

Abstract
We report a numerical study of the (un)folding routes of the truncated FBP28 WW domain at ambient conditions using a combination of four advanced rare event molecular simulation techniques. We explore the free energy landscape of the native state, the unfolded state, and possible intermediates, with replica exchange molecular dynamics. Subsequent application of bias-exchange metadynamics yields three tentative unfolding pathways at room temperature. Using these paths to initiate a transition path sampling simulation reveals the existence of two major folding routes, differing in the formation order of the two main hairpins, and in hydrophobic side-chain interactions. Having established that the hairpin strand separation distances can act as reasonable reaction coordinates, we employ metadynamics to compute the unfolding barriers and find that the barrier with the lowest free energy corresponds with the most likely pathway found by transition path sampling. The unfolding barrier at 300 K is ∼17 kBT ≈ 42 kJ/mol, in agreement with the experimental unfolding rate constant. This work shows that combining several powerful simulation techniques provides a more complete understanding of the kinetic mechanism of protein folding.
Introduction
Understanding the formation mechanism, stability, and kinetics of the simple but ubiquitous β-sheet secondary structure provides insight into how proteins are able to find their native state. A small three-strand β-sheet, namely, the formin binding protein 28 (FBP28), has been studied both in vitro and in silico (1–5), and is an excellent model system to use in studying β-sheet formation. FBP28 is a member of the WW domain protein family, which comprises a large number of small single-domain three-stranded anti-parallel β-sheet proteins (6–9) ranging from 35 to 40 amino acids. The family derives its name from the characteristic presence of two preserved tryptophans (W) separated by ∼20 amino acids along the domain sequence. WW domains can bind proline-rich ligands and the complexes they form have been implicated in a number of human diseases such as muscular dystrophy, cancer, hypertension, Alzheimer's, and Huntington's diseases (10). In addition, WW domains have been identified as a part of many signaling proteins (6). Being one of the smallest β-sheets, WW domains are attractive systems both from experimental and computational viewpoints, in particular because they do not contain groups that can complicate kinetic analysis and simulations, such as disulfide bonds or cis prolines. The WW domains have been the subject of several experimental (1,2,11–14) as well as computational (3–5,15–19) studies on the formation of β-sheet structures.
The native state structure of FBP28 has been resolved in solution by nuclear magnetic resonance (8,20) (see Fig. 1). Under the influence of denaturant and high temperatures, FBP28 unfolds reversibly (1,8,11,13,14,21) but can also form fibers at elevated temperatures (22). Temperature jump experiments indicated cooperative, two-state folding without any intermediate states, with a folding rate constant kf = (33 μs)−1 and an unfolding rate constant ku = (0.6 ms)−1 (1). Moreover, Φ-value analysis revealed the formation of the β-turn I as the rate-limiting step for the folding transition (3). Mu et al. (4) performed an extensive simulation of the FBP28 WW domain, and concluded that the transition state was characterized by a formed β-turn in the larger hairpin. This would imply that the rate-limiting step for folding is indeed the formation of the larger hairpin. Another laser-temperature jump experiment (2) suggests that the folding of the FBP28 WW domain is strongly biphasic at low temperatures, implicating three stable states. The two-state behavior can be regained by tuning temperature or by truncation of the termini. Karanicolas and Brooks (23) proposed that the biphasic behavior of FBP28 originates in the recently discovered dual binding specificity of the WW domain (10). In their implicit solvent simulation study, a close-to-native state was discovered, characterized by an alternative packing of the hydrophobic side chains, and the Trp-30 more solvent-exposed. The existence of this state was proposed as a link between the biphasic kinetics and the dual binding specificity of the FBP28 WW domain, as the residues involved belong to the WW domain-binding site.
Figure 1.
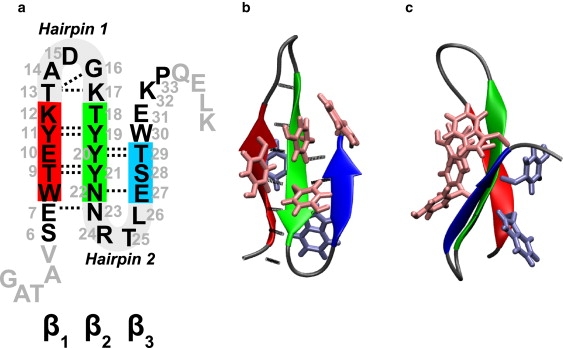
(a) Sequence of FBP28 and FBP28ΔNΔC. Amino acids are numbered according to their position in the sequence of original WW domain. The truncated amino acids are shown in gray font, with the turn regions and the strands β1 (residues 8–12), β2 (residues 18–22), and β3 (residues 27–29) as thick lines. Native H-bonds are plotted as dotted lines. Hairpin-1 comprises strands β1, β2, and turn I t1, whereas hairpin-2 comprises strands β2, β3, and turn II t2. (b) The structure of the native state N of the WW domain in cartoon representation. The hydrophobic core residues have been shown as licorice, with the upper hydrophobic core in a lighter shade than the lower core. (c) Side view of the structure in panel b.
In addition to the above-mentioned simulations on FBP28, several studies have focused on other members of the WW domain family, notably YAP65, Pin1, and Fip35 (a mutant of Pin1), which are sufficiently closely related to FBP28 to warrant a comparison. Early simulations by Ibragimova and Wade indicated that in contrast to FBP28, YAP65 is not stable in water, and unfolds (15). Luo et al. (16) performed discontinuous molecular dynamics on an all-atom Gō-model of the Pin1 domain, and observed that whereas the larger hairpin is more likely to be formed before the other, heterogeneity can exist in the folding mechanism depending on temperature. Freddolino et al. (17) performed a 10-μm-long molecular dynamics (MD) simulation of the Fip35 WW domain in explicit solvent. This simulation did not show correct folding to the native state, which was attributed to a force-field bias (18). Ensign and Pande (19) generated thousands of MD trajectories for Fip35 in implicit solvent using distributed computing. They found that in the few trajectories that showed folding, the mechanism was heterogeneous and included a hydrophobic collapse.
