemato-endothelial differentiation of DHFR-GFP hESCs in M210 stromal cell co-culture.
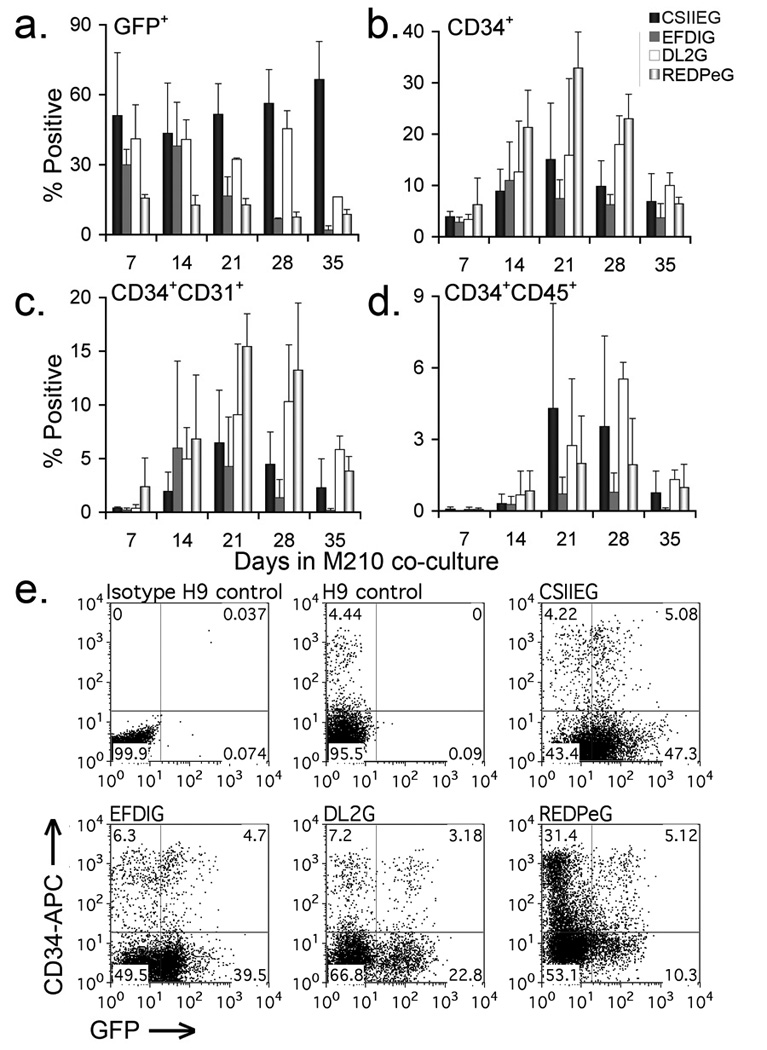
Flow cytometric analysis of gene-modified hESCs stimulated to differentiate by co-culture with M2-10B4 stromal cells was performed weekly for 5 weeks to assess differentiation potential and differences among cell populations. (a) Total GFP and (b) CD34 expression during differentiation. (c) Co-expression of CD34 and CD31, and (d) the pan-hematopoietic marker CD45. Results from each time point represent the mean +/− S.D. from 3 to 4 separate experiments. (e) Co-expression of CD34 and GFP in gene-modified cells after 21 days in M2-10B4 co-culture. Cell populations as indicated: GFP negative isotype control, CD34-APC stained H9, CSIIEG, REDPEG, DL2G, and EFDIG hESC-derived hematopoietic progeny. For flow cytometric analysis, gates were determined by isotype controls for each gene-modified cell population.
To further define hematopoietic potential and MTX resistance in DHFR+ hESC-derived progeny, the cell populations were evaluated for hematopoietic colony forming cells (CFC) in the presence of MTX. The nucleoside transport inhibitor dipyridamole (DP) was included with MTX to provide more stringent selective conditions 44. As previously demonstrated with other hESC populations, here we found that the gene-modified hESCs readily generate hematopoietic progenitor cells capable of hematopoietic colony formation as quantified in this CFC assay. Importantly and as expected, MTX treatment alone did not inhibit colony formation by CSIIEG-transduced cells (Fig. 4a). However, in the presence of both MTX and DP, CFCs were maintained for all Tyr22-DHFR-transduced cells and significantly reduced for CSIIEG-cells (Figure 4). Hematopoietic cells within the colonies retained GFP expression (Fig. 4b). These data demonstrate that CFCs derived from hESCs that express Tyr22-DHFR have a survival advantage over control CSIIEG-cells when both folate metabolism and nucleoside transport are inhibited (MTX/DP conditions).
Figure 4. MTXr-hematopoietic CFC from hESC-derived hematopoietic progenitors.

hESCs were induced to differentiate by co-culture with M2-10B4 stromal cells. At day 18 of co-culture, populations were evaluated for colony forming cells (CFC) using no drug (−/−), 30 nM methotrexate 31 (+/−) or both methotrexate and 5 µM dipyridamole (+/+) (a). The numbers represent the mean of three or four replicates per group +/− S.D. Statistical analysis was performed to determine whether colony formation was reduced in the presence of methotrexate (MTX) and dipyridamole (DP). CSIIEG-CFC were significantly inhibited in the presence of MTX/DP, compared to untreated CSIIEG-CFC (p=0.028). In (b), drug resistant colonies derived from DL2G expressing cells that formed in the presence and absence of selective conditions are shown (10× objective). Fluorescent images were captured at the same exposure (9 seconds) and then modified identically in Photoshop CS2 version 9 (brightness: −60, contrast: +35, smart sharpen).
