

Abstract
Pardaxin is a 33-amino-acid neurotoxin from the Red Sea Moses sole Pardachirus marmoratus, whose mode of action shows remarkable sensitivity to lipid chain length and charge, although the effect of pH is unclear. Here we combine optical spectroscopy and dye release experiments with laser scanning confocal microscopy and natural abundance 13C solid-state nuclear magnetic resonance to provide a more complete picture of how pardaxin interacts with lipids. The kinetics and efficiency of release of entrapped calcein is highly sensitive to pH. In vesicles containing zwitterionic lipids (PC), release occurs most rapidly at low pH, whereas in vesicles containing 20% anionic lipid (PG), release occurs most rapidly at high pH. Pardaxin forms stable or transient pores in PC vesicles that allow release of contents without loss of vesicle integrity, whereas the inclusion of PG promotes total vesicle collapse. In agreement with this, solid-state nuclear magnetic resonance reveals that pardaxin takes up a trans-membrane orientation in 14-O-PC/6-O-PC bicelles, whereas the inclusion of 14-0-PG restricts it to contacts with lipid headgroups, promoting membrane lysis. Pore formation in zwitterionic vesicles is more efficient than lysis of anionic vesicles, suggesting that electrostatic interactions may trap pardaxin in several suboptimal interconverting conformations on the membrane surface.
Introduction
The increasing occurrence of bacterial strains resistant to conventional antibiotics demands a steady output of new antimicrobial agents. The mode by which the bacteria gain resistance against a single antibiotic has a tendency to improve their tolerance to entire groups of antibiotics such as the quinolones, sulfonamides, penicillins, and cephalosporins. This means that even as the development of antibiotic compounds becomes more difficult and costly, their longevity for treating bacterial strains decreases. This makes antimicrobial peptides attractive alternatives as new antibiotic compounds. The rate with which bacteria can become resistant toward antimicrobial peptides is much lower, and the resistance against one type does not seem to induce a general increase in antimicrobial peptide resistance. A major challenge is to ensure specificity toward bacterial cells. The mechanism by which cationic linear antimicrobial peptides interacts and permeates membranes has been thoroughly studied and a consensus has been reached with regard to three different models—the barrel-stave (1), carpet (2), and toroidal pore models (3). The barrel-stave model is the only one that requires the peptide to interact with the hydrophobic core of the protein. For the two other models, the peptide is positioned on the surface or in the headgroup region of the lipid bilayer.
Pardaxin is a 33-amino-acid neurotoxin that was originally isolated from the Red Sea Moses sole Pardachirus marmoratus in 1986 (4). Over the years, many studies have been done on pardaxin, addressing its mode of action (5,6), its connection to peptide aggregation (7,8), and the effect of factors such as pH and lipid composition (8–12). At molar ratios of 1:50,000, pardaxin reduced the fluid lamellar-to-inverted hexagonal phase transition temperature of POPE vesicles; furthermore, the presence of anionic lipids or cholesterol was found to reduce the peptide's ability to disrupt bilayers (13). Solid-state 15N nuclear magnetic resonance (NMR) spectroscopy on mechanically aligned bilayers showed that pardaxin aligned in the plane of POPC bilayers but perpendicular to DMPC bilayers (13). Thus, pardaxin mode of action is sensitive to the acyl chain of the bilayer, leading to a barrel-stave mechanism in DMPC and the carpet mechanism in POPC. This was later supported by solid-state 2H NMR results that demonstrated that pardaxin induces considerable disorder in both the headgroups and the hydrophobic core of POPC (14). Note, however, that these experiments were done at pH 4.5, which is considerably different from the neutral to slightly alkaline conditions that are naturally encountered by pardaxin.
In another study, a combination of a vesicle permeation assay and fluorescently labeled pardaxin has been used to deduce that pardaxin forms pores with approximately six molecules per pore (7). The interpretation is complicated by the fact that the lipids used were a mix of naturally derived lipids consisting of a mix of carbon lengths from 16 to 20 with varying degree of saturation as well as phosphatidylcholine and phosphatidylserine headgroups. Given that pore formation appears to be highly lipid-specific, a combination of different peptide-membrane interactions might be taking place under these conditions. This makes it important to carry out studies under very well-defined conditions with simple lipid compositions.
Given that pardaxin interactions are sensitive to lipid composition, such as lipid headgroup charge, but that the mechanism of interaction is unclear, we decided to investigate the role of electrostatics in pardaxin-membrane interactions using simple synthetic lipids. Accordingly, we use two lipids with the same acyl (dioleoyl) chain but with different headgroups, namely zwitterionic DOPC or anionic DOPG. Our work was conducted using three different approaches: First, we investigated the effect of headgroup charge (lipid composition) and pardaxin protonation (at different pH values) on the kinetics of vesicle disruption. Second, we used this data to select a pH-value (8.0) to carry out real-time analysis of pardaxin's disruption of individual giant unilamellar vesicles (GUVs) using laser scanning confocal microscopy (LSCM). Third, we performed a structural analysis of the interaction between pardaxin and lipids using natural abundance 13C solid-state NMR spectroscopy on anionic and zwitterionic bicelles at pH 8.0. We find that the kinetics of vesicle permeation by pardaxin are affected by the pH and vesicle charge but are not simply linked to favorable electrostatic interactions. LSCM measurements clearly show that pardaxin forms pores in GUVs composed of zwitterionic vesicles whereas it causes complete membrane lysis when the GUVs contain anionic lipids. The solid-state NMR data reveal that pardaxin interacts with the zwitterionic lipids all along the lipid molecule, suggesting a trans-membrane conformation. In contrast, when anionic lipids are added, pardaxin interacts only with the upper layer of the lipid membrane.
Materials and Methods
Chemicals
The following lipids were used: DOPC (1,2-di-oleoyl-sn-glycero-3-phosphocholine); DOPG ((1,2-di-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]); 14-O-PC (1,2-di-O-tetradecyl-sn-glycero-3-phosphocholine); 6-O-PC (1,2-di-O-hexyl-sn-glycero-3-phosphocholine); and 14-O-PG (1,2-di-O-tetradecyl-sn-glycero-3-[phospho-rac-(1-glycerol)]). All lipids were from Avanti Polar Lipids (Alabaster, AL). Protected amino acids, EDT (1,2-ethane dithiole), TIPS (triisopropylsylane), DCM (di-chloromethane), DMF (dimethylformamide), DIPEA (n,n-diisopropylethylamine), HBTU ((1H-benzotriazolyl)-1,1,3,3-tetramethyl-uronium hexafluorophosphate), acetonitrile Chromasolv (Sigma-Aldrich) gradient grade, TFA (trifluoroacetic acid), sucrose 99.5% purity, and D(+) glucose 99.5% purity were from Sigma (St. Louis, MO). DiIC18 (3) and Alexa488 and Alexa633 hydrazide were from Invitrogen (Carlsbad, CA). Calcein disodium salt was from Fluka (Buchs, Switzerland).