Simulating the kinetics and folding mechanisms of β-sheets in explicit solvent still poses a major computational challenge. Some of the previous studies on the folding of the FBP28 WW domain have been performed either using an implicit solvent (23) or high temperature molecular dynamics unfolding simulations (3). The latter approach yields results that are not necessarily the same at room temperature. Room-temperature, straightforward MD simulations are hampered by the long timescales connected to the folding events. The current practical limit of explicit solvent MD simulation is approximately microseconds (17). Even if one observes a folding event in a microsecond simulation, it represents only one possible pathway out of many available to the protein. The long timescales are related to high free energy barriers separating the folded, unfolded, and intermediate states. Replica exchange MD (4,5,24) can overcome these free energy (FE) barriers, but has difficulties with convergence, and does not yield transition mechanisms for ambient conditions. The kinetics can be addressed, however, by established methods such as the reactive flux method (25,26). Such an approach requires the free energy as a function of a proper reaction coordinate. Biasing methods such as umbrella sampling (27) or metadynamics (28) can sample the free energy barrier as a function of a specified reaction coordinate. However, in a high dimensional and complex system such as a solvated protein, finding a proper coordinate is far from trivial. The transition path sampling (TPS) method avoids such problems by generating an ensemble of unbiased pathways that connect the unfolded with the folded state (29,30). This ensemble can then subsequently be analyzed to yield the rate constant, as well as the mechanism and transition states (30).
The aim of the current work is to:
-
1.
Establish the meta-stable states of the (truncated) FBP28 WW domain,
-
2.
Sample transition pathways between these states,
-
3.
Find the most likely folding mechanism, and
-
4.
Confirm this by estimating the free energy barriers.
No single rare event simulation technique can achieve all three goals at once. Hence, we combine the strengths of three approaches, i.e., replica-exchange MD (REMD), metadynamics, and TPS, to study the folding routes of the FBP28 WW domain. REMD can explore the free energy landscape (24), whereas bias exchange metadynamics (BE-Meta) (31) provides tentative transition pathways. Using these pathways as input trajectories, TPS subsequently collects an equilibrated path ensemble, yielding unbiased insight in the folding mechanism. Analyzing these pathways, we propose reasonable order parameters that can act as a reaction coordinate. This reaction coordinate is then used in metadynamics simulations to estimate free energy barriers (28). Both the pathways and the barrier free energies can be used to answer specific questions on the folding mechanism, such as whether the hairpins fold simultaneously in a cooperative fashion, or independently. In the latter case, what is the order of formation, and the rate-limiting step? In addition, the role of the hydrophobic core and the solvent in the formation of the hairpins can be addressed. In such a way, sampling MD pathways can lead to specific insight in the folding of this WW domain, and in β-sheet formation in general.
Methods
System preparation
Inspired by the work of Nguyen et al. (2) and of Periole et al. (5), we study the truncated form of the wild-type formin-binding domain FBP28 to keep the simulations tractable. The full-protein, nuclear magnetic-resonance structure (PDB entry 1E0L) containing 37 amino acids was truncated before Val-5 and after Pro-33, yielding the FBP28ΔNΔC sequence (see Fig. 1): SEWTE YKTAD GKTYY YNNRT LESTW EKP. Although removing the N-terminal domain (residues Gly-1 to Val-5) was found to have no observable effect on the stability of the domain (2), C-terminal domain truncation (residues Gln-34 to Lys-37) decreases the stability of the protein due to the removal of Leu-36, which is a part of the hydrophobic core Trp-8, Tyr-20, and Pro-33 in the wild-type native state (see Fig. 1). Nevertheless, the truncation does not result in significant structural change of the native state (2,5).
The polypeptide was solvated by 2994 water molecules in a cubic box in such way that the width of the water layer surrounding the protein was a least 12 Å. All simulations were performed using the GROMACS 3.3 molecular simulation package (32) in combination with the GROMOS96 (43a1) force-field (33) and simple point charge model of water (34). Using bond constraints allowed for a time step of 2 fs. Cubic periodic boundary conditions were applied. The van der Waals interactions were treated with a cutoff of 9 Å. Long-range electrostatic interactions were treated by fast particle-mesh Ewald (35) with a grid spacing of 1.2 Å.
After an energy minimization and a protein position restraint run of 100 ps, the system was equilibrated at ambient conditions (1 bar and 300 K) for 10 ns using the Nosé-Hoover thermostat and the Berendsen barostat. Adjusting the volume to the average of the isobaric simulation run, all of the subsequent MD simulations were performed in the NVT ensemble at constant volume of 97.24 nm3. Unless specified otherwise, the Nosé-Hoover thermostat (36,37) ensured constant temperature.
Order parameters
Order parameters (OPs) that describe conformational changes in the WW domain include the radius of gyration of the whole protein, rgα, based on the α-carbons, as well as rgXYZ, for residues X,Y,Z only. The value rg3YW is the radius of gyration for residues Tyr-11, Tyr-19, Tyr-21, and Trp-30 including all atoms. The values rmsd and rmsdca measure the root mean-square deviation (RMSD) from the native conformation using all atoms and α-carbons, respectively. The values rmsdt1 and rmsdt2 are, respectively, the α-carbon RMSD from the native structure of turn-1 residues (13–17 TADGK), and of turn-2 residues (23–26 NRTL). Several parameters measure hydrogen bonds (H-bonds). An H-bond is formed when the donor-acceptor distance is <3.5 Å and the O-H-N angle is >150°. A backbone H-bond occurs between acceptors and donors located on the peptide backbone. An H-bond is native if it also occurs in the native state. There is a maximum of seven native H-bonds between strand β1 and β2 and three bonds between strands β2 and β3 (Fig. 1). The value nhbpp denotes the number of native backbone H-bonds in the protein. The value nbb counts the broken native backbone H-bonds, with a donor-acceptor distance >0.7 nm. Backbone H-bond donors or acceptors can also form bonds with the solvent molecules. The number of solvated native H-bonds donors/acceptors is given by nhbps. As a measure for the effective solvation of the peptide, we compute Δ = 2nhbpp – nhbps, which is positive for low solvation and negative for high solvation. The factor of 2 appears because each backbone H-bond can be replaced by two peptide-solvent H-bonds. We also measure the sum of the O-H distances of the backbone native H-bonds, Roh, as it is a good OP for the GB1 β-hairpin unfolding transitions (38).
Denoting the larger hairpin by hairpin-1 and the smaller by hairpin-2 (see Fig. 1 a), we computed several OPs not only for the whole system (no superscript notation) but for hairpin-1 (denoted by superscript 1) and hairpin-2 (denoted by superscript 2) as well. These OPs include nhbpp, nhbps, nbb, Δ, and Roh. The continuous parameters and can characterize and distinguish the native and unfolded regions. We combine both Roh parameters into a single OP ξ, normalized per H-bond and the bond distance,
where d0 = 2 Å. Note that the parameters are divided by the maximum number of H-bonds, to weigh both parameters equally. To describe the packing of the polypeptide backbone and side chains we define nα and nγ, as the number of Cα and Cγ contacts, respectively. Smoothing the contact distances with a sigmoidal function f(x) = (1 – xN)/(1 – xM), with N = 8 and M = 10, makes the discrete parameters nα and nγ continuous (31). The contact parameters are given by
where the summations are over all Cα and Cγ pairs, respectively, and r0 is an average contact distance. We use r0 = 6 Å for Cα and r0 = 5 Å for Cγ contacts. Finally, the dihedral correlation ϕcorr is defined as
with ϕi the ith backbone dihedral angle.