Engraftment and differentiation of DHFR-GFP transduced hESC hematopoietic progenitors after transplantation into NOD/SCID-IL2Rγcnull mice
To further assess the effects of MTX administration and MTXr-DHFR expression by hESC-derived cells in vivo, NSG mice were injected intravenously with 4 × 106 EFDIG-hESC-derived cells after hematopoietic differentiation via M2-10B4 stromal cell co-culture for 10 days. This study consisted of three experimental groups: PBS treated mice (PBS, n=6), low dose (0.5 mg/kg) MTX treated mice (LDM, n=6) and DHFR marrow transplant recipients treated with high dose (2 mg/kg) MTX (HDM, n=8). Mice were treated 5 days per week for 4 weeks with either PBS or MTX. Two weeks after treatment cessation (6 weeks post transplantation), GFP+ cells and TRA-1-85+ cells (a monoclonal antibody that recognizes a cell surface antigen expressed by all human cells) in the blood of HDM and LDM mice were significantly higher than that observed in PBS treated animals (Fig. 5a). Twelve weeks after EFDIG-hESC-hematopoietic cell transplantation, GFP+TRA-1–85+ cells remained significantly higher in HDM mice, compared to PBS mice (Fig. 5b). CD34+ and CD45+ cells were also detected in some peripheral blood samples, though at low levels (<1%) (data not shown).
Figure 5. Detection of hESC-derived progeny in the peripheral blood of recipient mice.

After 10 days in co-culture with M210 stromal cells, EFDIG-hESC-derived hematopoietic cells were infused into DHFR-BMT mice (High dose MTX group [HDM]) or non-irradiated mice (low dose MTX [LDM], PBS groups). One week after transplantation, mice were treated daily with PBS, 0.5 mg/kg (LDM) or 2 mg/kg (HDM) MTX for 4 weeks. GFP marking and circulation of human cells (as detected by staining with TRA-1–85 antibody) in the peripheral blood was evaluated by flow cytometry at 6 (a) and 12 (b) weeks post cell transplantation. Mean percentages for each group are shown (bars). GFP percentages were also normalized to the total GFP marking (20%) associated with the input EFDIG hESC-differentiated population. Levels of statistical significance: * p<0.05, ** p< 0.005, *** p<0.0005
Twelve- to 16-weeks after EFDIG-cell transplantation, animals were sacrificed to evaluate engraftment in different organs. Cells harvested from bone marrow, liver and spleen were stained with combinations of antibodies specific for hematopoietic and endothelial lineages. Significantly more GFP+ cells were detected in the bone marrow of HDM mice in comparison with LDM or PBS-treated mice (p<0.05, Fig. 6a). QPCR analysis of bone marrow samples confirmed the GFP marking observed by flow cytometry with significantly higher GFP marking observed in LDM and HDM treated mice compared to PBS treated controls (p< 0.05). When GFP levels were normalized to the total percentage of GFP marking in the hESC-derived cells at the time of injection (20% GFP+ hESC-derived cells at day 10 of differentiation), the difference in GFP marking among treatment groups became more striking (Fig. 5 and Fig. 6a). There was significantly more TRA-1–85+ human cell engraftment in the bone marrow of mice from both MTX treatment groups compared to PBS administered mice (p<0.0005), with both CD34 and CD45 subsets represented at significantly higher levels in MTX treated groups (CD45: p<0.05) (Fig.6). LDM mice exhibited significantly higher CD34+ cell engraftment compared to both PBS (p<0.05) and HDM (p<0.005) administered mice, the latter perhaps resulting from high dose toxicity for both transduced as well as untransduced cells. Gene-modified human cells were also detected in the spleens and livers of transplanted mice (Fig. 6b,c). We conclude that MTX administration brought about the selective engraftment of DHFR+ hematopoietic cells derived from hESCs when transplanted into immunodeficient NSG mice.
Figure 6. Long-term engraftment of hESC-derived hematopoietic cells in mouse bone marrow, liver and spleen.

Animals were evaluated 12–16 weeks after EFDIG-hematopoietic cell transplantation (generated during 10 day co-culture with M210 stromal cells) and 8 weeks after withdrawal of MTX chemotherapy. Total GFP marking and engrafted cell phenotypes were evaluated by flow cytometry in the bone marrow (a), liver (b), and spleen (c) of PBS, LDM and HDM treated mice. GFP percentages in the bone marrow were also normalized to the total GFP marking (20%) associated with the input EFDIG hESC-differentiated population. GFP marking in the bone marrow was evaluated by qPCR for detection of GFP DNA sequences. Mean percentages are shown. * p<0.05, ** p< 0.005, *** p<0.0005
DISCUSSION
Our study is the first report of MTX resistance gene transfer to potentiate transplantation and selective engraftment of hESC-derived cells. We provide a model system to evaluate in vivo selection with antifolate-resistance genes in hESC-hematopoietic cells that may be applied to evaluate other chemotherapeutic drug resistance systems. In order to characterize expression of Tyr22-DHFR in hESCs, we first achieved stable DHFR-GFP expression in hESCs using three different genetic configurations. In contrast to control CSIIEG-hESCs, GFP expression decreased over time in cells transduced with DHFR-GFP bicistronic lentivirus vectors, possibly due to epigenetic silencing. We then used MTX chemotherapy and DHFR expression to establish teratomas with increased MTX resistance that were also enriched for gene-modified cells, as determined by the fold increase in transgene copies in the teratoma, compared to the GFP transgene copies detected in the undifferentiated input cell population. Finally, we demonstrated the feasibility of using Tyr22-DHFR and MTX chemotherapy to achieve in vivo selection and increased engraftment of hESC-hematopoietic cells in immunodeficient mice. These results extend our previous work on lentiviral-mediated chemoprotection in murine marrow-derived hematopoietic stem cells to demonstrate the effectiveness of this approach in ES-derived HSC and in the human system.