Peptide synthesis
Pardaxin (sequence GFFALIPKIISSPLFKTLLSAVGSALSSSGGQE) was synthesized on an automatic Liberty microwave-assisted peptide synthesizer (CEM, Tokyo, Japan) by solid-phase synthesis using standard Fmoc chemistry on Wang resin essentially as described in Nielsen et al. (15). Further details are provided in the Supporting Material.
Calcein release assay measured by fluorescence
Large unilamellar vesicles containing 40 mM calcein were extruded and desalted as described (16). All extruded vesicles were used within two days after production. All fluorescence measurements were conducted on a Cary Eclipse Fluorescence spectrophotometer (Varian, Palo Alto, CA). Release of free calcein from the vesicles and the subsequent increase in fluorescence was monitored by excitation at 490 nm, recording emission at 515 nm every second using a slit-width of 2.5 nm for both monochromators. The time delay between each measurement was set to 0.2 s. The vesicles were diluted in buffers with the desired pH values to a concentration of ∼0.017 g/L (20 μM), leading to a maximum emission signal of ∼200 units under these settings. A 10-mm quartz cuvette with magnetic stirring was used and the vesicle solution was allowed to equilibrate thermally for a minute before starting to record fluorescence. For each recording, pardaxin was injected and emission was followed until it reached a plateau (typically after several minutes). As calcein fluorescence is pH-sensitive and varies by approximately a factor of 5 within the pH range used here, we normalized the emission change at each pH with regard to maximum change, operationally defined as the fluorescence level obtained when 1% of Triton X-100 is added.
Laser scanning confocal microscopy (LSCM) measurements
Giant unilamellar vesicles (GUVs) were prepared by the electroformation method originally described by Angelova and Dimitrov (17). The GUVs were electroporated using a home-built electroformation chamber partly built on the specifications published by Bagatolli and Gratton (18). Further information is provided in the Supporting Material. Vesicles in 10 mM Tris pH 8.0 were equilibrated to room temperature on the microscope slide for 15–20 min before measuring. GUVs were observed with an LSM 510 scanning confocal microscope (Zeiss, Jena, Germany). Excitation of Alexa488, Alexa633, and DiI was done at 488, 633, and 543 nm, respectively. Fluorescence emission was measured at 505–548, 650–700, and 558–612 nm for Alexa488, Alexa633, and DiI, respectively. Ten microliters of 30 μM pardaxin dissolved in deionized water was injected into the microcopy chamber outside the field of vision, and the measurements started immediately afterward with images recorded every 15 s. Typically no changes were observed for the first 10–15 min after injection, probably due to slow diffusion of peptides into the field of vision in the viscous sucrose medium. Vesicle lysis was usually preceded by fluctuating vesicle movement associated with bulk solution changes, indicating the arrival of peptides diffusing in a lower-viscosity solution. Controls with addition of equivalent amounts of deionized water and buffer were performed but none of them showed signs of vesicle disruption.
Secondary structural changes measured by circular dichroism spectroscopy
Circular dichroism (CD) studies were performed on a model No. J-810 spectropolarimeter (JASCO, Hachioji City, Japan) with a model No. PTC-348W1 temperature control unit (JASCO). Scan speed was set to 100 nm/min, slit-width 2 nm. All experiments were carried out at 25°C using a 1-mm quartz cuvette. Each spectrum in titration experiments represents a single sample (mixed 2–5 min before measuring) and based on an average of three scans. Background spectra for the different buffer and additive solutions without peptide were measured and subtracted to yield the final spectra. Pardaxin concentrations were kept at 20 μM in either 20 mM phosphate buffer at pH 5.5, 20 mM Tris at pH 8, or 20 mM Glycine buffer at pH 10.
Changes in lipid chain conformation measured by solid-state NMR
Bicelles were made from 14-O-PC, 6-O-PC, and 14-O-PG. All lipids were solubilized in chloroform except for 14-O-PG, which was purchased as a dry powder. Two different bicelle samples were prepared, one containing only 14-O-PC and 6-O-PC at a molar ratio of 3.2:1 and the other containing 14-O-PC, 14-O-PG, and 6-O-PC at a molar ratio of 2.6:0.6:1. Each sample contained a total of 56 mg of lipids. The lipids were dried separately over a stream of nitrogen and left on vacuum overnight. Two-hundred microliters of a 10-mM phosphate buffer (pH = 6.8) was added to the dry 6-O-PC and briefly vortexed until solubilized. The clear solution of 6-O-PC was added to the dry 14-O-PC/14-O-PG and vortexed in an ice bath for 20 min. After a few heat-and-freeze cycles, the sample was transferred to a 3-cm-long, 5-mm tube and sealed. It was left at room temperature for 10 days. The final sample is clear and viscous at ambient temperatures and becomes fluid at <10°C. Pardaxin (peptide/lipids, 1:100) was added to the bicelle suspension as a lyophilized powder followed by agitation to allow solubilization. Details on solid-state NMR measurements (19) can be found in the Supporting Material.
Results
Conformational changes in pardaxin as function of solutes
The motivation for these measurements was to gain a greater understanding of the role of electrostatics in controlling pardaxin-membrane interactions. Here we focus on the effect of pH using two different lipid compositions. We initially look at the secondary structure of pardaxin by far-ultraviolet (UV) CD spectra for pardaxin in buffer and in the presence of 50% TFE (2,2,2-trifluoroethanol), 10 mM DPC (dodecylphosphocholine), 100% DOPC, and mixed vesicles containing 20% DOPG and 80% DOPC. The secondary structure was not significantly affected by pH. At pH 6–8, ∼20% of the pardaxin solution precipitated (according to Bradford assays on the supernatant left after centrifugation), probably due to the lack of overall charge in this pH range (see below). However, no differences in the spectral characteristics could be observed when we normalized for protein concentration (data not shown). Representative spectra are shown in Fig. 1. In both 50% TFE and 10 mM DPC, we observe spectra that resemble that of a classical α-helical spectrum with two local minima at 222 and 208 nm. For DOPC the local minimum at 208 nm is much less pronounced than for DPC and TFE, suggesting either a reduction in α-helicity, a change in oligomerization state, or a change in the relative orientation of the different helices (20–23). The change is even more dramatic when we introduce 20% of the anionic DOPG into the DOPC vesicles. The 208-nm minimum almost completely disappears, indicating a further loss in α-helicity.
Figure 1.
