

Abstract
RNA hairpin loops are one of the most abundant secondary structure elements and participate in RNA folding and protein-RNA recognition. To characterize the free energy surface of RNA hairpin folding at an atomic level, we calculated the potential of mean force (PMF) as a function of the end-to-end distance, by using umbrella sampling simulations in explicit solvent. Two RNA hairpins containing tetraloop cUUCGg and cUUUUg are studied with AMBER ff99 and CHARMM27 force fields. Experimentally, the UUCG hairpin is known to be significantly more stable than UUUU. In this study, the calculations using AMBER force field give a qualitatively correct description for the folding of two RNA hairpins, as the calculated PMF confirms the global stability of the folded structures and the resulting relative folding free energy is in quantitative agreement with the experimental result. The hairpin stabilities are also correctly differentiated by the more rapid molecular mechanics-Poisson Boltzmann-surface area approach, but the relative free energy estimated from this method is overestimated. The free energy profile shows that the native state basin and the unfolded state plateau are separated by a wide shoulder region, which samples a variety of native-like structures with frayed terminal basepair. The calculated PMF lacks major barriers that are expected near the transition regions, and this is attributed to the limitation of the 1-D reaction coordinate. The PMF results are compared with other studies of small RNA hairpins using kinetics method and coarse grained models. The two RNA hairpins described by CHARMM27 are significantly more deformable than those represented by AMBER. Compared with the AMBER results, the CHARMM27 calculated ΔGfold for the UUUU tetraloop is in better agreement with the experimental results. However, the CHARMM27 calculation does not confirm the global stability of the experimental UUCG structure; instead, the extended conformations are predicted to be thermodynamically stable in solution. This finding is further supported by separate unrestrained CHARMM27 simulations, in which the UUCG hairpin unfolds spontaneously within 10 ns. The instability of the UUCG hairpin originates from the loop region, and propagates to the stem. The results of this study provide a molecular picture of RNA hairpin unfolding and reveal problems in the force field descriptions for the conformational energy of certain RNA hairpin.
Introduction
RNA folding is believed to proceed in a hierarchical manner, as the initial rapid formation of the secondary structures is followed by slow development of tertiary structures (1). The latter involves the assembly and limited structural rearrangements of preformed secondary structure elements. The relative stabilities and the underlying energy surfaces of the secondary structures are of fundamental importance to the thermodynamics and kinetics of RNA folding (2).
Hairpin loops are a common secondary structural motif in RNA. It consists of several loop-forming nucleotides, flanked by a Watson-Crick basepaired helical stem. Hairpin loop has been suggested to act as initiation points in RNA folding (3). It is also found at recognition sites in the protein-RNA and RNA tertiary interactions (4). The stability and flexibility of RNA hairpin have been proposed as determining factors of its function. For example, the GNRA tetraloop (N: A, C, G or U; R: A or G) occurs more frequently in tertiary recognition sites than the UUCG tetraloop. One explanation for this is that the UUCG tetraloop is more rigid than the GNRA loop, and therefore it is less tolerant of the conformational changes important for binding (4,5).
Because of its importance in RNA structure and function, hairpin loop has been the subject of numerous experimental and theoretical studies aimed at understanding the atomic interactions involved in RNA folding. With a reasonable computational cost, all-atom molecular mechanics force fields provide currently the most detailed description of nucleic acids in ionic solutions (6–8). Computational studies using molecular dynamics (MD) simulation give important insights into the possible hairpin folding pathway (9–11), the effect of base substitutions (12) and the influence of loop-closing basepair on hairpin stability (13), and the role of water in hairpin folding (11). The results of these earlier theoretical investigations were summarized in Deng and Cieplak (14). In a recent study using replica exchange molecular dynamics in explicit solvent, Garcia and Paschek (15) observed the reversible folding of an 8-nt RNA hairpin UUCG starting from extended conformations. The authors also obtained the pressure temperature (P-T) free energy diagram for the hairpin loop formation and found that the RNA hairpin folding is destabilized by the increase of hydrostatic pressure. However, the calculated folding free energy of 0.65 kcal/mol at 310 K is different from the corresponding experimental value of ≈−1.0 kcal/mol. Villa et al. (16) studied the folding mechanisms of two 14-nt hairpin loops uCACGg and cUUCGg using replica exchange molecular dynamics. Their simulations confirmed the structural similarity of the two hairpins, and reproduced the relative melting temperature, but the absolute values of Tm are found to be 20% too high. The study showed that the folding of the uCACGg hairpin is more cooperative than that of the cUUCGg hairpin.