Historically, in vivo selection at the level of slowly dividing stem cells has not been achieved by MTX administration alone, because the inhibitory activity of MTX affects primarily highly proliferative cells, such as myeloid and lymphoid progeny. In vivo selection has been achieved using the anti-folate trimetrexate (TMTX) when administered along with the nucleoside transport inhibitor nitrobenzylmercaptopurine ribose phosphate (NBMPR-P) 14,45,46. Although TMTX/NBMPR-P treatment was sufficient to achieve in vivo selection of gene-modified HSCs in mice, selection was only transient in a study of non-human primates14,45. In contrast to the DHFR/antifolate system, expression of P140K-O6-methylguanine DNA methyltransferase (P140K-MGMT), which confers resistance to O6-benzylguanine (O6-BG) and alkylating agents such as 1,3-bis-(2-chloroethy)-1-nitrosourea (BCNU) and temozolomide (TMZ), has been shown to support significant, long-term in vivo selective expansion of gene-modified HSCs in dogs 7,47. However, in a clinical study using retroviral vector to transduce MGMT activity in hematopoietic cells, the gene transfer frequency remained low 8. The differential outcomes between the canine studies and the clinical trial highlight the importance of evaluating drug resistance genes and chemoprotection in alternative human cell sources that can be expanded ex vivo, in order to anticipate potential challenges that may arise in future studies of large animals and humans. The differential outcomes between the antifolate studies and the MGMT studies highlight the importance of evaluating drug resistance genes and chemoprotection in alternative cell sources that can be expanded ex vivo, in order to anticipate potential challenges that may arise in future studies of large animals and humans.
Drug resistance gene therapy models in human-mouse xenografts have been developed to determine whether high dose chemotherapy inhibits human tumor progression and the extent of protection provided by drug resistance genes (DHFR, MGMT) expressed in the mouse marrow 48,49. Human CD34+ cells, either from umbilical cord blood (UCB) or mobilized peripheral blood (MPB) have been evaluated in mice after transfer of P140K-MGMT 50,51. Pollok and colleagues showed that O6-BG/BCNU treatment increased long-term engraftment of CD45+GFP+ cells in the marrow of NOD/SCID mice two to eightfold 14 weeks after transplantation of UCB or MPB CD34+ cells, respectively 50. In Pollok’s studies, CD34-enriched cell populations that were 60% GFP+ were infused into mice that were irradiated immediately before transplantation. Four treatments of O6-BG and BCNU had a potent selective effect in vivo, increasing CD45+ cell engraftment in the bone marrow from 30% to 80% for UCB-derived cells and from 20% to 90% for MPB-derived cells. These results demonstrate the potential effectiveness of in vivo human stem cell selection as xenografts in immunodeficient animals, also applicable to human ES cells as demonstrated in this article.
In our study, we transplanted an unselected population of mixed, differentiated cells that was only 20% GFP+ at the time of infusion. By normalizing GFP marking of engrafted cells to this initial percentage (i.e., by dividing the percent of GFP+ cell detected in the bone marrow by the percent of GFP+ cells in the input cell population), the selective effect of MTX treatment becomes even more apparent at twelve weeks post transplant (Figure S3). Twelve times more GFP marking (6% vs. 0.5% mean GFP+ cells) was detected in the bone marrow of HDM compared to PBS treated mice. In the peripheral blood, 5 times more GFP marking (22 % vs. 4 % mean GFP+ cells) was detected in HDM treated mice vs. PBS-treated mice. We thus achieved significantly higher long-term engraftment of hESC-hematopoietic cells in the bone marrow of treated mice, compared to untreated mice. It is also striking that the levels of CD34+/CD45+ cells were similar in LDM and HDM treatment groups. These results demonstrate the long-term selective effects of MTX on hESC-hematopoietic cell engraftment and show that MTX supports in vivo selection that persists long after treatment cessation.
iPS cells and/or hESC-derived hematopoietic cells provide candidate sources for allogeneic or autologous HSCT, so it is important to consider the conditions of this clinical translation and the potential challenges that may arise in achieving effective engraftment of these cells after transplantation. Drug resistance gene transfer represents an important complement to such cell-based therapies as a means of supporting selective engraftment of gene-modified cells for hematopoietic reconstitution and prevention of allograft rejection. In this study, we show that MTX administration provided a selective benefit to the engraftment of MTXr-DHFR hESC-hematopoietic cells in the bone marrow of NSG mice. Other conditions may improve hematopoietic differentiation 26,42,52 coupled with the evaluation of more potent drug resistance genes (such as MGMT) that also support stem cell selection will better define the potential roles of drug resistance genes in hESC-derived hematopoietic function in vivo.
MATERIALS AND METHODS
Embryonic Stem Cell Culture
The hESC line H9 (WiCell Research Institute, Inc., Madison, WI) was maintained on mitotically inactive mouse embryonic fibroblast (MEF) feeder layers (Chemicon/Millipore, Temecula, CA) that were irradiated at 5500 cGy using a Cesium source as previously described 17,41. MEFs and the stromal cell line M2-10B4 were maintained as described 17,41.