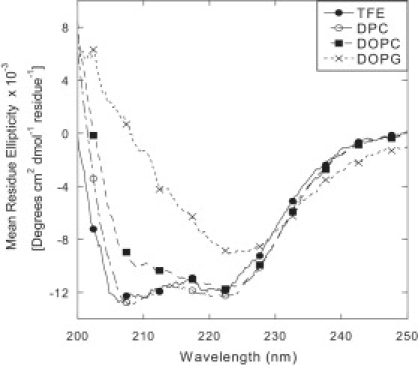
CD spectra of 20 μM pardaxin in 20 mM phosphate-buffered saline, pH 7.5 in the presence of TFE, DPC, 100% DOPC vesicles, and DOPG/DOPC (20:80) vesicles.
The degree and kinetics of vesicles disruption depends on pH and lipid charge
Having established that the ensemble structure of pardaxin is not affected by changes in pH and thereby peptide protonation, but is more sensitive to the solvent or amphiphile, we next investigated how the protonation of pardaxin affects its ability to permeate vesicles of both zwitterionic and anionic composition. We measured the release of the fluorescent probe calcein from vesicles consisting of DOPC and DOPG/DOPC (20:80) at pH 5.5, 8, and 10 where pardaxin has a net charge of + 2, 0, and −1, respectively, according to simple side-chain titration calculations. In the same pH range, DOPC has a net charge of 0 and DOPG a net charge of −1. Typical release profiles are shown in Fig. 2.
Figure 2.
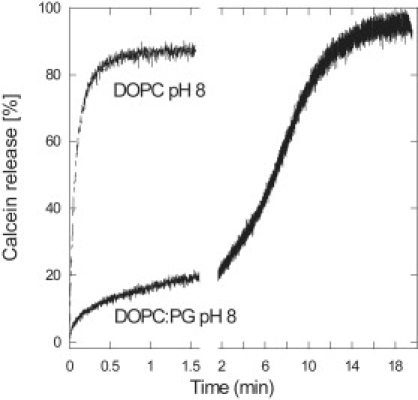
Two representative time profiles from the calcein release experiments. The addition of pardaxin to DOPC/PG vesicles at pH 8 shows a step-type time profile that can be fitted to a triple exponential decay, whereas the time profile at all other conditions (pH 5.5–10 and both vesicle types) follows a double-exponential decay.
In contrast to the pH invariance displayed by the far-UV CD spectra, the kinetics of calcein release are highly sensitive to both pH and lipid type. Comparison of the two lipid types reveals that 5–10-fold less pardaxin is required for maximum disruption of DOPC vesicles (∼20 nM) compared to DOPC/DOPG vesicles (∼100 nM at pH 5.5, ∼240 nM at pH 8, and ∼180 nM at pH 10) (Fig. 3, A and B). This indicates a substantial difference in the efficacy of permeabilization. Furthermore, the protein concentration needed for 50% calcein release from vesicles ([pardaxin]50%) varies by a factor of 2–3 for DOPC vesicles in the order pH 5 > pH8 > pH10. In contrast, the DOPC/DOPG vesicles are permeabilized two-to-threefold more efficiently at pH 5.5 than at pH 8 and 10. We attribute this preference for low pH to electrostatic interactions between pardaxin (which is only positively charged at pH 5.5) and the partially anionic lipids. However, the pH preference with regard to the zwitterionic DOPC vesicles is unexpected, and cannot be rationalized from simple electrostatic considerations.
Figure 3.
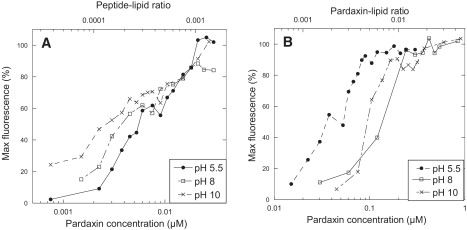
Maximum release of calcein by pardaxin from (A) DOPC vesicles and (B) DOPG/DOPC (20:80) vesicles at pH 5.5, 8, and 10. In all experiments, the lipid concentration was 20 μM.
The release of calcein from DOPC vesicles at all three pH values could be fitted to an equation involving two exponential decays (which fitted significantly better than a single bimolecular reaction) (Fig. 2). This suggests that at least two processes are occurring, leading in both cases to calcein release. A similar behavior is observed for the DOPC/DOPG vesicles, except at pH 8, where we consistently observe a step-type function in which two exponential phases are separated by a plateau (Fig. 2). This can fitted as a three-phase process involving three exponential decays, in which the second (intermediate) phase has an amplitude that is opposite in sign to the first (fast) and last (slow) phase, leading to an apparent lag phase. The step-type appearance only occurred at low pardaxin concentrations (<∼10–15 μM). Simulation using the program KinTek (24) indicated that the concentration-dependence was consistent with the following minimalist scheme:
where A is the nonbound state and B, B2, and B3 represent different bound and membrane-permeabilizing states of pardaxin (note that membrane binding is not incorporated specifically into the simulation). In this scheme, the bimolecular interaction 2B⇔B2 is rate-limiting at low pardaxin concentrations but not at higher concentrations. However, the intrinsic singularity problems associated with the fitting of multiple exponential decays reduced reproducibility in the determination of rate constants and amplitudes significantly, compared to the two-exponential fits used for the other kinetic profiles. Accordingly, we do not pursue a more quantitative analysis of the kinetic behavior, but restrict ourselves to concluding that at pH 8 with DOPG/DOPC vesicles we observe an additional phase in the dye release mechanism, which contributes to slowing the permeabilization process. This process may be promoted by the overall neutrality of pardaxin at pH 8.0, which, in combination with the orientation, it assumes in the membrane (see below)—allowing it to aggregate into higher-order structures in the membrane environment. The complex kinetics at pH 8.0 highlight yet another difference in pardaxin's DOPC/DOPG interactions compared to pure DOPC vesicles.
Several interesting features are exposed upon plotting the half-lives of the different exponential phases versus pardaxin concentration for DOPC vesicles (Fig. 4, A and C). It is evident that release occurs much more rapidly at pH 10 than at pH 5.5 and 8. Furthermore, the half-life of release reaches a plateau at pH 10 at two-to-threefold lower pardaxin concentrations than at pH 5.5 and 8. The half-life plateau at pH 5.5 and 8 (reached at ∼20 nM) coincides nicely with the amplitude plateau (Fig. 3 A), whereas the half-life plateau at pH 10 is reached at ∼7 nM in contrast to ∼30 nM for the amplitude plateau. As well as highlighting the difference in the mechanism of interaction at the three pH values, it demonstrates a remarkable attraction between negatively charged pardaxin and zwitterionic lipids. The half-life plateau presumably reflects a situation in which the rate-limiting step changes from diffusion-controlled protein association to the vesicle to structural rearrangements on the vesicle.
Figure 4.