In this study, we are interested in characterizing the free energy profile of RNA hairpin and determining the free energy of folding. Theoretical calculation of free energy is central to our understanding of the conformational transitions between unfolded and compact states in biomolecules. The free energy of folding may be estimated using the molecular mechanics-Poisson Boltzmann-surface area (MM-PB/SA) method (17). This approach has been adopted in studying the thermal equilibrium of RNA secondary structures such as duplex, hairpin loop, and single strand (14), and the calculation correctly described the temperature dependence of the conformational equilibrium. Whereas MM-PB/SA allows rapid estimation of the fee energy, the conformational entropy associated with large structural changes is not adequately taken into account by the method. Moreover, the small folding free energy is obtained from the difference between two end point free energies, which are of large magnitudes. In this study, we compare the folding free energies calculated using MM-PB/SA and those obtained using the potential of mean force (PMF). The PMF is calculated by umbrella sampling simulations (18) in explicit solvent, with the end-to-end distance chosen as the order parameter. In contrast to the MM-PB/SA approach, here the conformational entropy is not estimated separately. Also, in the pathway approach, the free energy of folding (of the order of kcal/mol) is not calculated from the difference between two large numbers.
In recent years, the kinetics and thermodynamics of RNA hairpin folding have been the focus of a number of statistical mechanical calculations using coarse-grained models (19–21). In a joint experimental and theoretical study of several 8-nt RNA hairpins (21), Ma et al. showed that the thermodynamics and the temperature-dependent kinetics data can be explained by using a four-state model, derived from a sequence-specific 2-D lattice representation of RNA hairpin. Even for the short 8-nt RNA, the free energy landscape described by the four-state model is rugged, with barrier heights ranging from 2 to 9 kBT. It is of interest to compare our atomistic simulation results with the statistical mechanical calculations using low-resolution representations of RNA.
To study how free energy surface depends on hairpin sequence and thermal stability, we calculate the PMF of two RNA tetraloop sequences cUUCGg and cUUUUg. The UUCG hairpin is one of the best characterized RNA tetraloop and is considerably more stable than UUUU (2). We analyze the results from umbrella sampling to estimate folding free energy, to identify major transitions during hairpin folding, and compare the results with other theoretical and experimental studies of RNA hairpin. The folding free energies are also calculated using the more rapid MM-PB/SA approach, and the free energy contributions breakdown in this method gives additional insights into the energetics of hairpin folding. To examine the influence of different force fields on the calculated free energies, we compare the results given by the two widely used nucleic acids force fields AMBER ff99 (22) and CHARMM27 (23).
Materials and Methods
RNA hairpins
The conformational equilibrium of two RNA tetraloops 5′-GCC(UUCG)GGC-3′ and 5′-CGC(UUUU)GCG-3′ are studied in this work (Fig. 1). The native structure of UUCG was taken from the hairpin segment (residues 6–15) in the crystal structure PDB 1F7Y. The coordinates of the UUUU hairpin were built by using the hairpin segment of the RNA structure PDB 1MME as the template. To compare with the experimental results at the same temperature, all the simulations were carried out at 37°C. The melting temperature for a similar 10-nt UUCG tetraloop is 69.0°C (13), whereas the Tm for a 12-nt UUUU loop is between 59.2°C and 60.4°C (2).
Figure 1.
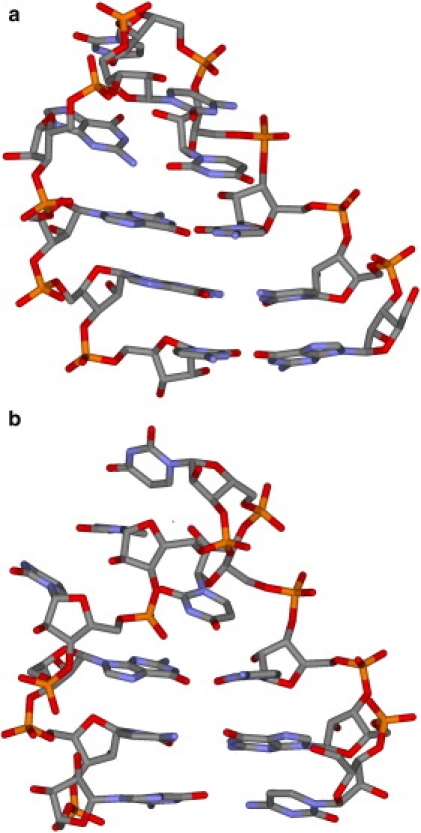
Native structures of (a) 5′-GCC(UUCG)GGC-3′ and (b) 5′-CGC(UUUU)GCG-3′ hairpin loops.