Replica exchange
We employ REMD (24) to explore the FE surface as well as to identify the metastable regions for TPS (39). Each REMD simulation comprises 56 replicas covering the temperature range 280–555 K. Based on short trial simulations at the extreme temperatures, we chose the temperatures for intermediate replicas such that the exchange probability would be ∼20–30%. These temperature gaps were ΔT0 = 3K for the T0 = 280K replica and ΔT55 = 10K for the T55 = 555K replica, respectively. We pick the intermediate temperatures from a quadratic function, in such a way that the temperature gap is 3K for the first two replicas and 10K for the last two replicas.
From the 300K replica we can compute any two-dimensional FE landscape from βF(s1, s2) = − ln P(s1, s2), the negative logarithm of the probability histogram P(s1, s2) to observe an arbitrary pair of OPs s1, s2. Here, β = 1/kBT is the reciprocal temperature, with kB denoting Boltzmann's constant. Two-dimensional FE landscapes have the advantage of giving insight into correlation between OPs.
Metadynamics and bias-exchange metadynamics
Metadynamics (28) adds a history-dependent biasing potential to the molecular dynamics, constructed as a sum of Gaussian hills, which discourages the system from revisiting the same regions of a priori chosen low-dimensional collective variable space. The height h and the width of w, and the deposition frequency τg−1 of these Gaussian hills, are important simulation parameters. It has been shown that in the limit of long times the biasing potential converges to the negative of the FE surface, within an accuracy determined by the parameters h, w, and τg (40). Recently, several methods have been developed to improve convergence properties of metadynamics. Min et al. (41) introduced a recursive Wang-Landau scheme that systematically reduces the height of the Gaussian for optimized sampling. Barducci et al. (42) developed well-tempered metadynamics to achieve the same goal. To improve on the efficiency of free energy computations, Zheng et al. (43) recently presented a novel method that performs a random walk in a space orthogonal to the reaction coordinate.
In this work, we employ basic, one-dimensional (i.e., biasing in a single OP) metadynamics for computing the free energy barriers after the establishment of the important transitions and approximate reaction coordinate from the TPS simulation. We deposit relatively small hills (h = 0.05 kJ/mol, w = 0.05 nm) every τg = 5 ps, to reach sufficient accuracy. These values are similar to the ones used in Piana and Laio (31) and are a good trade-off between accuracy and efficiency.
For more than three collective variables, metadynamics becomes unpractical (31). To overcome this problem, Piana and Laio developed bias-exchange metadynamics, which combines metadynamics with a replica exchange method (31). Instead of using temperature to accelerate sampling, BE-Meta performs a different metadynamics simulation in each of the replicas. Allowing for regular exchange of the biasing potential between the replicas enhances the exploration of phase space (31). An additional neutral replica, in which the system equilibrates without a bias, enables FE projection in the same way as in the REMD simulations. We use an eight replica BE-Meta simulation to explore the phase space of the WW domain and to obtain room temperature initial paths for the TPS. The first replica was set up as neutral. Six of the seven remaining replicas performed metadynamics in the order parameters , , d3YW, nα, nγ, and ϕcorr. The eighth replica applied a bias in two dimensions, and simultaneously. The height and width of the Gaussian hills was h = 0.3 kJ/mol, and w = 0.1 nm, respectively, and we deposited these hills every τg−1 = 1 ps. The exchanges of the biasing potentials were attempted every 10 ps, and the overall acceptance ratio was ∼30%.
Transition path sampling (TPS)
The transition path sampling (TPS) algorithm collects an ensemble of reactive trajectories leading from a predefined initial state to a known final region (30). Our version of the TPS algorithm, previously implemented, tested, and successfully applied to protein systems (39,44), requires stochastic dynamics. These are ensured by an Andersen thermostat coupled to the center-of-mass motion of water molecules only, and allows for a varying path length through an appropriately modified acceptance rule (45). The TPS algorithm requires a definition of initial and final states, which we infer from REMD simulations. In addition, TPS needs an initial trajectory to bootstrap the simulation. In previous work, we took high temperature unfolding pathways from REMD as initial pathways for TPS (39,44). During the path sampling initialized from this artificial trajectory, the pathways will relax to the true unbiased path ensemble. Here, BE-Meta simulations provide room temperature trajectories that undergo a transition due to the bias potential. The advantage of using initial BE-Meta trajectories over high temperature REMD pathways is that the first are already at room temperature and equilibrate faster toward the unbiased trajectories.
Results
Replica Exchange
We performed two independent 56-replica REMD simulations of the FBP28ΔCΔN WW domain for 20 ns per replica, one (REMD-fol) initialized from a native state N and one (REMD-unf) started from a completely unfolded structure U obtained from a 550 K trajectory. Fig. 2, rows 1 and 2, shows the resulting FE contour maps for different combinations of relevant OPs. The REMD-fol simulation reveals an intermediate state I2, separated from the native state by a relatively low barrier. This state I2 is characterized by ≈ 2 nm and rmsdt2 ≈ 0.2 nm, broken native H-bonds in hairpin 2 (with donor-acceptor distances larger than 7 Å on average) and a solvated turn t2. The metastability of this state is mainly due to hydrophobic interactions of the Trp-30 and Pro-33 with other aromatic and hydrophobic residues in hairpin-1. The FE plot in the – plane shows that from an unfolding perspective, unraveling of the shorter hairpin-2 is much easier than the unfolding of the large hairpin-1. This would seem to imply that during unfolding, hairpin-2 unfolds before hairpin-1. However, as we cannot draw trustworthy conclusions about the sequence of events based on REMD simulations, we will attempt to confirm this hypothesis using metadynamics and TPS in the following sections. Another conclusion from the REMD-fol simulation is that unfolding of the larger hairpin-1 is initiated at the turn, or equivalently, that the turn forms last in the process of folding, suggesting the so-called hydrophobic-collapse mechanism (46) in the formation of the large hairpin. This can be clearly seen in the (, rmsdt1) plane (Fig. 2, row 1 c), where the FE contour distinctly curves in the direction of increasing rmsdt1. Starting from the native state, rmsdt1 reaches a high value of 0.2 nm, whereas the per residue is only doubled compared to the native state. For hairpin-2, when the rmsdt2 reaches a value of 0.2 nm, the per residue is already tripled (see Fig. 2, row 1 d).
Figure 2.