Lentivirus vector construction and vector preparation
The self-inactivating HIV-1-based lentivirus plasmids CCDG, CSIIMCS, CSIIEG, DL2G, and EFDIG have been previously described 12. Briefly, in these vectors the human elongation factor-1 alpha (hEF-1α) promoter regulates transcription of the murine tyrosine-22 dihydrofolate reductase (Tyr22-DHFR) variant and enhanced green fluorescent protein (eGFP) coding sequences. EFDIG contains a picornaviral internal ribosomal entry site (IRES), DL2G constitutes a genetic fusion between the two transgenes, and CSIIEG encodes GFP alone. To construct REDPeG, the Tyr22-DHFR coding sequence was amplified from pCCDG 12 using sense (5'-TCTCGAGGATTCTCTAGAGCAAGCTTTTA-3') and anti-sense (5'-GGCGGTACCGATAAGCTGATCCTCTA-3') primers (BamHI and KpnI sites underlined). The PCR product was cloned between BamHI and KpnI sites in pRSCEMPGw2 (a construct identical to REDPeG containing a methylguanine DNA methyltransferase sequence at the site into which DHFR was inserted, kindly provided by Dr. Hans-Peter Kiem, Fred Hutchinson Cancer Research Center, Seattle, Washington) to yield REDPeG, in which the hEF-1α promoter regulates transcription of Tyr22-DHFR, and the human phosphoglycerate kinase (hPGK) promoter regulates expression of GFP. To construct the Tyr22-DHFR-expressing lentivirus vector plasmid CSIID, Tyr22-DHFR was excised from CCDG 12 and cloned into the EcoRI and BamHI sites of CSIIMCS 40. Lentiviral vectors were packaged by DNA-calcium phosphate mediated transfection of human HEK 293 cells, collected, concentrated and titered as previously described 53.
Transduction of human embryonic stem cells
Confluent hESCs growing on Matrigel (BD Biosciences, San Jose, CA) were treated with 0.05% trypsin plus 2% chicken serum (Sigma-Aldrich) for 7 minutes at 37°C with or without a 1-hour pretreatment with 10 µM Rho Kinase (Rock) inhibitor Y-27632 (CalBiochem, EMD Biosciences, San Diego, CA,). Cells were collected by centrifugation and then suspended in transduction medium (conditioned medium described above, plus 8 ng/mL bFGF, 8 µg/mL polybrene, with or without 10 µM Rock inhibitor). One million cells (in small aggregates) were mixed with lentivirus vector to achieve a multiplicity of infection (M.O.I) of 2.5 (based on functional titer [DHFR or GFP transducing units/mL] on 3T3TK- fibroblasts) and replated on Matrigel. Cell suspensions were gently agitated every hour for three hours to facilitate virus-cell contact. Twenty-four hours later, the cells were gently washed with Dulbecco’s modified phosphate buffered saline (DPBS) and covered with fresh conditioned medium plus 8 ng/mL bFGF. Two weeks and two passages later, confluent transduced cell populations were subcultured 2:1 onto MEF feeders.
Karyotype analysis
The University of Minnesota Cytogenetics Core laboratory performed the karyotype analysis. Gene-modified undifferentiated hESCs were cultured in 6-well plates as described above. Cells in metaphase were evaluated by G-banding. For each gene-modified cell population, 20 metaphase cells were analyzed at a 400–475 band resolution.
MTXr-DHFR enzyme assay
Transduced, undifferentiated hESCs were collected by centrifugation and stored at −20°C in DPBS. Cell extracts were prepared and protein content determined as described 12. Cell extracts were incubated with or without 0.25 µM MTX for 10 minutes. The addition of 120 µM NADPH (β-nicotinamide adenine dinucleotide phosphate reduced tetrasodium salt) and 20 µM dihydrofolic acid started the enzyme reaction. The change in absorbance (A340) on a DU40 spectrophotometer (Beckman Coulter, Fullerton, CA) was monitored and recorded after reaction initiation. One unit of enzyme activity is defined as the amount of enzyme required to reduce 1 nmol of dihydrofolic acid per minute (ε = 12,300 M−1 cm−1) 54.
Hematopoietic differentiation of hESCs
Undifferentiated hESCs were subcultured onto M210 cells in RPMI media with 15% FBS and allowed to differentiate as described 41. Between days 7 and 35, wells were harvested every three to seven days for flow cytometric analysis of phenotype and GFP expression. For colony forming assays (CFU), differentiation co-cultures were collected 16 to 19 days after plating on M210 stromal cells. 2.5 × 105 cells were replated into Methocult GF+ medium (Stem Cell Technologies, Vancouver, BC, Canada) supplemented with or without 5 µM dipyridamole (Sigma-Aldrich) and 30 nM MTX. Hematopoietic colonies were scored 16 days later for computation of colony-forming cells (CFC) in the plated cell population.
Animals
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice (The Jackson Laboratory, Bar Harbor, ME) were bred and maintained in microisolator cages and provided with autoclaved food and water containing antibiotics ad libetum. All procedures were reviewed and approved by the University of Minnesota Institutional Animal Care and Use Committee.
Bone marrow transduction and transplantation in NSG mice
Bone marrow was flushed from the hind limbs of eight- to twelve-week-old male NSG donor mice processed as described 12. Cells were suspended in complete StemPro medium (Invitrogen) supplemented with 2 mM L-glutamine (Gibco), 1% P/S, 8 µg/mL polybrene (Simga-Aldrich), and cytokines (100 ng/mL each of human IL-3, IL-6, and mouse SCF [all from R&D Systems, Minneapolis, MN]) 12. Lentivirus was added to cell suspensions to achieve a M.O.I. of 10 and incubated overnight in a humidified atmosphere containing 5% CO2 in air at 37°C. Eight- to twelve-week old syngeneic female recipient mice were irradiated using a cesium source (320 cGy) and then intravenously infused with 2 to 3 × 106 transduced marrow cells.