PMF calculation
Choosing a suitable reaction coordinate is the first step in a free energy profile study. Experimentally, solution NMR studies of RNA hairpins (24) indicate that RNA tetraloops adopt hairpin conformation under native conditions and become single stranded under denatured conditions. The end-to-end distance therefore provides a measure for the progress of hairpin folding and is used as the order parameter in the umbrella sampling calculation of PMF. Details of the umbrella sampling and MD simulation protocol are provided in the Supporting Material.
To ascertain the statistical significance of the calculated PMF, three independent umbrella sampling simulations were carried out for each of the UUCG and UUUU hairpins using the AMBER force field. Different initial velocities were used in these simulations. A total of ≈2.3 μs explicit solvent simulations have been carried out for calculating the PMF of two hairpins using two force fields.
Results and Discussions
To investigate the folding energetics of small RNA hairpins, we computed the PMF of end-to-end distance for the RNA hairpins UUCG and UUUU, using AMBER ff99 and CHARMM27 force fields. In the following sections, we first discuss the results obtained using AMBER force field. We then analyze the result given by CHARMM27 force field.
Before moving to the results section, here we discuss the caveats of the calculations. First, the umbrella sampling simulations are started from the folded structures; hence there may be some bias toward the native state in the calculated PMF. Second, the sampling may be insufficient for the characterization of free energy surface. Although the use of umbrella potentials in multiple windows facilitates uphill movement in the free energy surface along the reaction coordinate, the sampling in other degrees of freedom remains hindered by energy or entropy barriers in those directions. These limitations in the umbrella sampling will affect the accuracy of the PMF and may have contributed to the overestimation in the absolute folding free energy as discussed below.
Free energies calculated using AMBER ff99
Figs. 2 and 3 shows the PMF and the representative snapshots along the reaction coordinate obtained with AMBER ff99. To examine the statistical significance of the results, three independent sets of PMF were computed with different velocity distributions for both UUCG and UUUU. The variations between different runs are ≤2.5 kcal/mol for UUCG and ≤1.2 kcal/mol for UUUU. Although the deviations between the different sets of PMF are substantial, especially in the case of UUCG, the main features of the PMF are preserved in the different simulations. As discussed below, these features include the location and depth of the native minimum, the lack of barriers in the shoulder region, the heights of the unfolded state plateau, and the relative stability of the two hairpins.
Figure 2.
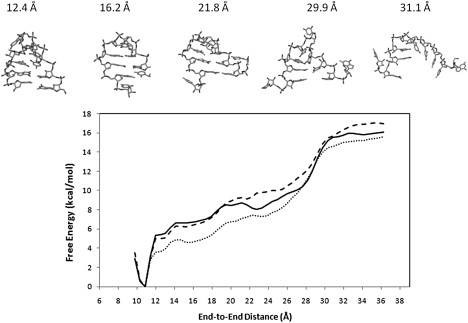
Potential of mean force (PMF) as a function of the end-to-end distance for UUCG, obtained with different initial velocities using AMBER ff99. Solid: set 1; dotted: set 2; dashed: set 3. The structures shown along with the PMF are snapshots of structures sampled by the umbrella sampling simulation. Note that because the hairpin becomes more disordered at large end-to-end distance, a significantly larger ensemble was sampled than is suggested by these snapshots at large r. The sampled conformations are better represented by these snapshots at short end-to-end extensions.
Figure 3.
PMF of UUUU obtained with different initial velocities using AMBER ff99. Solid: set 1; dotted: set 2; dashed: set 3. The snapshots of representative structures sampled by the umbrella sampling simulation are also displayed.
The free energy profiles of UUCG (Fig. 2) shows a dominant free energy minimum centered at 10.8 Å, a shoulder region between 12.4 and 27.6 Å, and a plateau region beyond 30.3 Å. The nature of the three regions is identified by examining the conformations sampled along the chosen reaction coordinate. Those three regions are discussed in more detail:
-
1.
The global free energy minimum at 10.8 Å is associated with the fully folded conformations. Thus, the overall thermodynamic stability of the experimental structure is predicted by the calculated PMF. The native energy well ends at r ≈ 12.4 Å, where the terminal G-C basepair frays. Based on the PMF profiles, the free energy cost of breaking the terminal basepair is estimated to be between 3.7 kcal/mol to 5.3 kcal/mol. To verify that global stability of the folded structure is not caused by the umbrella biasing potential, we carried out an unrestrained simulation with AMBER ff99 for 100 ns starting from the crystal structure. In this simulation, the average RMS deviation from the initial structure is ≈ 1.46 Å (Fig. S9, Fig. S10, and Fig. S12). The helical stem is stable throughout the trajectory and the key structural features in the loop region (i.e., U5 and C6 have C2′-endo ribose puckering; G7 in syn conformation) are reproduced. Thus, the result from this extended simulation supports the use of AMBER ff99 in modeling RNA hairpin.