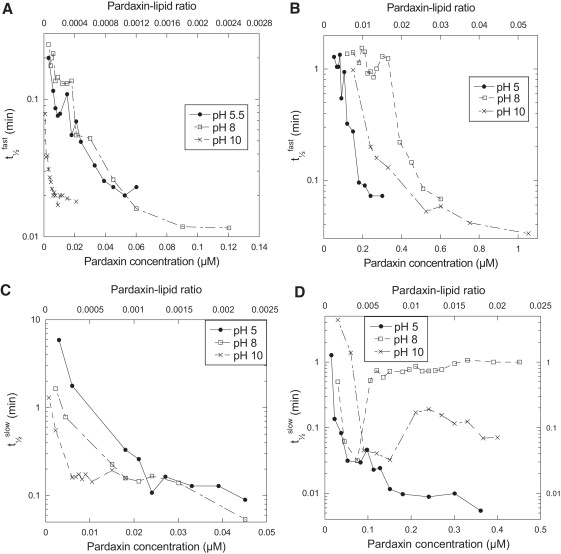
Half-lives for both the fast (A and C) and the slow phase (B and D) of the calcein release plotted for DOPC (A and B) and DOPG/DOPC (20:80) (C and D) vesicles as a function of the protein concentration.
For the DOPC/DOPG vesicles, the situation is reversed. For the fast phase, kinetics is fastest at pH 5.5 for all pardaxin concentrations, followed by pH 10 and then pH 8 (Fig. 4 B). Furthermore, the plateau level is reached first at pH 5.5 followed by pH 10 and then pH 8, but in all cases the plateau level is reached at an approximately twofold higher pardaxin concentration than the amplitude plateau (Fig. 3 B). The same trend is seen for the slow phase, although it is less distinct (data at pH 8 are complicated by the triple exponential decay). Overall, it appears that pH and lipid composition combine to make the fine-tuning kinetics of pardaxin-lipid interactions very delicate and complex.
Pardaxin displays a charge-dependent disruption of giant unilamellar vesicles
The use of LSCM together with GUVs allows us to visualize how pardaxin disrupts individual lipid vesicles. This is achieved by loading the GUVs with a water-soluble fluorophore and incorporating a lipid-specific dye in the vesicles. Using LSCM, we then monitor how the release of entrapped fluorophore correlates with disruption of the GUVs and thus distinguish between disruption by pore formation and vesicle lysis. For simplicity, we restrict ourselves to pH 8 for these studies, where DOPC vesicles are permeabilized much more rapidly and efficiently than DOPC/PG vesicles (Fig. 4).
In DOPC vesicles, the release of vesicle contents over a time course of roughly 15 min (between t = 25 min and t = 42 min) does not lead to substantial loss of vesicle integrity (Fig. 5). This nondisruptive efflux of vesicle contents indicates formation of pores. When the GUVs contain 20% DOPG, the effects are quite different. We observe the same lag time as with DOPC vesicles but pardaxin binding causes release of fluorophore with simultaneous loss of membrane integrity. It takes ∼50 min from the addition of pardaxin to disrupt >95% of the vesicles (Fig. 5). These two distinct modes of vesicle disruption are also apparent when we have a mixture of the two types of vesicles that can be distinguished using different lipid fluorophores (Fig. S1 in the Supporting Material). This implies that we can exclude the possibility that what we observe is an effect of small differences in experimental conditions. Furthermore, the two processes happen at comparable rates in the lipid mixture, indicating that pardaxin shows no preference for one type of vesicle. This contrasts with the antimicrobial peptide Novicidin, which shows a much stronger preference for anionic lipids under these conditions (B. S. Vad and D. E. Otzen, unpublished).
Figure 5.
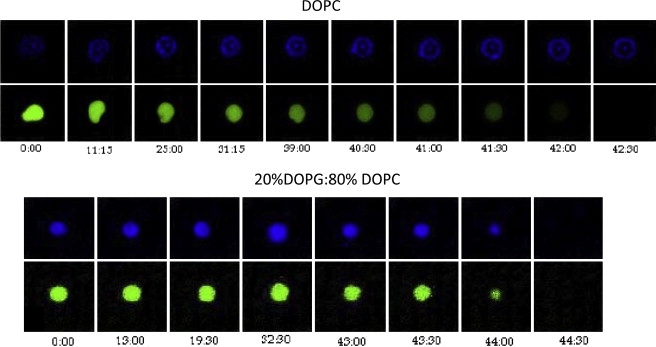
LSCM investigations of pardaxin disruption of GUVs. For each lipid, we show the time course (in minutes) of the fluorescence from a water-soluble fluorophore (Alexa 488, encapsulated within the vesicle interior) in the top row and a membrane-bound dye (DiI) in the bottom row after the addition of 5 mM pardaxin to GUV composed of either DOPC or DOPG/DOPC (20:80). For DOPC GUVs we start to observe a decrease in the water-soluble fluorophore fluorescence after ∼29 min, reaching a minimum after 45 min, whereas the DiI fluorescence is unchanged. For the DOPG/DOPC (20:80) GUVs, we observe a decrease in the GUV size after roughly 40 min, leading to vesicle disruption within 5 min indicated by the disappearance of both fluorophores. Note that there is a slight inhomogeneity in the dye distribution in the DOPC vesicle. However, the same type of behavior, indicative of pore-formation, is observed for a large range of different vesicle sizes for DOPC irrespective of the dye distribution.
13C solid-state NMR reveals that pardaxin interacts with the lipid acyl chain in a charge-dependent fashion
To complement our biophysical studies, we use solid-state NMR to analyze the conformation/orientation of pardaxin when incorporated into or binding to the two lipid types. As with mimics of vesicles and phospholipid bilayers, we use bicelles (25) composed of a mixture of long-chain (14-O-PC with or without 14-O-PG) and short-chain (6-O-PC) phospholipids. In these phospholipids, the ester bonds linking the two acyl chains to the glycerol backbone are replaced with a nonhydrolyzable ether bond (26,27). This increases the chemical stability of the lipids but does not affect the secondary structure of pardaxin according to far-UV CD spectroscopy (data not shown). Bicelles mixed using appropriate stoichiometry of short- and long-chain lipids will align when placed in a strong magnetic field, making them useful for oriented-sample solid-state NMR. In this study we take advantage of the macroscopical alignment to determining the orientation of anisotropic nuclear spin interactions (e.g., 13C-1H dipole-dipole couplings that depend on the angle between the internuclear axis and the external magnetic field) and through these establish information about the conformation of the lipid molecules upon interacting with the peptides. These studies may be conducted with lipids in natural abundance (i.e., without incorporating 15N or 13C isotope labels).