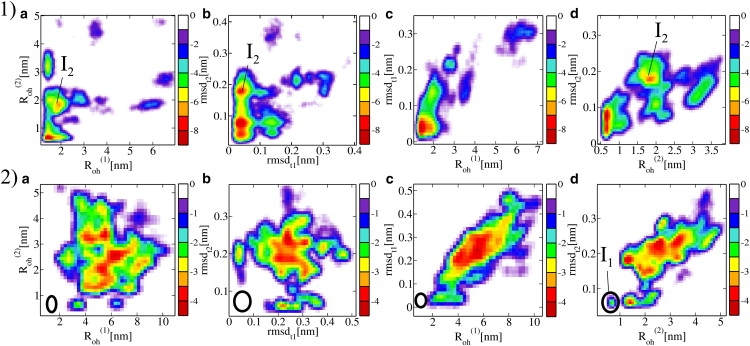
FE contour maps for the (row 1) REMD-fol and (row 2) REMD-unf simulations of FBP28ΔCΔN in four different planes (a) (, ); (b) (rmsdt1, rmsdt2); (c) (rmsdt1, ); and (d) (rmsdt2, ). All distances and RMSDs are in nm. The legend gives the free energy in kBT units. The native state area in the REMD-unf plots from row 2 is indicated by an ellipse. Note that in the (, rmsdt2) plane the native state overlaps with the intermediate I1.
In the REMD-unf simulation, several unfolded structures were sampled, and many folding events took place. The FE landscapes in Fig. 2, row 2, show the folding of hairpin-2 ( < 1, rmsdt2 < 0.6, Fig. 2, row 2 d), whereas hairpin-1 remains unfolded ( > 2, Fig. 2, row 2, a and c). We denote this partially unfolded state as I1. reaches the minimum value of ∼2.0 nm only, corresponding to a hairpin structure with some nonnative H-bonds, or even to an unfolded state with mispacked hydrophobic side chains. Note that state I1 was reached in the REMD-unf simulation, whereas in the REMD-fol simulation I2 was formed. We thus hypothesize that the rate-limiting barrier is the formation of the large hairpin, and corresponds either to the I2 → U or N → I1 transition.
Fig. 2, row 2 b, shows the FE landscape for the formation of both nativelike hairpin turns as a function of rmsdt1 and rmsdt2. Both turns form during the REMD-unf simulation, but never simultaneously. Only two of all the REMD-trajectories reach very low values of rmsdt1 and rmsdt2 simultaneously, corresponding to a state similar to I2, but with Trp-8 stacking with the upper instead of the lower hydrophobic core. This suggests that folding to the native state can occur only if, after the collapse, the hydrophobic core is packed correctly.
Our results are in agreement with those of Mu et al. (4), who performed REMD simulation on the full FBP28 WW domain. They found that in the transition state based on Φ-analysis, only turn-1 was formed. This corresponds to the formation of the large hairpin being the rate-limiting step.
The two REMD simulations have not yet converged after 20 ns per replica. We estimate that at least hundreds of nanoseconds are needed to reach convergence, as argued in Rhee and Pande (47). We have not attempted to do so, as our aim is to study the kinetic folding mechanism. Instead, we use the insight obtained from the REMD to define the native state, and the (partially) unfolded states for the TPS simulation. In addition, bootstrapping the TPS requires an initial path at room temperature, which is obtained here using BE-Meta.
Initial path by bias-exchange metadynamics
We performed a BE-Meta simulation on the WW domain for 20 ns per replica. In most of the replicas, the protein unfolded after a few nanoseconds, and subsequently sampled multiple clusters belonging to the unfolded region. No refolding was observed. In the neutral (unbiased) replica, we find configurations belonging to the native state N, intermediate state I2 (unfolded hairpin 2), intermediate state I1 (unfolded hairpin 1), and a host of completely unfolded configurations. The N → I1 barrier turns out to be very high. The large I2 population in the neutral replica agrees with the REMD-fol simulation.
From our BE-Meta simulation, we extracted three different generic unfolding scenarios (see Fig. 3). These routes cannot be considered equilibrium pathways, as metadynamics is, in principle, a nonequilibrium method due to its time-dependent biasing potential. Nonetheless, the biased pathways are interesting because two of them (Scenario 2 and 3) resemble previously observed elevated temperature (373K), unfolding pathways of the original, nontruncated, WW domain (3).
Figure 3.
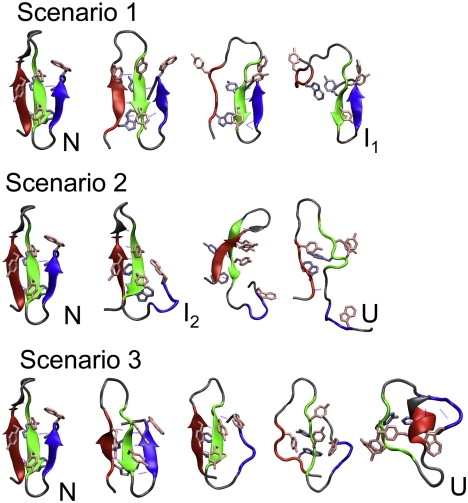
BE-Meta unfolding trajectories of the truncated FBP28 WW domain, described in the main text as unfolding Scenarios 1, 2, and 3. The protein structures are depicted as in Fig. 1.
Scenario 1
In this scenario Tyr-21, belonging to the upper hydrophobic core, splits from the rest of the aromatic residues. This move results in the decrease of interaction between the hydrophobic core and Tyr-11. The latter residue then detaches, allowing for the solvation of the large hairpin-1. The hairpin maintains its U-shape, mainly due to the presence of the lower hydrophobic core. That core eventually also breaks up, resulting in an entirely solvated hairpin-1. Hairpin-2 stays intact in this scenario.
Scenario 2
In several of the BE-Meta replicas, the WW domain unfolded by first solvating the smaller hairpin-2. During this process, the hairpin hydrophobic core remains compact, sustained by the interaction of the Trp-30 with the Tyrosine side chains. RgW3Y decreases because the core becomes more compact than in the native state, due to breaking the three H-bonds that stabilize hairpin-2. At the same time hairpin-1 remains in its native configuration. The seven H-bonds fluctuate more as hairpin-1 gains some extra twist, but stay intact. Next, the detachment of Trp-30 from the Tyrosines 19 and 21 destabilizes the protein and leads to the solvation of hairpin-2, which loses its U-shape. Subsequently, the H-bonds in hairpin-1 break and are solvated as well. A larger hydrophobic core made of both upper and lower subcores forms, but eventually dissolves.