-
2.
The shoulder region spans from 12.4 to 27.6 Å in the end-to-end distance space. In this region the RNA molecule adopts various native-like structures, in which the terminal stem basepair is frayed, but the UUCG loop and the other two stem basepairs remain largely intact. Although the basepairing between the two terminal nucleotides is broken, their bases continue to interact with the rest of the hairpin unit through stacking and hydrogen bonding (see Fig. 2 and Fig. S1 for the snapshots of the sampled structures). These nonbonded interactions are weakened and eventually broken by the progressive increase of the end-to-end distance. The loss of these interactions contributes to the gradual energy increase within the shoulder region, which is partially offset by the conformational entropy gains due to the liberation of the two end nucleotides from stacking and hydrogen bonding. Another important factor is the solvation free energy change, which contains two opposite contributions: the nonpolar component favors compact conformations due to the hydrophobicity of the nucleotide bases, whereas the electrostatic component prefers open conformations that are better solvated by the polar solvent. The net effect of the above contributions is a 5.2 kcal/mol increase of free energy in the shoulder region. As seen from Fig. 2, none of the three free energy profiles contain major barriers. The height of the small barrier near r = 22.3 Å in the PMF “set 1” is of the order of 1 kBT, which can be easily surmounted by thermal fluctuation. This points to the lack of kinetic traps in the native like shoulder region.
-
3.
Further extension in the end-to-end distance beyond 27.6 Å induces the unfolding of the remaining hairpin structure, which includes the middle and closing stem basepairs and the UUCG loop. The fraying of these structures results from a small increase (∼3 Å) in the chain extension, causing a relatively steep rise in the free energy (≥ 4.0 kcal/mol). The reverse process is strongly downhill in free energy, indicating that the formation of the tetraloop and its nearest two stem basepairs is cooperative. Intermediate states such as hairpin loop closed by a single basepair are therefore unstable. This result is in agreement with the experimental observation that stable hairpin formation requires at least two basepairs in the stem (25). Beyond the narrow transition region (27.6 Å < r < 30.6 Å), the RNA molecule exists predominantly in the single stranded form, where all the native interactions are lost.
The PMF and the sampled structures of the UUUU hairpin are shown in Fig. 3 (see also Fig. S2). To facilitate comparisons with the UUCG result shown in Fig. 2, the same vertical scale is used in Fig. 3 for plotting the PMF. Broadly speaking, the free energy profiles also contain three regions: a native state basin (r < 11.6 Å), a shoulder region (11.6 Å < r < 27.8 Å) and an unfolded region (r > 30 Å). On the other hand, the boundary between the native basin and the shoulder region is not defined as clearly as in UUCG's case (see Figs. 2 and 3). As with UUCG, the folded structure of UUUU is also found at the global free energy minimum (r ≈ 10.5 Å). A second free energy minimum was found at 8.8 Å in the PMF “set 3”. Because this second minimum is located close to the global free energy minimum at 10.5 Å and because it was only observed in one data set, we speculate that it might eventually merge with the global minimum with a longer simulation time. Within the intermediate region, the conformational fluctuations are dominated by the large movement of the two terminal nucleotides, whereas the rest of the hairpin structure experiences limited structural changes. The transition between the unfolded states and the native-like conformations occurs cooperatively within a narrow range between 27.8 Å and 30.1 Å. The free energy profiles of the UUUU hairpin are essentially barrierless.
The PMF results (Figs. 2 and 3) as a function of the end-to-end distance are comparable to a hairpin unfolding pathway in which the unfolding is started at the terminal basepair and then propagates to the loop region. Because different pathways exist and compete with each other, the PMF results do not represent the full picture of reversible unfolding and folding. In general, the unfolding pathway obtained from umbrella sampling will be dependent on the choice of reaction coordinate. Because the hairpin was pulled at the two ends in our simulation, the umbrella sampling is, to some extent, related to the single molecule end-to-end pulling experiments at slow pulling rate. Therefore the results (Figs. 2 and 3) may be interpreted as a force extension induced unfolding that proceeds in two unit steps, i.e., the breaking of the terminal basepair, followed by the unfolding of the loop and its two nearest basepairs. The main differences between the PMF of the two hairpins are: 1), the free energy cost of deformation in UUCG is higher than that in UUUU; and 2), the intermediate region of UUCG is better defined than that of UUUU, as the transitions in the former are sharper than those in the latter. These observations are consistent with the fact that the UUCG hairpin is more stable than the UUUU hairpin (2).