We recorded 13C natural abundance PISEMA (19) NMR spectra of the two types of bicelles, and record changes upon addition of pardaxin. In the present context, the PISEMA spectrum correlates 13C chemical shifts with the 1H-13C heteronuclear dipole-dipole coupling, reflecting the binding of the carbon to directly attached protons. The dipole-dipole coupling (and to a smaller extent the chemical shift) is very sensitive to reorientation/dynamics of the lipids and thereby to changes in lipid order upon interaction with pardaxin. The information is highly localized, in the sense that signals from different carbons in the lipid chain provide separate responses, rendering the experiment useful to distinguish trans-membrane and surface orientations of the peptide in the membrane (28,29). The change in the lipid conformation may conveniently be expressed in terms of an order parameter S2CH, which is extracted from the experimental spectra from the relationship (28)
We assume that Do = 21.5 kHz and β = 90°. The measured coupling is the sum of the dipole dipole- and the J-coupling, and the J-coupling is assumed to be 150 Hz for all sites.
Fig. 6 shows a two-dimensional PISEMA spectrum (Fig. 6 A) and one-dimensional slices obtained for a bicelle composed of 14-O-PC/6-O-PC (molar ratio 3.2:1.0) lipids (Fig. 6, B–D) and 14-O-PC/14-O-PG/6-O-PC (molar ratio 2.6:0.6:1,0) lipids (Fig. 6, E–G). The slices (extracted from the two-dimensional spectra) provide measures of 1H-13C dipole-dipole couplings observed for three representative carbon sites along the 14-O-PC molecule, namely Cα (Fig. 6, D and E), C3 (Fig. 6, C and F), and C14 (Fig. 6, D and G). The slice in red represents pure lipids whereas the slice in black represents the case where pardaxin is incorporated (molar ratio: peptide/lipids, 1:100). It is evident that pardaxin interacts differently with lipid bicelles depending on their surface charge. Fig. 6, B–D, reveals that the order parameter in PC bicelles changes at carbon sites all along the lipid molecule, indicating that pardaxin is incorporated in a trans-membrane configuration. This agrees very well with the formation of pores according to GUV analyses (Fig. 5). A different pattern is observed for bicelles containing a mixture of 14-O-PC/14-O-PG/6-O-PC (Fig. 6, E–G). Here only the order parameters for the headgroup region (C3 (Fig. 6 E) and Cα (Fig. 6 D)) change upon 1:100 molar ratio addition of pardaxin, whereas that of the membrane buried C14 site remains invariant (Fig. 6 F). Accordingly, in this case the NMR data suggest that pardaxin is positioned on the surface of the PC/PG bicelle and only interacts with the headgroup. This also correlates very well with our GUV data, which indicate membrane lysis rather than pore formation (Fig. 5). Fig. 6, H and I, provides a color-code representation of the relative changes in order parameters when pardaxin is added to PC bicelles (Fig. 6 H) and PC/PG mixed bicelles (Fig. 6 I). Red indicates an increase in the order parameter, blue represents a decrease, and white represents no measured change in order parameters. The diameters of the spheres in Fig. 6, H and I, are inversely proportional to the measured order parameter in bicelles without pardaxin. Thus, a small order parameter is designated with a circle with large radius (indicating geometrical variability) whereas a larger order parameter is represented by a smaller radius (indicative of less flexibility). It is not surprising that the order parameter for the headgroup generally decreases as peptide is added to the bicelles, due to perturbation by accommodation of the peptide. The increase in the order parameter for the very disordered C14 site that occurs in DOPC vesicles (where pardaxin is trans-membrane) may be attributed to a restriction of the free rotation of the acyl methyl group by pardaxin. Similar arguments apply to γ1 (the first carbon in the glycerol linker region of the lipid molecule), which also becomes slightly more ordered upon addition of pardaxin to PC bicelles (Fig. 6 H).
Figure 6.
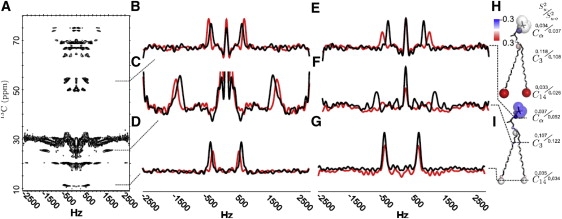
Static-sample, natural abundance 13C solid-state NMR spectra for oriented PC or PC/PG bicelles with and without pardaxin. (A) Two-dimensional PISEMA spectrum of a PC bicelle with pardaxin. (B–G) One-dimensional slices from two-dimensional PISEMA spectra of PC bicelles with (black) and without (red) pardaxin in bicelles composed of (B–D) 14-O-PC/6-O-PC (molar ratio 3.2:1.0) and (E–G) 14-O-PC/14-O-PG/6-O-PC (molar ratio 2.6:0.6:1,0) lipids. From top (B and E) to bottom (D and G), the slices represent traces through the Cα, C3, and C14 resonances of the lipid molecules, respectively. (H and I) Summary of measured order parameters for 14-O-PC/6-O-PC bicelles (H) and for 14-O-PC/14-O-PG/6-O-PC bicelles (I). The radii of the spheres are inversely proportional to the measured order parameters of atomic sites along the lipid molecule in bicelles with no pardaxin (see text). The order parameters measured both with and without pardaxin are given beside the relevant carbon. The color of the spheres indicate the relative change in measured order parameters upon addition of pardaxin, with red indicating that the order parameter increases; blue that it decreases; and white indicating no change.
For the PG-containing bicelles, we observe no change in the conformation order of the acyl chains in the deeper parts of the bicelles. This is indicated by the generally white color of the C4–C14 in Fig. 6 I, reflecting the data in Fig. 6, E–G, where only small changes in the dipolar splitting (and thereby the order parameter) are observed.
Discussion
Calcein release patterns are dependent on lipid headgroup charge
Numerous studies have addressed the interaction of pardaxin with lipid membranes (7–14,30–36), but have not been based on detailed kinetic studies—focusing instead on endpoint permeation (8–10,31,33). We extend these studies by a methodical investigation of how the rate of vesicle permeabilization (calcein release) is affected by solution pH and lipid headgroup charge. Our data show that pardaxin-mediated lipid permeations are dependent on lipid headgroup charge and show subtle kinetic effects. We observe a dependence of pardaxin protonation both with vesicles consisting of DOPC/DOPG and those composed of only DOPC. Permeabilization of DOPC/PG vesicles occurred more efficiently when pardaxin had a net charge of +1 (pH 5.5) compared to higher pH. Nevertheless, 5–12 times less pardaxin was needed to permeate purely zwitterionic vesicles, and this permeation efficiency was more efficient at higher pH. The CD spectra indicate a higher degree of α-helical structure in PC vesicles than in PC/PG vesicles, which suggest a more intimate association of pardaxin with PC vesicles. There is a large difference in the concentrations needed to permeate vesicles by either pore formation or the carpet mechanism (the latter being the most protein-demanding (37)). These findings suggest that the differences in pardaxin concentration needed for permeation reflect differences in the mode of action with zwitterionic and anionic vesicles. Similar conclusions have been reached for the antimicrobial peptide Melittin, which was found to form pores in zwitterionic vesicles and induce aggregation/fusion of anionic vesicles (38).