Teratoma formation in NSG mice
CSIIEG and EFDIG hESCs were harvested and aggregates suspended in DMEM:F12 plus 10% FBS. Equal volumes of CSIIEG- and EFDIG-hESC aggregates were injected intramuscularly (I.M.) into the right and left hind limbs, respectively, of untransplanted and CSIID-marrow transplanted NSG mice (transplant described in preceding section). One month after cell aggregate injection, mice were treated 5 to 7 days per week with PBS or MTX, starting at 2 mg/kg/day on days 1–7 and then escalating to 4 mg/kg/day on days 8–30. Animals were euthanized and teratomas excised, diameter measured, and sections prepared for real time qPCR, immunofluorescence, or histopathologic analysis.
Transplantation of hESC-derived hemato-endothelial cells in NSG mice
EFDIG-hESCs co-cultured with M2-10B4 stromal cells for 10 days were harvested by trypsinization as described above. 4 × 106 cells were injected intravenously into 8- to 12-week old female NSG mice. In some cases, the mice were transplanted with CSIID transduced bone marrow 1 to 3 months (as described above) before hESC-derived cell transplantation. One week after hESC-hematopoietic cell infusion, animals were treated with PBS or MTX 5 days per week for 4 weeks (see Results). Blood samples were taken 6 and 12 weeks after cell transplantation and analyzed by flow cytometry for GFP marking and human cell content.
Fluorescence microscopy, immunofluorescence and histopathology
Gene-modified hESCs were visualized using a Zeiss inverted fluorescence microscope (Axiovert 200M). Images were captured using a digital camera (Zeiss AxioCam) with AxioVision version 4.6 software interface (all from Carl Zeiss MicroImaging GmbH, Müchen, Germany). Fluorescent images were captured (10–20× objective) at the same exposure and modified identically in Adobe Photoshop CS2 Version 9. Fluorescent images of hematopoietic colonies were captured in a 9 second exposure time.
Teratomas were embedded in optimal cutting temperature medium (O.C.T.) and frozen or fixed in 10% formalin. For immunofluorescence, teratomas were sectioned, fixed onto slides with 10% formalin and permeabilized with 0.25% Triton-X 100 in DPBS. 10 µm sections were incubated with 10% donkey serum to block non-specific antibody binding and incubated with either 1:200 rabbit-anti-GFP in 10% donkey serum (Molecular Probes, Invitrogen) or rabbit isotype control antibody (Zymed, Invitrogen) overnight at 4°C. Slides were washed several times with DPBS, incubated for 1 hour at room temperature with FITC-conjugated donkey anti-rabbit secondary antibody (1:200 in 10% donkey serum), washed and set with Prolong gold antifade reagent plus DAPI (Invitrogen). Sections were visualized and images captured (20× objective, 4 second exposure time) by fluorescence microscopy as described above. For histopathologic analysis, formalin fixed teratomas were embedded in paraffin, sectioned, mounted, stained with hematoxylin and eosin and analyzed by a veterinary pathologist without prior knowledge of sample identity or treatment (MTX, PBS).
Real-time quantitative PCR
DNA was extracted from human teratomas or mouse tissue samples using the Puregene DNA purification system (Gentra Systems, Inc., Minneapolis, MN). DNA was quantitated spectrophotometrically (A260) and qPCR reactions carried out as previously described without prior knowledge of sample identity or treatment (MTX, PBS).
Flow cytometry
Flow cytometric analysis of differentiating hESCs in stromal cell co-culture was performed as previously described 17,41. To assess engraftment of hESC-derived progeny and gene marking in vivo, blood, bone marrow and spleen were harvested from mice after euthanasia and treated with ammonium chloride solution (Stem Cell Technologies). Liver was treated with collagenase mixture (1 mg/mL collagenase IV, 50 µg/mL DNase I (Gibco, Invitrogen), 10% FBS in RPMI). Cells were resuspended in Iscove’s modified DMEM plus 5% FBS (I-5) medium and layered onto Ficoll Paque Plus (GE Healthcare Biosciences AB, Uppsala, Sweden). After centrifugation, the ficoll layer was collected. Blood, bone marrow and liver cells were suspended in I-5 medium, filtered, collected and incubated in blocking solution (5% human AB serum [Valley Biomedical, Winchester, VA] 5% FBS in DPBS) for 30 minutes on ice. Cells stained with PE-conjugated TRA-1–85 (R&D systems, Minneapolis, MN) were also treated with 4 µg of human IgG1 protein (R&D Systems) for 15 minutes at room temperature. Cells were suspended in flow cytometry buffer (0.1% NaN3, 2% FBS in DPBS) and stained with pairs of APC- and PE- conjugated mouse anti-human antibodies for 20 minutes at 4°C in the dark. Cells were suspended in buffer plus 2 µg/mL 7-amino actinomycin D (7-AAD, Sigma-Aldrich).
All flow cytometry data were collected using a FACSCalibur Instrument with CellQuest Pro software interface (BD Biosciences, San Jose, CA). Depending on the cell or tissue sample, 5,000–20,000 live events (7-AAD negative gate) were collected for analysis. Data were analyzed using FlowJo software, version 8.6.1. (Tree Star, Inc., Ashland, OR). All antibodies were obtained from BD Pharmingen (San Diego, CA) except PE-conjugated Flk-1, TRA-1–85 (both from R&D Systems) and CD235A (GlyA) (from Becton Coulter [Fullerton, CA]).