Next we compare the calculated PMF with related studies of RNA hairpin folding. Ma et al. (21) studied the kinetics of small RNA hairpins using temperature jump experiments. They found that the folding kinetics are sensitive to the temperature as well as the loop and stem sequence. At temperatures below Tm, a UUCG hairpin shows double-exponential folding kinetics, whereas a UUUU hairpin shows single exponential relaxation. These experimental observations are to some extent compatible with the calculated PMF in this study, which are also determined at T < Tm. Whereas the PMF of UUCG resembles that of a three-state system (Fig. 2), containing the native basin, the intermediate shoulder region and the unfolded ensemble, the PMF of UUUU is intermediate between a two-state and a three-state model, as the boundary between the intermediate states and the native minimum is less clear-cut. In our calculations (Figs. 2 and 3), the hairpin folding are essentially downhill in free energy, as no major free energy barriers are apparent in the transition regions. The reason for the smooth free energy profiles may have to do with the nature of the reaction coordinate: with a 1-D reaction coordinate, different conformations having the same end-to-end distance are combined together and appear at the same point on the reaction coordinate; hence the roughness of the conformational free energy surface may be hidden. In principle, the use of higher dimensional reaction coordinates could provide better resolution to show the energy barriers in the transition regions, but the computing cost associated with the multidimensional umbrella sampling can be prohibitive. Related to these observations, we note that in a recent replica exchange MD on a 14-nt UUCG hairpin (15), free energy barriers were indeed observed in a projected 2-D reaction coordinates surface (only at temperatures T ≥ 400 K).
In the study of Ma et al. (21), the authors found that the temperature-dependent kinetics data can be explained by a 2-D lattice model. The distinct microstates in the 2-D lattice model are further simplified to generate a four-state model that comprises a native state (denoted as N), a state having a frayed terminal stem basepair and a native-like loop (E), a unfolded state (U) and a state containing a native stem and a nonnative loop (state S) (Fig. S8). Comparing the four-state model with our atomistic simulation results, we note that the state N, E, and U correspond approximately to the native basin, the intermediate shoulder and the unfolded state plateau shown in Fig. 2. However, structures that resemble the state S is not observed in our umbrella sampling simulations. The reason for this could be the following: our simulations are carried out at low temperature, which favors compact structures with low energy and low entropy. Under such conditions the state S is unstable because it contains a disordered loop.
Using the information from the PMF, we now estimate the free energy of hairpin formation by assuming the two-state model. The calculated folding free energy depends on how the reaction coordinate space is partitioned between the folded and unfolded states. We use the mid-point of the transition between the shoulder and plateau region to define folded and unfolded states (Figs. 2 and 3). This location coincides with the disruption of the central stem basepair. There is an experimental basis to this criterion for folded and unfolded states because in both ultraviolet absorbance and solution NMR, the signature for the melting transition arises directly from the formation/disruption of H-bonding in basepairing. It therefore allows a meaningful comparison with experimental free energy to assess the quality of the force field and extent of sampling. The ΔGfold is estimated as
| (1) |
where K is the equilibrium constant and p(ri) is the probability distribution of the end-to-end distance ri. The results of the calculation are summarized in Table 1. The free energies calculated with AMBER ff99 are −13.5 ± 2.0 kcal/mol (UUCG) and −10.0 ± 0.7 kcal/mol (UUUU). The experimentally determined thermodynamic data for two closely related RNA hairpin sequences are −3.7 ± 0.2 kcal/mol (UUCG) and −1.5 ± 0.1 kcal/mol (UUUU) (2,13). Therefore, the calculation significantly overestimates the absolute folding free energies for both hairpins. However, the calculated relative free energy ΔΔGfold of −3.5 kcal/mol between the two hairpins is in good agreement with the experimental value of −2.2 kcal/mol (2). We note that, if the native state basins in the PMF (Figs. 2 and 3) are chosen to define the folded and unfolded states, then the calculated absolute free energies ΔGfold would be significantly less overestimated, whereas the relative free energy ΔΔGfold would be essentially unaffected.
Table 1.
Folding free energies of RNA hairpin UUCG and UUUU calculated with AMBER ff99
| PMF∗ | MM-PB/SA | Experiment | |
|---|---|---|---|
| ΔGfold: UUCG | −13.5 ± 2.0 | −12.6 | −3.7 ± 0.2† |
| ΔGfold: UUUU | −10.0 ± 0.7 | −2.8 | −1.5 ± 0.1† |
| ΔΔGfold: UUCG–UUUU | −3.5 | −9.8 | −2.2‡ |
Units: kcal/mol.