The kinetics of the membrane disruption by pardaxin reveals two distinct processes—an initial fast process and a subsequent slow process. The measured release time profiles represent the permeabilization activity and only indirectly report on structural changes. The multiple decays indicate multiple ways of perturbing the membrane. The kinetics of membrane disruption by the antimicrobial peptide Cecropin also displays two distinct kinetic processes (39). The fast step is coupled to initial peptide binding whereas the slower process is linked to a more specific mode of action, possibly pore formation or shedding of membrane fragments. Remarkably, the kinetics of vesicle disruption is markedly slower for a Cecropin mutant operating through the carpet mechanism as opposed to the pore formation by wild-type Cecropin (39). We see a similar scenario for pardaxin with regard to the kinetics of membrane disruption of zwitterionic and anionic vesicles.
The half-life values for disruption of DOPC stabilize at a minimum level at 0.1 μM pardaxin, well below the concentration where pardaxin starts to disrupt DOPC/PG vesicles. Gouy-Chapman theory describes how negative charge on the acidic membrane creates a membrane potential that causes cationic peptides to redistribute to the membrane (40,41). This passive accumulation of peptides near an anionic membrane surface will result in a high apparent binding constant. If the initial fast process relates to peptide binding, it should be expected that this would be even faster on anionic vesicles. The fact that this is not the case indicates that electrostatic attraction to anionic vesicles does not lead to more efficient permeabilization. Instead, we speculate that these strong interactions may in fact trap pardaxin in conformations that are not optimal for vesicle permeabilization. The complex kinetics on DOPC/PG vesicles at pH 8 support this view, indicating an extra step that may constitute a less efficient way to organize the peptide in the membrane compared to the rapid pore formation observed on DOPC vesicles.
GUV experiments reveal two distinct vesicle disruption patterns
The mode of action of pardaxin is known to be sensitive to the charge of the lipid groups, with clear indications of pore formation in pure POPC vesicles that appeared to change upon the inclusion of anionic lipids (13). Based on our GUV experiments and supported by solid-state NMR data, we unequivocally conclude that pardaxin forms pores in pure DOPC vesicles and disrupts DOPC/PG vesicles in a all-or-none fashion, suggesting a carpet mechanism. Previous studies combining GUVs and LSCM to study antimicrobial peptides such as Maculatin, Citropin, Aurein, and Magainin 2 were similarly able to resolve the permeabilization mechanism in zwitterionic and anionic vesicles (42,43). They found that for these peptides the mode of action was independent of the lipid composition. This study of pardaxin is the first to our knowledge, that is able to demonstrate, visually, that the mode of action can be dependent on the lipid composition of the vesicles. Furthermore, we were able to reproduce the specific modes of action in a heterogeneous mixture composed of anionic vesicles and zwitterionic vesicles. These two different modes of action were observed on the same timescale, showing there was no preference for either lipid composition.
The interaction of pardaxin with membranes changes the lipid
Two-dimensional solid-state 13C NMR experiments of DMPC/DHPC bicelles with or without pardaxin allow us to probe the local interactions of the antimicrobial peptide with lipid membranes (both with 13C in natural abundance) without the addition of exotic probes to the sample, which might, in turn, effect the molecule-lipid interactions. This method, which relies on magnetic orientation of the bicelles by the strong static magnetic field of the NMR spectrometer, has previously been used for studying the membrane interactions of the antidepressant desipramine (44) as well as the antimicrobial peptide pexiganan (28,45). Pexiganan has been shown by various techniques (fluorescence assays, calorimetric techniques, microscopy, solid-state NMR spectroscopy, and neutron diffraction) to work by a toroidal pore mechanism. In the natural abundance 13C NMR experiments, this resulted in an increase in the disorder of DMPC mainly at the headgroup region and the upper part of the acyl chain. Given that pexiganan works by the toroidal pore mechanism, the NMR signal is a mixture of the signal from the bilayer-forming lipids and the lipids that line the toroidal pore, making specific interpretations of peptide-induced changes in the lipid conformation difficult.
The calcein release measurements and the LSCM results for pardaxin revealed two distinct vesicle disruption patterns, compatible with pore formation and the carpet mechanism for zwitterionic and anionic vesicles, respectively. This is remarkably similar to the study by Ladokhin and White (46) on the 26-residue peptide melittin, where calcein release assays and oriented CD measurements suggested a trans-membrane orientation in POPC vesicles and an orientation parallel to the membrane plane in POPG vesicles. Interestingly, the two peptides show weak N-terminal homology although melittin is highly positively charged (+6 at neutral pH). These two modes of action will lead to different local membrane perturbations that may be probed by oriented-bicelle, natural abundance, 13C two-dimensional NMR measurements. For bicelles composed only of zwitterionic vesicles, we observed a slight disordering of the lipid headgroup and an increase in the order of the acyl chain upon addition of pardaxin. This is the opposite of what was observed for pexiganan, indicating that pardaxin does not form toroidal pores.
The increase in acyl-chain order is instead what would be expected if the pardaxin was trans-membrane, and thereby restricted the movement of the acyl chains. This ordering effect on lipid acyl chains by trans-membrane peptides has been reported previously (47,48), corresponding to the formation of pores by the barrel- stave mechanism (49). For the bicelles containing anionic lipids, a disordering of the headgroup region is observed when pardaxin is added. From the LSCM measurements we have shown that pardaxin works by an all-or-none mechanism, as would be expected from a peptide that worked by the carpet mechanism. It has been shown that the toroidal pore formation is followed by membrane disruption at higher concentration (50), but we do not believe that this is pardaxin's mode of action, as no pore formation was observed by lowering the pardaxin concentration. These results indicate that there is a significant difference in the impact on acyl-chain order induced by the toroidal pore formation and the carpet mechanism. This is interesting from the standpoint that in both processes the peptide is lying in the lipid headgroup and acyl-chain interface (51–54). If the difference in order induced by the two binding modes is a general phenomenon, this would allow for an easy way of distinguishing between the two binding modes. This can only be proved by further studies on other peptide-lipid systems.
Supporting Material
Materials and methods and a figure are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01690-7.
Supporting Material
Acknowledgments
We are very grateful to Professor L. Bagatolli, Dr. Jeppe L. Nielsen, and Associate Professor T. Vosegaard for assistance with GUV production, LSCM, and solid-state NMR setup.
B.S.V. is supported by a grant from the Villum Kann Rasmussen Foundation (BioNET) and Aalborg University. This work is supported by the Danish Research Foundation (inSPIN).