Statistical analysis
To test for differences among treatment groups or gene-modified cell populations, data were evaluated using an unpaired, two-tailed student t-test in Microsoft Excel, Version 11.5. Differences were considered statistically significant if p values were less than 0.05.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health Grants CA060803 (R.S.M.), HL77923 (D.S.K), and CA34196 (L.D.S.). We thank Andrea Mohs, Melinda Hexum, Katie Hill, Julie Morris, Jeremy Allred and Jon Linehan for assistance with these studies.
Footnotes
Presented in oral abstract form at the 11th Annual Meeting of the American Society of Gene Therapy, Boston MA, May 31– June 1 2008
REFERENCES
- 1.Halene S, Kohn DB. Gene therapy using hematopoietic stem cells: Sisyphus approaches the crest. Human Gene Therapy. 2000;11:1259–1267. doi: 10.1089/10430340050032366. [DOI] [PubMed] [Google Scholar]
- 2.Hacein-Bey-Abina S, Le Deist F, Carlier F, Bouneaud C, Hue C, De Villartay JP, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. The New England Journal of Medicine. 2002;346:1185–1193. doi: 10.1056/NEJMoa012616. [DOI] [PubMed] [Google Scholar]
- 3.Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. The New England Journal of Medicine. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 4.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 5.Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410–2413. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 6.Gerull S, Beard BC, Peterson LJ, Neff T, Kiem HP. In vivo selection and chemoprotection after drug resistance gene therapy in a nonmyeloablative allogeneic transplantation setting in dogs. Human Gene Therapy. 2007;18:451–456. doi: 10.1089/hum.2006.039. [DOI] [PubMed] [Google Scholar]
- 7.Neff T, Horn PA, Peterson LJ, Thomasson BM, Thompson J, Williams DA, et al. Methylguanine methyltransferase-mediated in vivo selection and chemoprotection of allogeneic stem cells in a large-animal model. The Journal of Clinical Investigation. 2003;112:1581–1588. doi: 10.1172/JCI18782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cornetta K, Croop J, Dropcho E, Abonour R, Kieran MW, Kreissman S, et al. A pilot study of dose-intensified procarbazine, CCNU, vincristine for poor prognosis brain tumors utilizing fibronectin-assisted, retroviral-mediated modification of CD34+ peripheral blood cells with O6-methylguanine DNA methyltransferase. Cancer Gene Therapy. 2006;13:886–895. doi: 10.1038/sj.cgt.7700963. [DOI] [PubMed] [Google Scholar]
- 9.Nash RA, Antin JH, Karanes C, Fay JW, Avalos BR, Yeager AM, et al. Phase 3 study comparing methotrexate and tacrolimus with methotrexate and cyclosporine for prophylaxis of acute graft-versus-host disease after marrow transplantation from unrelated donors. Blood. 2000;96:2062–2068. [PubMed] [Google Scholar]
- 10.Ratanatharathorn V, Nash RA, Przepiorka D, Devine SM, Klein JL, Weisdorf D, et al. Phase III study comparing methotrexate and tacrolimus (prograf, FK506) with methotrexate and cyclosporine for graft-versus-host disease prophylaxis after HLA-identical sibling bone marrow transplantation. Blood. 1998;92:2303–2314. [PubMed] [Google Scholar]
- 11.Bertino JR. Karnofsky memorial lecture. Ode to methotrexate. J Clin Oncol. 1993;11:5–14. doi: 10.1200/JCO.1993.11.1.5. [DOI] [PubMed] [Google Scholar]
- 12.Gori JL, Podetz-Pedersen K, Swanson D, Karlen AD, Gunther R, Somia NV, et al. Protection of mice from methotrexate toxicity by ex vivo transduction using lentivirus vectors expressing drug-resistant dihydrofolate reductase. The Journal of Pharmacology and Experimental Therapeutics. 2007;322:989–997. doi: 10.1124/jpet.107.123414. [DOI] [PubMed] [Google Scholar]
- 13.Allay JA, Galipeau J, Blakley RL, Sorrentino BP. Retroviral vectors containing a variant dihydrofolate reductase gene for drug protection and in vivo selection of hematopoietic cells. Stem Cells. 1998;16(Suppl 1):223–233. doi: 10.1002/stem.5530160827. [DOI] [PubMed] [Google Scholar]
- 14.Persons DA, Allay JA, Bonifacino A, Lu T, Agricola B, Metzger ME, et al. Transient in vivo selection of transduced peripheral blood cells using antifolate drug selection in rhesus macaques that received transplants with hematopoietic stem cells expressing dihydrofolate reductase vectors. Blood. 2004;103:796–803. doi: 10.1182/blood-2003-05-1572. [DOI] [PubMed] [Google Scholar]
- 15.Kaufman DS, Hanson ET, Lewis RL, Auerbach R, Thomson JA. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10716–10721. doi: 10.1073/pnas.191362598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian X, Morris JK, Linehan JL, Kaufman DS. Cytokine requirements differ for stroma and embryoid body-mediated hematopoiesis from human embryonic stem cells. Experimental Hematology. 2004;32:1000–1009. doi: 10.1016/j.exphem.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 17.Tian X, Kaufman DS. Hematopoietic development of human embryonic stem cells in culture. Methods in molecular biology. 2008;430:119–133. doi: 10.1007/978-1-59745-182-6_8. [DOI] [PubMed] [Google Scholar]