Scenario 3
This route starts with the perturbation of the turn area of the hairpin-1. The H-bonds with Thr-8 break and the turn t1 becomes solvated, allowing for an extra twist in the large hairpin. This is followed by breaking of the H-bonds of hairpin-2. During this process, there is no detachment of residues forming the hydrophobic cores. Eventually, water solvates the larger hairpin as well. The protein remains in the overall topology of the native protein, with the hydrophobic cores practically nativelike. At the end of the trajectory, the cores reorganize, and the N-terminus forms a small α-helix.
These three unfolding routes are most likely not equilibrium pathways, but can act as initial pathways in a TPS simulation.
Transition path sampling
TPS requires a definition of both the initial native and the final unfolded state, which we derive from REMD simulations. We note that the labels initial-state and final-state are arbitrary, as the TPS pathways are microscopically reversible and describe the folding as much as the unfolding process. The REMD simulations indicate that the native state is well characterized by a small RMSD, whereas a large value of and/or is typical for states with unfolded hairpins. From the REMD results, it is not clear whether each of the hairpins (un)folds independently or a simultaneous (un)folding is preferred. To avoid trapping in long-lived intermediate states, the final state should allow for either or both hairpins being unfolded. Using a definition ξ > 3.0, which includes the combination of and , allows the pathways to switch freely between the various (un)folding routes. The initial state is defined by rmsd < 0.20 nm and ξ < 1.41. Using RMSD alone to identify a (meta)stable state can be dangerous because it is then possible to lump two kinetically very distinct states together (see, for example, (48–50)). The hydrogen-bond parameter ξ (based on ROH) ensures that only nativelike structures are included in the initial state definition.
Starting from each of the qualitatively different types of trajectories obtained from the BE-Meta simulation, we ran three independent TPS simulations with a total simulation time of 2.8 μs. The number of trial shots, accepted pathways, and acceptance ratio were 745, 268, and 0.36, respectively. The number of decorrelated pathways was 15, and the average path length was 2.6 ns. Although the three initial pathways are different, switches between pathways are allowed in TPS, and we expect the three TPS runs to converge to the same path ensemble. Paths that follow Scenario 2 (for (un)folding of small hairpin-2 via the N → I2 route, see, e.g., blue line in Fig. 4) show the breaking and solvation of the three H-bonds of hairpin-2. Detachment of Trp-30 from the rest of the upper hydrophobic core leads to formation of nonnative hydrophobic contacts. Waters may be trapped between lower and upper hydrophobic cores. The two H-bonds between Tyr-20 and Thr-29 break simultaneously when Trp-30 rearranges, pulling Thr-29 away from its H-bond partner Tyr-20. H-bonds in the vicinity of the turn persist longer, but eventually also break in the I2 state.
Figure 4.
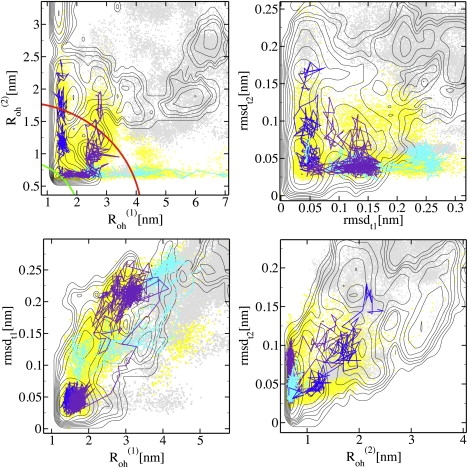
Three typical TPS pathways presented in four planes: (Top left) – ; (Top right) rmsdt1 – rmsdt2; (Bottom left) – rmsdt1; and (Bottom right) – rmsdt2. The whole TPS ensemble is plotted as yellow and the REMD-unf ensemble as gray points. The contours give the FE landscape of the REMD-fol simulation (compare to Fig. 2, row 1), and are separated by 1 kBT. In panel a, the initial and the final state boundaries are indicated with green and red lines. The blue trajectory represents a typical N → I2 transition, the cyan trajectory an N → I1 transition, and the violet depicts a switching pathway.
The TPS runs initiated with Scenario 1 and Scenario 3 pathways relax to the route represented in Fig. 4 as the violet line. After equilibration of the TPS ensemble, the contact between Trp-30 and Tyr-19 temporarily breaks in all paths, to form a more compact upper core, or to detach the Trp-30 from the core. In addition, the short hairpin-2 is always disturbed, even when in the initial trajectories this hairpin is untouched. Although initially both runs sample the unfolding of hairpin-1 only, or simultaneously unfold with hairpin-2, they eventually sample pathways in which only hairpin-2 unravels—indicated by the fact that does not cross the boundary between the folded and unfolded hairpin, estimated at ∼ ≈ 4 nm. Such a switching from one route to another is evidence for the hypothesis that the unfolding barrier for hairpin-1 is higher than for hairpin-2. Moreover, we believe that the entire path ensemble would finally relax to the N → I2 route, because the endpoint of all trajectories resembles the I2 state, except for some additional solvation around turn I. Indeed, Fig. 4, top left, shows that the pathways tend to switch to the most preferred unfolding channel, following the FE landscape. The I1 state, although not sampled by the REMD-fol ensemble, was found in REMD-unf (Fig. 2, row 2), suggesting that I1 is in fact committed to the unfolded state. The TPS simulation did not show reverse switching from I2 → N trajectories to I1 → N paths. This probably means the difference between the free energy barriers of the I2 → N and I1 → N routes is too large for spontaneous switching to occur during the path sampling.
Unfortunately, the TPS simulations do not resolve the question of the most preferred folding route. The I2 → U barrier might still be higher than the free energy barrier on the N → I1 → U path. The TPS simulation can only handle one barrier between the initial and final state at a time, and as we restrict the initial state to be the native state, we cannot expect to sample the I2 → U or the I1 → U barrier. Nevertheless, the final structures of all trajectories point toward the I2 state, as the on-pathway intermediate. In principle, we could perform TPS for all possible transitions, and compute the rate constant and free energy using transition interface sampling (44), but this is beyond the scope of this work.
The TPS results suggest that the four transitions N → I2, N → I1, I2 → U, and I1 → U, dominate the unfolding and folding mechanism of the WW-domain. For these transitions, the Roh parameters are reasonable reaction coordinates. Therefore, we will try to test the hypothesis that the (un)folding occurs via I2 by calculating free energy barriers for all four transitions with metadynamics in the following section.