References
- 1.Ehrenstein G., Lecar H. Electrically-gated ionic channels in lipid bilayers. Rev. Biophys. 1977;10:1–34. doi: 10.1017/s0033583500000123. [DOI] [PubMed] [Google Scholar]
- 2.Shai Y., Oren Z. From “carpet” mechanism to de-novo designed diastereomeric cell-selective antimicrobial peptides. Peptides. 2001;22:1629–1641. doi: 10.1016/s0196-9781(01)00498-3. [DOI] [PubMed] [Google Scholar]
- 3.Yang L., Weiss T.M., Huang H.W. Crystallization of antimicrobial pores in membranes: magainin and protegrin. Biophys. J. 2000;79:2002–2009. doi: 10.1016/S0006-3495(00)76448-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lazarovici P., Primor N., Loew L.M. Purification and pore-forming activity of two hydrophobic polypeptides from the secretion of the Red Sea Moses sole (Pardachirus marmoratus) J. Biol. Chem. 1986;261:16704–16713. [PubMed] [Google Scholar]
- 5.Shai Y., Bach D., Yanovsky A. Channel formation properties of synthetic pardaxin and analogues. J. Biol. Chem. 1990;265:20202–20209. [PubMed] [Google Scholar]
- 6.Oren Z., Shai Y. A class of highly potent antibacterial peptides derived from pardaxin, a pore-forming peptide isolated from Moses sole fish Pardachirus marmoratus. Eur. J. Biochem. FEBS. 1996;237:303–310. doi: 10.1111/j.1432-1033.1996.0303n.x. [DOI] [PubMed] [Google Scholar]
- 7.Rapaport D., Peled R., Shai Y. Reversible surface aggregation in pore formation by pardaxin. Biophys. J. 1996;70:2502–2512. doi: 10.1016/S0006-3495(96)79822-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rapaport D., Shai Y. Interaction of fluorescently labeled pardaxin and its analogues with lipid bilayers. J. Biol. Chem. 1991;266:23769–23775. [PubMed] [Google Scholar]
- 9.Shai Y., Hadari Y.R., Finkels A. pH-dependent pore formation properties of pardaxin analogues. J. Biol. Chem. 1991;266:22346–22354. [PubMed] [Google Scholar]
- 10.Epand R.F., Ramamoorthy A., Epand R.M. Membrane lipid composition and the interaction of pardaxin: the role of cholesterol. Protein Pept. Lett. 2006;13:1–5. [PubMed] [Google Scholar]
- 11.Kolusheva S., Lecht S., Lazarovici P. Pardaxin, a fish toxin peptide interaction, with a biomimetic phospholipid/polydiacetylene membrane assay. Peptides. 2008;29:1620–1625. doi: 10.1016/j.peptides.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 12.Pouny Y., Shai Y. Interaction of D-amino acid incorporated analogues of pardaxin with membranes. Biochemistry. 1992;31:9482–9490. doi: 10.1021/bi00154a022. [DOI] [PubMed] [Google Scholar]
- 13.Hallock K.J., Lee D.K., Ramamoorthy A. Membrane composition determines pardaxin's mechanism of lipid bilayer disruption. Biophys. J. 2002;83:1004–1013. doi: 10.1016/S0006-3495(02)75226-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porcelli F., Buck B., Veglia G. Structure and orientation of pardaxin determined by NMR experiments in model membranes. J. Biol. Chem. 2004;279:45815–45823. doi: 10.1074/jbc.M405454200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nielsen J.T., Bjerring M., Nielsen N.C. Unique identification of supramolecular structures in amyloid fibrils by solid-state NMR. Angew. Chem. Int. Ed. 2009;48:2118–2121. doi: 10.1002/anie.200804198. [DOI] [PubMed] [Google Scholar]
- 16.Mogensen J.E., Ferreras M., Otzen D.E. The major allergen from birch tree pollen, Bet v 1, is a membrane binding protein. Biochemistry. 2007;46:3356–3365. doi: 10.1021/bi062058h. [DOI] [PubMed] [Google Scholar]
- 17.Angelova M.L., Dimitrov D.S. Liposome electroformation. Faraday Discuss. Chem. Soc. 1986;81:303–311. [Google Scholar]
- 18.Bagatolli L.A., Gratton E. Two-photon fluorescence microscopy observation of shape changes at the phase transition in phospholipid giant unilamellar vesicles. Biophys. J. 1999;77:2090–2101. doi: 10.1016/S0006-3495(99)77050-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu C.H., Ramamoorthy A., Opella S.J. High-resolution heteronuclear dipolar solid-state NMR spectroscopy. J. Magn. Reson. 1994;109:270–272. [Google Scholar]
- 20.Fung B.M., Khitrin A.K., Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000;142:97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- 21.Zhou N.E., Kay C.M., Hodges R.S. Synthetic model proteins. Positional effects of interchain hydrophobic interactions on stability of two-stranded α-helical coiled-coils. J. Biol. Chem. 1992;267:2664–2670. [PubMed] [Google Scholar]
- 22.Lau S.Y., Taneja A.K., Hodges R.S. Synthesis of a model protein of defined secondary and quaternary structure. Effect of chain length on the stabilization and formation of two-stranded α-helical coiled-coils. J. Biol. Chem. 1984;259:13253–13261. [PubMed] [Google Scholar]
- 23.Lazarova T., Brewin K.A., Robinson C.R. Characterization of peptides corresponding to the seven transmembrane domains of human adenosine A2a receptor. Biochemistry. 2004;43:12945–12954. doi: 10.1021/bi0492051. [DOI] [PubMed] [Google Scholar]
- 24.Johnson K.A., Simpson Z.B., Blom T. Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 2009;387:20–29. doi: 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 25.Prosser R.S., Hwang J.S., Vold R.R. Magnetically aligned phospholipid bilayers with positive ordering: a new model membrane system. Biophys. J. 1998;74:2405–2418. doi: 10.1016/S0006-3495(98)77949-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Losonczi J.A., Prestegard J.H. Improved dilute bicelle solutions for high-resolution NMR of biological macromolecules. J. Biomol. NMR. 1998;12:447–451. doi: 10.1023/a:1008302110884. [DOI] [PubMed] [Google Scholar]
- 27.Aussenac F., Lavigne B., Dufourc E.J. Toward bicelle stability with ether-linked phospholipids: temperature, composition, and hydration diagrams by 2H and 31P solid-state NMR. Langmuir. 2005;21:7129–7135. doi: 10.1021/la050243a. [DOI] [PubMed] [Google Scholar]
- 28.Dvinskikh S., Dürr U., Ramamoorthy A. A high-resolution solid-state NMR approach for the structural studies of bicelles. J. Am. Chem. Soc. 2006;128:6326–6327. doi: 10.1021/ja061153a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gross J.D., Warschawski D.E., Griffin R.G. Dipolar recoupling in MAS NMR: a probe for segmental order in lipid bilayers. J. Am. Chem. Soc. 1997;119:796–802. [Google Scholar]