- 18.Vodyanik MA, Bork JA, Thomson JA, Slukvin Human embryonic stem cell-derived CD34+ cells: efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood. 2005;105:617–626. doi: 10.1182/blood-2004-04-1649. [DOI] [PubMed] [Google Scholar]
- 19.Qiu C, Olivier EN, Velho M, Bouhassira EE. Globin switches in yolk sac-like primitive and fetal-like definitive red blood cells produced from human embryonic stem cells. Blood. 2008;111:2400–2408. doi: 10.1182/blood-2007-07-102087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chadwick K, Wang L, Li L, Menendez P, Murdoch B, Rouleau A, et al. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood. 2003;102:906–915. doi: 10.1182/blood-2003-03-0832. [DOI] [PubMed] [Google Scholar]
- 21.Woll PS, Martin CH, Miller JS, Kaufman DS. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J Immunol. 2005;175:5095–5103. doi: 10.4049/jimmunol.175.8.5095. [DOI] [PubMed] [Google Scholar]
- 22.Woll PS, Grzywacz B, Tian X, Marcus RK, Knorr DA, Verneris MR, et al. Human embryonic stem cells differentiatiate into a homogeneous population of natural killer cells with highly potent in vivo anti-tumor activity. Blood. 2009;113:6094–6101. doi: 10.1182/blood-2008-06-165225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma F, Ebihara Y, Umeda K, Sakai H, Hanada S, Zhang H, et al. Generation of functional erythrocytes from human embryonic stem cell-derived definitive hematopoiesis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13087–13092. doi: 10.1073/pnas.0802220105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiu C, Hanson E, Olivier E, Inada M, Kaufman DS, Gupta S, et al. Differentiation of human embryonic stem cells into hematopoietic cells by coculture with human fetal liver cells recapitulates the globin switch that occurs early in development. Experimental Hematology. 2005;33:1450–1458. doi: 10.1016/j.exphem.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 25.Lu SJ, Feng Q, Park JS, Vida L, Lee BS, Strausbauch M, et al. Biologic properties and enucleation of red blood cells from human embryonic stem cells. Blood. 2008;112:4475–4484. doi: 10.1182/blood-2008-05-157198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ledran MH, Krassowska A, Armstrong L, Dimmick I, Renstrom J, Lang R, et al. Efficient hematopoietic differentiation of human embryonic stem cells on stromal cells derived from hematopoietic niches. Cell Stem Cell. 2008;3:85–98. doi: 10.1016/j.stem.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 27.Tian X, Woll PS, Morris JK, Linehan JL, Kaufman DS. Hematopoietic engraftment of human embryonic stem cell-derived cells is regulated by recipient innate immunity. Stem Cells. 2007;24:1370–1380. doi: 10.1634/stemcells.2005-0340. [DOI] [PubMed] [Google Scholar]
- 28.Wang L, Menendez P, Shojaei F, Li L, Mazurier F, Dick JE, et al. Generation of hematopoietic repopulating cells from human embryonic stem cells independent of ectopic HOXB4 expression. The Journal of Experimental Medicine. 2005;201:1603–1614. doi: 10.1084/jem.20041888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Narayan AD, Chase JL, Lewis RL, Tian X, Kaufman DS, Thomson JA, et al. Human embryonic stem cell-derived hematopoietic cells are capable of engrafting primary as well as secondary fetal sheep recipients. Blood. 2006;107:2180–2183. doi: 10.1182/blood-2005-05-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfeifer A, Ikawa M, Dayn Y, Verma IM. Transgenesis by lentiviral vectors: lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:2140–2145. doi: 10.1073/pnas.251682798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilber A, Linehan JL, Tian X, Woll PS, Morris JK, Belur LR, et al. Efficient and stable transgene expression in human embryonic stem cells using transposon-mediated gene transfer. Stem Cells. 2007;25:2919–2927. doi: 10.1634/stemcells.2007-0026. [DOI] [PubMed] [Google Scholar]
- 32.Giudice A, Trounson A. Genetic modification of human embryonic stem cells for derivation of target cells. Cell Stem Cell. 2008;2:422–433. doi: 10.1016/j.stem.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 33.Ben-Dor I, Itsykson P, Goldenberg D, Galun E, Reubinoff BE. Lentiviral vectors harboring a dual-gene system allow high and homogeneous transgene expression in selected polyclonal human embryonic stem cells. Mol Ther. 2006;14:255–267. doi: 10.1016/j.ymthe.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 34.Xu XQ, Zweigerdt R, Soo SY, Ngoh ZX, Tham SC, Wang ST, et al. Highly enriched cardiomyocytes from human embryonic stem cells. Cytotherapy. 2008;10:376–389. doi: 10.1080/14653240802105307. [DOI] [PubMed] [Google Scholar]
- 35.Gallo P, Grimaldi S, Latronico MV, Bonci D, Pagliuca A, Gallo P, et al. A lentiviral vector with a short troponin-I promoter for tracking cardiomyocyte differentiation of human embryonic stem cells. Gene Therapy. 2008;15:161–170. doi: 10.1038/sj.gt.3303017. [DOI] [PubMed] [Google Scholar]