ΔGfold is the average result calculated using three independent PMF. The error was estimated from calculations using the first and second half of the trajectory of each umbrella sampling window.
Experimental result is for the 5′-CGC(UUCG)GCG-3′ tetraloop (13).
Estimated from the experimental results of ΔΔGfold between two 12-nt UUCG and UUUU tetraloops (2).
What is the underlying cause for the overestimation of absolute folding free energies calculated by the PMF approach (Table 1)? Because ΔGfold = ΔHfold − TΔSfold, there are two possibilities: 1), the intramolecular interaction given by the force field may be too attractive, which will give a overly negative enthalpy of folding ΔHfold; or 2), the limited sampling in our simulations may cause the configurations of the unfolded states to be inadequately explored and underweighted, and this will lead to a too small TΔSfold. At this point, it is not yet clear which of the two factors is most responsible for the overestimated absolute folding free energy.
The folding free energies were also estimated using the MM-PB/SA method. With this approach, two unrestrained 6-ns MD simulations in explicit TIP3P water were carried out, one starting from the folded structure and the other from single stranded form. During these simulations, the RNA molecules remained in their respective initial conformational ensembles. The main contribution to the folding free energy comes from the difference between ensemble averaged effective energies (the sum of intramolecular energy and solvation free energy, with the latter determined by solving the Poisson-Boltzmann equation plus a solvent accessible surface area term) of the two trajectories. The vibrational entropy contribution estimated by the normal mode analysis is also included. The detailed breakdown of contributions is given in Table S1 and Table S2. The ΔGfold predicted by the MM-PB/SA are −12.6 kcal/mol and −2.8 kcal/mol for UUCG and UUUU, respectively (Table 1). Thus, the calculations also correctly differentiate the thermodynamic stabilities of the two hairpins, while overestimating the relative stability. We noted that the electrostatic solvation free energy component is sensitive to the atomic radii used in the Poisson-Boltzmann calculation. The results given in Table 1 are obtained with the van der Waals radii from the force field. Using the PARSE radii set (26) destabilizes both hairpins by ∼9 kcal/mol (data not shown), which would predict a positive folding free energy for the UUUU hairpin. Because the force field van der Waals radii yields the correct sign for folding free energy, it is more appropriate for characterizing the electrostatic solvation effect in these RNA hairpins.
Besides estimating the free energy difference, the MM-PB/SA calculations also provide insights into the energetics of hairpin folding. As seen from Table S1 and Table S2, the folding is driven by the solute van der Waals interactions, and to a lesser extent, by the nonpolar solvation term. Both the solute strain energy and vibrational entropy are unfavorable to the hairpin formation. The reduction of the vibrational entropy on folding indicates that the folded molecule resides in narrow and steep energy wells compared with the unfolded molecule. The contribution from the overall electrostatics varies with molecular system: the overall electrostatics contribution ΔG(total_elec) stabilizes the native conformation in UUCG, but disfavors the folding of UUUU. It should be noted that the entropy associated with jumping between energy wells are not accounted for in MM-PB/SA. This is because the only solute entropy term considered by the MM-PB/SA calculation is the vibrational entropy contribution, which is calculated by assuming that the solute molecule moves within a single harmonic energy well, which is clearly incorrect. The inadequate account for entropy in this method could be the main reason for the overestimation of the relative folding free energy between the two hairpins.
PMF calculated using CHARMM27
The PMF and the associated structural snapshots obtained with the CHARMM27 force field are presented in Figs. 4 and 5. For a better comparison with the AMBER results, the PMF are shown with the same vertical scale as those of Figs. 2 and 3. The structural snapshots sampled during the umbrella sampling simulations are also shown with more detail in Fig. S3 and Fig. S4. These results indicate that the RNA molecules described by CHARMM27 are more flexible than those modeled by AMBER, as the free energy cost of deformation calculated by CHARMM27 is significantly smaller. As seen from Figs. 4 and 5, the free energies are within 4.5 kcal/mol from the ground state level, whereas in Figs. 2 and 3 the free energies of the unfolded states are 10 kcal/mol higher than those of the folded states. This suggests that the two force fields differ in the balance of intrasolute interaction and solute-solvent interaction in the RNA molecules studied here.
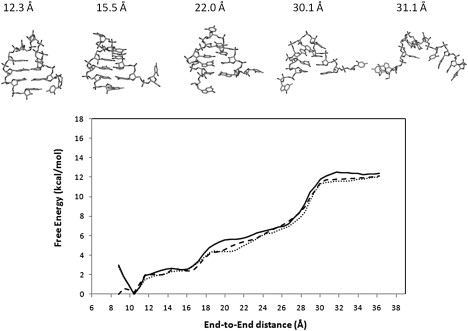
Figure 4.