- 30.Rapaport D., Nir S., Shai Y. Capacities of pardaxin analogues to induce fusion and leakage of negatively charged phospholipid vesicles are not necessarily correlated. Biochemistry. 1994;33:12615–12624. doi: 10.1021/bi00208a012. [DOI] [PubMed] [Google Scholar]
- 31.Rapaport D., Hague G.R., Shai Y. pH- and ionic strength-dependent fusion of phospholipid vesicles induced by pardaxin analogues or by mixtures of charge-reversed peptides. Biochemistry. 1993;32:3291–3297. doi: 10.1021/bi00064a011. [DOI] [PubMed] [Google Scholar]
- 32.Rapaport D., Shai Y. Aggregation and organization of pardaxin in phospholipid membranes. A fluorescence energy transfer study. J. Biol. Chem. 1992;267:6502–6509. [PubMed] [Google Scholar]
- 33.Lelkes P.I., Lazarovici P. Pardaxin induces aggregation but not fusion of phosphatidylserine vesicles. FEBS Lett. 1988;230:131–136. doi: 10.1016/0014-5793(88)80656-2. [DOI] [PubMed] [Google Scholar]
- 34.Thennarasu S., Nagaraj R. Solution conformations of peptides representing the sequence of the toxin pardaxin and analogues in trifluoroethanol-water mixtures: analysis of CD spectra. Biopolymers. 1997;41:635–645. doi: 10.1002/(SICI)1097-0282(199705)41:6<635::AID-BIP4>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 35.Shi Y.L., Edwards C., Lazarovici P. Ion selectivity of the channels formed by pardaxin, an ionophore, in bilayer membranes. Nat. Toxins. 1995;3:151–155. doi: 10.1002/nt.2620030306. [DOI] [PubMed] [Google Scholar]
- 36.Primor N. Pharyngeal cavity and the gills are the target organs for the repellent action of pardaxin in shark. Experientia. 1985;41:693–695. doi: 10.1007/BF02007726. [DOI] [PubMed] [Google Scholar]
- 37.Steiner H., Andreu D., Merrifield R.B. Binding and action of cecropin and cecropin analogues: antibacterial peptides from insects. Biochim. Biophys. Acta. 1988;939:260–266. doi: 10.1016/0005-2736(88)90069-7. [DOI] [PubMed] [Google Scholar]
- 38.van den Bogaart G., Mika J.T., Poolman B. The lipid dependence of melittin action investigated by dual-color fluorescence burst analysis. Biophys. J. 2007;93:154–163. doi: 10.1529/biophysj.107.106005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen H.M., Clayton A.H., Sawyer W.H. Kinetics of membrane lysis by custom lytic peptides and peptide orientations in membrane. Eur. J. Biochem. FEBS. 2001;268:1659–1669. doi: 10.1046/j.1432-1327.2001.02039.x. [DOI] [PubMed] [Google Scholar]
- 40.Matsuzaki K., Harada M., Miyajima K. Physicochemical determinants for the interactions of magainins 1 and 2 with acidic lipid bilayers. Biochim. Biophys. Acta. 1991;1063:162–170. doi: 10.1016/0005-2736(91)90366-g. [DOI] [PubMed] [Google Scholar]
- 41.Kuchinka E., Seelig J. Interaction of melittin with phosphatidylcholine membranes. Binding isotherm and lipid head-group conformation. Biochemistry. 1989;28:4216–4221. doi: 10.1021/bi00436a014. [DOI] [PubMed] [Google Scholar]
- 42.Ambroggio E.E., Separovic F., Bagatolli L.A. Direct visualization of membrane leakage induced by the antibiotic peptides: maculatin, citropin, and aurein. Biophys. J. 2005;89:1874–1881. doi: 10.1529/biophysj.105.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tamba Y., Yamazaki M. Single giant unilamellar vesicle method reveals effect of antimicrobial peptide magainin 2 on membrane permeability. Biochemistry. 2005;44:15823–15833. doi: 10.1021/bi051684w. [DOI] [PubMed] [Google Scholar]
- 44.Dvinskikh S.V., Dürr U.H., Ramamoorthy A. High-resolution 2D NMR spectroscopy of bicelles to measure the membrane interaction of ligands. J. Am. Chem. Soc. 2007;129:794–802. doi: 10.1021/ja065536k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gottler L.M., Ramamoorthy A. Structure, membrane orientation, mechanism, and function of pexiganan—a highly potent antimicrobial peptide designed from magainin. Biochim. Biophys. Acta. Biomembr. 2008;1788:1680–1686. doi: 10.1016/j.bbamem.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ladokhin A.S., White S.H. “Detergent-like” permeabilization of anionic lipid vesicles by melittin. Biochim. Biophys. Acta. 2001;1514:253–260. doi: 10.1016/s0005-2736(01)00382-0. [DOI] [PubMed] [Google Scholar]
- 47.Marcotte I., Wegener K.L., Separovic F. Interaction of antimicrobial peptides from Australian amphibians with lipid membranes. Chem. Phys. Lipids. 2003;122:107–120. doi: 10.1016/s0009-3084(02)00182-2. [DOI] [PubMed] [Google Scholar]
- 48.Koenig B.W., Ferretti J.A., Gawrisch K. Site-specific deuterium order parameters and membrane-bound behavior of a peptide fragment from the intracellular domain of HIV-1 gp41. Biochemistry. 1999;38:6327–6334. doi: 10.1021/bi982800g. [DOI] [PubMed] [Google Scholar]
- 49.Yang L., Harroun T.A., Huang H.W. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 2001;81:1475–1485. doi: 10.1016/S0006-3495(01)75802-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sengupta D., Leontiadou H., Marrink S.J. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta. 2008;1778:2308–2317. doi: 10.1016/j.bbamem.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 51.Murzyn K., Pasenkiewicz-Gierula M. Construction of a toroidal model for the magainin pore. J. Mol. Model. 2003;9:217–224. doi: 10.1007/s00894-003-0127-z. [DOI] [PubMed] [Google Scholar]
- 52.Wi S., Kim C. Pore structure, thinning effect, and lateral diffusive dynamics of oriented lipid membranes interacting with antimicrobial peptide protegrin-1: 31P and 2H solid-state NMR study. J. Phys. Chem. 2008;112:11402–11414. doi: 10.1021/jp801825k. [DOI] [PubMed] [Google Scholar]
- 53.Wieprecht T., Dathe M., Bienert M. Influence of the angle subtended by the positively charged helix face on the membrane activity of amphipathic, antibacterial peptides. Biochemistry. 1997;36:12869–12880. doi: 10.1021/bi971398n. [DOI] [PubMed] [Google Scholar]
- 54.Oren Z., Shai Y. Selective lysis of bacteria but not mammalian cells by diastereomers of melittin: structure-function study. Biochemistry. 1997;36:1826–1835. doi: 10.1021/bi962507l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.