Metadynamics free energies
From the equilibrated TPS trajectories, it follows that the N → I2, N → I1, I2 → U, and I1 → U transitions are important for the folding mechanism. To estimate the unfolding barriers for these transitions we performed one-dimensional metadynamics simulations, biasing the system in the order parameters, , and . The one-dimensional metadynamics simulations yield more precise unfolding barriers than the BE-Meta simulations, due to smaller hills and a lower deposition frequency. We performed six metadynamics unfolding simulations, initialized in different states and using as a biasing parameter either or (see Table 1). In addition to the four unfolding transitions N → I1, N → I2, I1 → U, and I2 → U, we perform two more simulations: a direct N → U transition and the I2 → N transition. The N → U simulation could check the barrier heights calculated for the N → I2 → U route, whereas the I2 → N simulation could confirm the relative stability of the I2 and N state. Metadynamics of the refolding from the U state and from the I1 state failed, possibly due to the lack of a correct reaction coordinate for folding the hydrophobic core. Each of the six metadynamics simulations was stopped after the transition of interest happened, and hills started to be added to the final state. The only exception was the N → I2 transition, which was run longer, to estimate the relative stability of the N and I2 states, and to compare to the I2 → N results. The estimated barrier free energies are summarized in Table 1. Final and initial structures, free energy profiles, and FE barriers are summarized in Fig. 5.
Table 1.
Summary of all metadynamics simulations
| Transition | Bias OP | Simulation time (ns) | ΔF‡[kBT] | |
|---|---|---|---|---|
| N → I2 | 27 | 11 | ||
| N → I1 | 69 | 25 | ||
| N → U | Roh | 27 | 17 | |
| I2 → U | 13 | 10 | ||
| I1 → U | 40 | 14 | ||
| I1 → N | 56 | 7 |
FE barriers ΔF‡ were measured between the top of the barrier and initial state.
Figure 5.
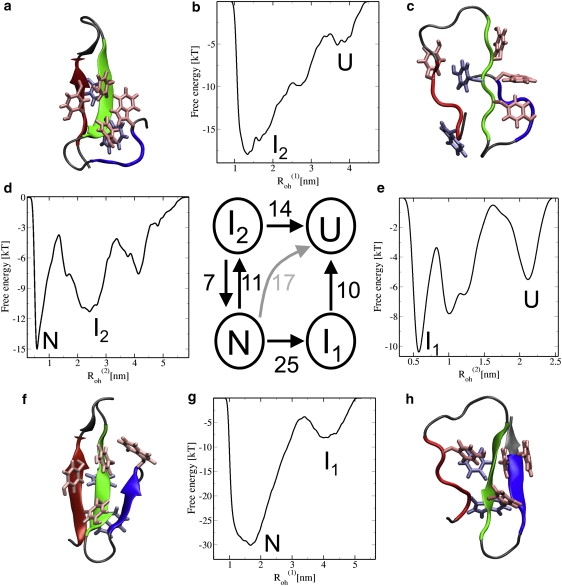
(g), (d), (e), and (b) FE barriers based on metadynamics simulations for the N → I1, N → I2, I1 →U, and I2 → U transitions, respectively. The simulations biasing in are represented as horizontal and in as vertical arrows. One metadynamics simulation was done using Roh = + , and is depicted with a bent arrow, indicating that these trajectories pass I2 without relaxing in it. The numbers indicate the approximate FE barriers in kBT associated with every transition. Protein configurations are depicted as in Fig. 1. Configurations: (a) intermediate state I2 with unfolded hairpin-2; (c) unfolded state, with a roughly native topology; (f) native state N; and (h) intermediate state I1 with unfolded hairpin-1.
The results confirm the N → I1 unfolding barrier is much higher than the N → I2 barrier, as expected, considering the number of H-bonds involved. The I2 → U and I1 → U unfolding barriers are 14 kBT and 10 kBT, respectively. The folding barrier I2 – N was estimated to be 7 kBT. In addition, the barrier height for the direct unfolding transition was found to be 17 kBT, whereas the total barrier height for the N → I2 → U path equals ΔFN–‡ = 11 − 7 + 14 = 18 kBT. The difference of 1 kBT is within the error of our metadynamics simulations, which is roughly of the order of 2 kBT (40). The I2 → N metadynamics simulation was performed to test the previously estimated relative free energy of the I2 and N states, and was indeed confirmed to be 4 kBT (plot not shown in Fig. 5). The metadynamics simulations thus indicate that the I2 → N barrier is lower than the I2 → U barrier. This also agrees with the finding that I2 is accessible in the REMD-fol simulations.
From the metadynamics simulation, we can draw some conclusions concerning the rate-limiting barriers for the (un)folding process of the WW domain. The difference of the barrier heights between the N → I2 → U and N → I1 → U routes is ∼7 kBT, making the latter transition improbable with respect to the first, during unfolding. Hence, the proposed hypothesis is confirmed: The unfolding of the large hairpin-1 is the rate-limiting unfolding transition. State I1 belongs to the unfolded basin whereas I2 is already committed to the native state. Moreover, as I2 is probably more populated than I1, both unfolding and folding are more likely to occur via the intermediate I2.
Discussion and Conclusions
In this study, we have made use of several different methods. None of these techniques separately was able to completely reveal the mechanism of the WW domain folding and unfolding. Nevertheless, all these methods combined yield an interesting picture. The simulations initiated in the folded state, including REMD-fol and BE-meta-fol, indicate the existence of a close-to-native intermediate state I2, characterized by a disrupted hairpin-2 and nonnative side-chain interactions of Trp-30. The folding barrier between this intermediate and the native state was estimated to be 7 kBT. The BE-Meta unfolding scenarios 2 and 3 resemble previously observed elevated temperature (373K) unfolding pathways of the original, nontruncated, WW domain (3). The TPS simulations were able to sample the conformational transitions and eventually relaxed to the same route (resembling Scenario 2), choosing the transition toward the I2 state, over crossing the N → I1 barrier. Metadynamics confirmed that the direct unfolding of hairpin-1 without disruption of hairpin-2 is very improbable (ΔF‡ ≈ 25 kBT), and that unfolding of hairpin-2 lowers the barrier for unfolding of hairpin-1 to ∼14 kBT, probably due to the accompanying disruption of the upper hydrophobic core. The barrier for the sequential unfolding of both hairpins, first the small and then the large one, without relaxing into the state I2 was estimated to be ∼17 kBT. The most likely path is thus via I2 with a rate-limiting barrier of 17 kBT (at ∼42 kJ/mol). Estimating the error committed in metadynamics of ∼2 kBT (40), this value in agreement with the experimental unfolding barrier, which is ∼15 kBT (2). Thus, the metadynamics simulations indicate I2 as the on-pathway intermediate in the process of folding of the WW domain. To complete the description, future research might aim at computing the barriers, in particular the I1 → N and U → I2 barriers.