PMF of UUCG calculated using CHARMM27 and the snapshots of representative structures sampled by the umbrella sampling simulations.
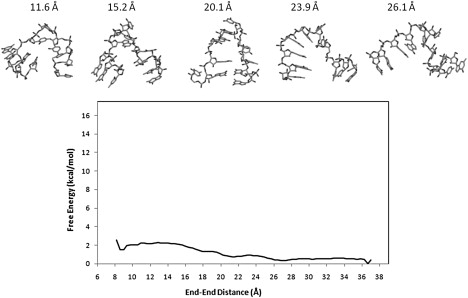
Figure 5.
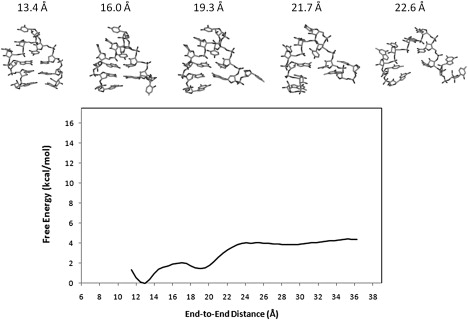
PMF of UUUU calculated using CHARMM27 and the snapshots of representative structures sampled by the umbrella sampling simulation.
More surprisingly, the PMF of UUCG indicates that the compact states are less stable than the extended states by ∼2 kcal/mol. It should also be noted that the folded structures observed in the umbrella sampling with CHARMM27 differ significantly from the experimental structure: the computed structure lacks both the correct stem basepairing and the loop conformation (see Fig. S3). To investigate whether this instability in the UUCG hairpin is caused by the umbrella potential, separate unrestrained simulations of UUCG were carried out in explicit solvent starting from the experimental structure and with different initial velocities. In the two CHARMM27 simulations (Fig. 6), one 7 ns and another 10 ns, the RMSD from the native structure reached 9.05 Å and 6.49 Å, respectively. The hairpin unfolds to produce structures with frayed stem and nonnative loop (Fig. S5 and Fig. S6). For comparison purpose, we also carried out a 100-ns unrestrained simulation using AMBER ff99 on UUCG. In this extended simulation, the average RMSD from the crystal structure is ≈1.46 Å (see Fig. S9, Fig. S10, and Fig. S12 for details).
Figure 6.
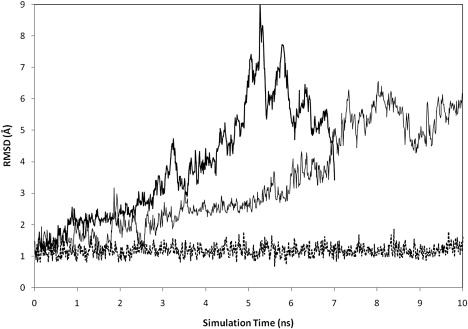
Time series of the heavy atom RMS difference relative to the native structure of UUCG. The three unrestrained MD simulations were started from the experimental structure and lasted for 7 ns and 10 ns. Solid lines: two CHARMM27 trajectories (7 ns and 10 ns); dotted line: an AMBER ff99 trajectory (10 ns).
The snapshots of the structures sampled in the two CHARMM27 simulations on UUCG are shown in Fig. S5 and Fig. S6. These and the time history of the RMSD for the loop and stem (Fig. 7) provide more details of the hairpin instability in the CHARMM27 simulations. The structural instability seems to originate from the loop region, which unfolds ∼3 ns before the fraying of the three stem basepairs. Therefore, in the CHARMM27 description of the UUCG hairpin, the helical stem is more stable than the loop. In the experimental native structure (Fig. 1 a), the loop is stabilized by the following interactions: a), U4/C3 stacking; b), C6/U4 stacking; c), G7/G8 stacking; and d), hydrogen bonds between G7/U4. During the 7 ns CHARMM27 simulation, all these interactions in the loop region are lost after 3 ns, as U4 and G7 looped out whereas C6 extends into the solvent. Similarly, in the 10 ns CHARMM27 trajectory, only the U4/C3 stacking remains after 3 ns. These results suggest that the force field problem may be corrected by modifying only those parameters pertaining to the loop region, where the structural instability is most pronounced.
Figure 7.