- 36.Davis RP, Ng ES, Costa M, Mossman AK, Sourris K, Elefanty AG, et al. Targeting a GFP reporter gene to the MIXL1 locus of human embryonic stem cells identifies human primitive streak-like cells and enables isolation of primitive hematopoietic precursors. Blood. 2008;111:1876–1884. doi: 10.1182/blood-2007-06-093609. [DOI] [PubMed] [Google Scholar]
- 37.Huber I, Itzhaki I, Caspi O, Arbel G, Tzukerman M, Gepstein A, et al. Identification and selection of cardiomyocytes during human embryonic stem cell differentiation. FASEB J. 2007;21:2551–2563. doi: 10.1096/fj.05-5711com. [DOI] [PubMed] [Google Scholar]
- 38.Lavon N, Yanuka O, Benvenisty N. The Effect of Overexpression of Pdx1 and Foxa2 on the Differentiation of Human Embryonic Stem Cells into Pancreatic Cells. Stem Cells. 2006;24:1923–1930. doi: 10.1634/stemcells.2005-0397. [DOI] [PubMed] [Google Scholar]
- 39.Zwaka TP, Thomson JA. Homologous recombination in human embryonic stem cells. Nat Biotechnol. 2003;21:319–321. doi: 10.1038/nbt788. [DOI] [PubMed] [Google Scholar]
- 40.Agarwal S, Nikolai B, Yamaguchi T, Lech P, Somia NV. Construction and use of retroviral vectors encoding the toxic gene barnase. Mol Ther. 2006;14:555–563. doi: 10.1016/j.ymthe.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 41.Hill KL, Kaufman DS. Hematopoietic differentiation of human embryonic stem cells by cocultivation with stromal layers. Chapter 1: Unit 1F 6. Current protocols in stem cell biology. 2008 doi: 10.1002/9780470151808.sc01f06s6. [DOI] [PubMed] [Google Scholar]
- 42.Woll PS, Morris JK, Painschab MS, Marcus RK, Kohn AD, Biechele TL, et al. Wnt signaling promotes hematoendothelial cell development from human embryonic stem cells. Blood. 2008;111:122–131. doi: 10.1182/blood-2007-04-084186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zambidis ET, Park TS, Yu W, Tam A, Levine M, Yuan X, et al. Expression of ACE (CD143) identifies and regulates primitive hemangioblasts derived from human pluripotent stem cells. Blood. 2008 doi: 10.1182/blood-2008-03-144766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warlick CA, Sweeney CL, McIvor RS. Maintenance of differential methotrexate toxicity between cells expressing drug-resistant and wild-type dihydrofolate reductase activities in the presence of nucleosides through nucleoside transport inhibition. Biochem Pharmacol. 2000;59:141–151. doi: 10.1016/s0006-2952(99)00311-1. [DOI] [PubMed] [Google Scholar]
- 45.Allay JA, Persons DA, Galipeau J, Riberdy JM, Ashmun RA, Blakley RL, et al. In vivo selection of retrovirally transduced hematopoietic stem cells. Nature Medicine. 1998;4:1136–1143. doi: 10.1038/2632. [DOI] [PubMed] [Google Scholar]
- 46.Allay JA, Spencer HT, Wilkinson SL, Belt JA, Blakley RL, Sorrentino BP. Sensitization of hematopoietic stem and progenitor cells to trimetrexate using nucleoside transport inhibitors. Blood. 1997;90:3546–3554. [PubMed] [Google Scholar]
- 47.Neff T, Beard BC, Peterson LJ, Anandakumar P, Thompson J, Kiem HP. Polyclonal chemoprotection against temozolomide in a large-animal model of drug resistance gene therapy. Blood. 2005;105:997–1002. doi: 10.1182/blood-2004-08-3169. [DOI] [PubMed] [Google Scholar]
- 48.Kreklau EL, Pollok KE, Bailey BJ, Liu N, Hartwell JR, Williams DA. Hematopoietic expression of O6-methylguanine DNA methly-transferase-P140K allows intenstive treatment of human glioma xenografts with combination O6-benzylguanine and 1,3-bis-(2-chloroethyl)-1-nitrosourea. Molecular Cancer Therapeutics. 2003;2:1321–1329. [PubMed] [Google Scholar]
- 49.Budak-Alpdogan T, Alpdogan O, Banerjee D, Wang E, Moore MA, Bertino JR. Methotrexate and cytarabine inhibit progression of human lymphoma in NOD/SCID mice carrying a mutant dihydrofolate reductase and cytidine deaminase fusion gene. Mol Ther. 2004;10:574–584. doi: 10.1016/j.ymthe.2004.06.115. [DOI] [PubMed] [Google Scholar]
- 50.Pollok KE, Hartwell JR, Braber A, Cooper RJ, Jansen M, Ragg S, et al. In vivo selection of human hematopoietic cells in a xenograft model using combined pharmacologic and genetic manipulations. Human Gene Therapy. 2003;14:1703–1714. doi: 10.1089/104303403322611728. [DOI] [PubMed] [Google Scholar]
- 51.Cai S, Hartwell JR, Cooper RJ, Juliar BE, Kreklau E, Abonour R, et al. In vivo effects of myeloablative alkylator therapy on survival and differentiation of MGMTP140Ktransduced human G-CSF-mobilized peripheral blood cells. Mol Ther. 2006;13:1016–1026. doi: 10.1016/j.ymthe.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 52.Ng ES, Davis RP, Azzola L, Stanley EG, Elefanty AG. Forced aggregation of defined numbers of human embryonic stem cells into embryoid bodies fosters robust, reproducible hematopoietic differentiation. Blood. 2005;106:1601–1603. doi: 10.1182/blood-2005-03-0987. [DOI] [PubMed] [Google Scholar]
- 53.Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nature Protocols. 2006;1:241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- 54.Carr F, Medina WD, Dube S, Bertino JR. Genetic transformation of murine bone marrow cells to methotrexate resistance. Blood. 1983;62:180–185. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.