Previous work using high temperature simulations (3) already suggested that the WW domain can fold via two routes. Our room temperature simulations confirm this. In addition, we have shown that one of the routes, N → I2 → U, is the most probable folding path at ambient conditions. Our prediction that the formation of hairpin 1 is the rate-limiting step, agrees with previous REMD simulation by Mu et al. (4), who predict the turn-1 formation as the transition state. Moreover, this finding is in agreement with the experimental Φ-value analysis results (3). In addition, our simulations are qualitatively consistent with the Gō-model simulation results by Luo et al. (16) on Pin1, which showed initial folding of the larger hairpin as well as the presence of heterogeneity in the mechanism. Our findings are also consistent with the simulation results by Ensign and Pande (19) on the Fip35 in implicit solvent, in which it was found that the mechanism is heterogeneous, but that it is more likely that the larger hairpin is formed first. Our TPS simulations allow a more detailed discussion on the hydrophobic side-chain interactions during the (un)folding pathways.
The existence of the I2 state was proposed to be the source of the bimodality observed in experiments for the FBP28 WW domain (10). We think the truncation of the termini may have an effect on state I2, making it less stable, and thus less visible on the experimental timescales. This could be the reason of the experimentally observed two-state behavior of truncated FBP28 (1,2).
Periole et al. (5) recently performed an extensive simulation on the FBP28 WW domain using all-atom as well as coarse-grained simulations. Although they did not observe spontaneous folding to the native state, their general conclusion is similar to ours here: to gain insight into complex folding problems, one requires a combination of multiple techniques. In particular, we find that the combination of TPS with replica exchange and metadynamics (or other efficient FE methods) is fruitful. REMD can explore free energy landscapes, but is not effective in establishing barriers or mechanisms. The BE-metadynamics cannot only be used for exploring phase space faster, but also for generating tentative transition routes at room temperature, which can act as initial trajectories for path sampling. Subsequently, TPS finds the equilibrium path ensemble for rare transitions, yielding correct unbiased mechanisms and reaction coordinates. Finally, metadynamics (or other potential biasing methods) can efficiently estimate the transitions barrier heights for a particular reaction coordinate. This article shows that a single rare event method is not sufficient to elucidate the (un)folding transitions of a protein with several metastable states. Instead, a combination of several sampling methods provides a more complete picture.
Acknowledgments
We thank A. Laio for kindly providing us with the BE-Meta GROMACS code. We also acknowledge X. Periole for helpful discussions and suggesting the use of the truncated version of the WW domain.
The work was financially supported by the “Nederlandse Organisatie voor Wetenscappelijk Onderzoek (NWO)” and the “Stichting Nationale Computerfaciliteiten”.
References
- 1.Ferguson N., Johnson C.M., Fersht A. Ultrafast folding of WW domains without structured aromatic clusters in the denatured state. Proc. Natl. Acad. Sci. USA. 2001;98:13002–13007. doi: 10.1073/pnas.221467198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nguyen H., Jager M., Kelly J.W. Tuning the free-energy landscape of a WW domain by temperature, mutation, and truncation. Proc. Natl. Acad. Sci. USA. 2003;100:3948–3953. doi: 10.1073/pnas.0538054100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petrovich M., Jonsson A.L., Fersht A.R. Phi-analysis at the experimental limits: mechanism of β-hairpin formation. J. Mol. Biol. 2006;360:865–881. doi: 10.1016/j.jmb.2006.05.050. [DOI] [PubMed] [Google Scholar]
- 4.Mu Y.G., Nordenskiöld L., Tam J.P. Folding, misfolding, and amyloid protofibril formation of WW domain FBP28. Biophys. J. 2006;90:3983–3992. doi: 10.1529/biophysj.105.076406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Periole X., Allen L.R., Paci E. Probing the free energy landscape of the FBP28WW domain using multiple techniques. J. Comput. Chem. 2009;30:1059–1068. doi: 10.1002/jcc.21128. [DOI] [PubMed] [Google Scholar]
- 6.Sudol M. Structure and function of the WW domain. Prog. Biophys. Mol. Biol. 1996;65:113–132. doi: 10.1016/s0079-6107(96)00008-9. [DOI] [PubMed] [Google Scholar]
- 7.Huang K., Johnson K.D., Bresnick E.H. A HECT domain ubiquitin ligase closely related to the mammalian protein WWP1 is essential for Caenorhabditis elegans embryogenesis. Gene. 2000;252:137–145. doi: 10.1016/s0378-1119(00)00216-x. [DOI] [PubMed] [Google Scholar]
- 8.Macias M.J., Gervais V., Oschkinat H. Structural analysis of WW domains and design of a WW prototype. Nat. Struct. Biol. 2000;7:375–379. doi: 10.1038/75144. [DOI] [PubMed] [Google Scholar]
- 9.Kanelis V., Rotin D., Forman-Kay J.D. Solution structure of a Nedd4 WW domain-ENaC peptide complex. Nat. Struct. Biol. 2001;8:407–412. doi: 10.1038/87562. [DOI] [PubMed] [Google Scholar]
- 10.Hu H., Columbus J., Herrero J.J. A map of WW domain family interactions. Proteomics. 2004;4:643–655. doi: 10.1002/pmic.200300632. [DOI] [PubMed] [Google Scholar]
- 11.Jäger M., Nguyen H., Gruebele M. The folding mechanism of a β-sheet: the WW domain. J. Mol. Biol. 2001;311:373–393. doi: 10.1006/jmbi.2001.4873. [DOI] [PubMed] [Google Scholar]
- 12.Koepf E.K., Petrassi H.M., Kelly J.W. WW: an isolated three-stranded antiparallel β-sheet domain that unfolds and refolds reversibly; evidence for a structured hydrophobic cluster in urea and GdnHCl and a disordered thermal unfolded state. Protein Sci. 1999;8:841–853. doi: 10.1110/ps.8.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crane J.C., Koepf E.K., Gruebele M. Mapping the transition state of the WW domain β-sheet. J. Mol. Biol. 2000;298:283–292. doi: 10.1006/jmbi.2000.3665. [DOI] [PubMed] [Google Scholar]
- 14.Ferguson N., Pires J.R., Fersht A. Using flexible loop mimetics to extend ϕ-value analysis to secondary structure interactions. Proc. Natl. Acad. Sci. USA. 2001;98:13008–13013. doi: 10.1073/pnas.221467398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ibragimova G.T., Wade R.C. Stability of the β-sheet of the WW domain: a molecular dynamics simulation study. Biophys. J. 1999;77:2191–2198. doi: 10.1016/S0006-3495(99)77059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo Z., Ding J., Zhou Y. Temperature-dependent folding pathways of Pin1 WW domain: an all-atom molecular dynamics simulation of a Gō model. Biophys. J. 2007;93:2152–2161. doi: 10.1529/biophysj.106.102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freddolino P.L., Liu F., Schulten K. Ten-microsecond molecular dynamics simulation of a fast-folding WW domain. Biophys. J. 2008;94:L75–L77. doi: 10.1529/biophysj.108.131565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freddolino P.L., Park S., Schulten K. Force field bias in protein folding simulations. Biophys. J. 2009;96:3772–3780. doi: 10.1016/j.bpj.2009.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ensign D.L., Pande V.S. The Fip35 WW domain folds with structural and mechanistic heterogeneity in molecular dynamics simulations. Biophys. J. 2009;96:L53–L55. doi: 10.1016/j.bpj.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan D.C., Bedford M.T., Leder P. Formin binding proteins bear WWP/WW domains that bind proline-rich peptides and functionally resemble SH3 domains. EMBO J. 1996;15:1045–1054. [PMC free article] [PubMed] [Google Scholar]
- 21.Sudol M., Hunter T. NeW wrinkles for an old domain. Cell. 2000;103:1001–1004. doi: 10.1016/s0092-8674(00)00203-8. [DOI] [PubMed] [Google Scholar]
- 22.Ferguson N., Berriman J., Fersht A.R. Rapid amyloid fiber formation from the fast-folding WW domain FBP28. Proc. Natl. Acad. Sci. USA. 2003;100:9814–9819. doi: 10.1073/pnas.1333907100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karanicolas J., Brooks C.L. Integrating folding kinetics and protein function: biphasic kinetics and dual binding specificity in a WW domain. Proc. Natl. Acad. Sci. USA. 2004;101:3432–3437. doi: 10.1073/pnas.0304825101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugita Y., Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999;314:141–151. [Google Scholar]
- 25.Bennett, C. H. 1977. Algorithms for chemical computations. In ACS Symposium Series No. 46. R. Christofferson, editor. American Chemical Society, Washington, DC.
- 26.Chandler D. Statistical mechanics of isomerization dynamics in liquids and the transition state. J. Chem. Phys. 1978;68:2959–2970. [Google Scholar]
- 27.Torrie G.M., Valleau J.P. Monte-Carlo free energy estimates using non-Boltzmann sampling. Application to the subcritical Lennard-Jones fluid. Chem. Phys. Lett. 1974;28:578–581. [Google Scholar]
- 28.Laio A., Parrinello M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA. 2002;99:12562–12566. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dellago C., Bolhuis P.G., Chandler D. Transition path sampling and the calculation of rate constants. J. Chem. Phys. 1998;108:1964–1977. [Google Scholar]
- 30.Bolhuis P.G., Chandler D., Geissler P.L. Transition path sampling: throwing ropes over rough mountain passes, in the dark. Annu. Rev. Phys. Chem. 2002;53:291–318. doi: 10.1146/annurev.physchem.53.082301.113146. [DOI] [PubMed] [Google Scholar]
- 31.Piana S., Laio A. A bias-exchange approach to protein folding. J. Phys. Chem. B. 2007;111:4553–4559. doi: 10.1021/jp067873l. [DOI] [PubMed] [Google Scholar]
- 32.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 33.Scott W.R.P., Hünenberger P.H., van Gunsteren W.F. The GROMOS biomolecular simulation program package. J. Phys. Chem. 1999;103:3596–3607. [Google Scholar]
- 34.Berendsen H.J.C., Postma J.P.M., Hermans J. Interaction models for water in relation to protein hydration. In: Pullman B., editor. Intermolecular Forces. Reidel, Dordrecht; The Netherlands: 1981. [Google Scholar]
- 35.Essman U., Perera L., Pedersen L. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8592. [Google Scholar]
- 36.Nosé S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984;52:255–268. [Google Scholar]
- 37.Hoover W.G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 38.Bolhuis P.G. Kinetic pathways of β-hairpin (un)folding in explicit solvent. Biophys. J. 2005;88:50–61. doi: 10.1529/biophysj.104.048744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Juraszek J., Bolhuis P.G. Sampling the multiple folding mechanisms of Trp-cage in explicit solvent. Proc. Natl. Acad. Sci. USA. 2006;103:15859–15864. doi: 10.1073/pnas.0606692103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laio A., Rodriguez-Fortea A., Parrinello M. Assessing the accuracy of metadynamics. J. Phys. Chem. B. 2005;109:6714–6721. doi: 10.1021/jp045424k. [DOI] [PubMed] [Google Scholar]
- 41.Min D., Liu Y., Yang W. On the convergence improvement in the metadynamics simulations: a Wang-Landau recursion approach. J. Chem. Phys. 2007;126:194104. doi: 10.1063/1.2731769. [DOI] [PubMed] [Google Scholar]
- 42.Barducci A., Bussi G., Parrinello M. Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys. Rev. Lett. 2008;100:020603. doi: 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- 43.Zheng L., Chen M., Yang W. Random walk in orthogonal space to achieve efficient free-energy simulation of complex systems. Proc. Natl. Acad. Sci. USA. 2008;105:20227–20232. doi: 10.1073/pnas.0810631106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Juraszek J., Bolhuis P.G. Rate constant and reaction coordinate of Trp-cage folding in explicit water. Biophys. J. 2008;95:4246–4257. doi: 10.1529/biophysj.108.136267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Erp T.S., Moroni D., Bolhuis P.G. A novel path sampling method for the calculation of rate constants. J. Chem. Phys. 2003;118:7762–7774. [Google Scholar]
- 46.Muñoz V., Ghirlando R., Eaton W.A. Folding and aggregation kinetics of a β-hairpin. Biochemistry. 2006;45:7023–7035. doi: 10.1021/bi052556a. [DOI] [PubMed] [Google Scholar]
- 47.Rhee Y.M., Pande V.S. Multiplexed-replica exchange molecular dynamics method for protein folding simulation. Biophys. J. 2003;84:775–786. doi: 10.1016/S0006-3495(03)74897-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swope W., Pitera J., Suits F. Describing protein folding kinetics by molecular dynamics simulations. 1. Theory. J. Phys. Chem. B. 2004;108:6571. [Google Scholar]
- 49.Swope W.C., Pitera J.W., Zhou R. Describing protein folding kinetics by molecular dynamics simulations. 2. Example applications to alanine dipeptide and α-hairpin peptide. J. Phys. Chem. B. 2004;108:6582. [Google Scholar]
- 50.Chodera J.D., Singhal N., Swope W.C. Automatic discovery of metastable states for the construction of Markov models of macromolecular conformational dynamics. J. Chem. Phys. 2007;126:155101. doi: 10.1063/1.2714538. [DOI] [PubMed] [Google Scholar]