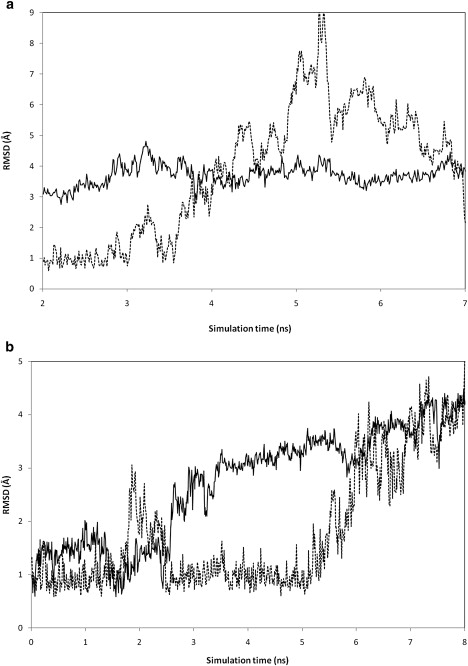
CHARMM27 simulation time series of RMS difference relative to the crystal structure of UUCG. (a) 7 ns trajectory; (b) 10 ns trajectory. Solid line: loop region; dotted line: stem.
In contrast to the UUCG results, the PMF of the UUUU calculated with CHARMM27 is well-behaved (Fig. 5). Here, the folded state coincides with the global free energy minimum. The PMF contains the native minimum, an intermediate basin, and a plateau of unfolded states. The barrier height in the intermediate region is 1.0 kBT. The absolute folding free energy calculated using the same criteria as discussed earlier is −2.8 ± 0.2 kcal/mol. Compared with the corresponding AMBER result, the CHARMM27 result is in better agreement with the experimental folding free energy of −1.5 kcal/mol (Table 1). On the other hand, significant force field dependency also exists for the UUUU hairpin. The free energy minimum in the CHARMM27 PMF is located at 12.9 Å, whereas the global minimum in the AMBER profile is found at 10.5 Å (Figs. 3 and 5). Thus, the native state in the CHARMM27 simulations has a different equilibrium loop structure compared with that in the AMBER calculation (Fig. S7). In particular, although the loop structure obtained with CHARMM27 is stabilized by a long lived hydrogen bond between U4 base and the phosphate linking U6/U7, the AMBER structure shows no intramolecular hydrogen bond involving phosphate in the loop region (Fig. S7). The sugar puckering for the UUUU tetraloop obtained from CHARMM27 and AMBER simulations are compared with those determined by NMR study (24). As seen from Table S3, the AMBER results are more consistent with the NMR measurements. Other notable differences in the PMF include the fact that the unfolding of the UUUU hairpin observed in the CHARMM27 calculation occurs at an end-to-end distance of 22.5 Å, which is substantially smaller than the corresponding distance of 29.0 Å seen in the AMBER calculation (Figs. 3 and 5).
Conclusion
In this study, the potential of mean force of RNA hairpins UUCG and UUUU has been determined in explicit ionic solvent using the end-to-end distance as the order parameter. The energetics of the hairpin folding has also been studied using the MM-PB/SA approach. We have also investigated the force field dependency of the free energy calculation by comparing the results obtained from AMBER ff99 and CHARMM27.
The AMBER results described correctly the physical interactions in the two hairpins because both the native state stability and the relative free energy are reproduced. The PMF profiles show that the native state basin is separated from the unfolded state plateau by a wide shoulder region, which samples a variety of native-like structures with frayed terminal basepair. The presence of the unfolded state plateau and the intermediate shoulder region can be viewed as entropic barriers that need to be overcome by diffusional search (biased toward the native state) in a hairpin folding pathway in which the formation of the loop region occurs before the formation of the stem. There are differences in the details of the PMF between the two hairpins: 1), the free energy cost of unfolding for UUCG is higher than that for UUUU; and 2), the boundary between the native states and the intermediate shoulder region in the UUCG PMF is clearly defined, whereas the same boundary in the UUUU PMF is less clear-cut. These properties of the calculated PMF are consistent with the thermodynamic data on the relative stability of the two hairpins and some aspects of the kinetic experiments. For example, at low temperatures, the free energy profile of UUCG is more compatible with a three-state system, whereas that of UUUU resembles a two-state system. However, the calculated PMF lacks major barriers that could be expected in transition regions, and we think that this is related to the use of 1-D reaction coordinate, which does not have the resolution to fully describe the roughness of the underlying free energy surface. Additional insights into the energetics of hairpin folding are gained by MM-PB/SA calculations through its breakdown of energy contributions. Here, information provided by MM-PB/SA is mainly enthalpic because it is very limited in correctly estimating the conformational entropy change associated with large structural transitions. MM-PB/SA gives correct ranking order of stability, but its estimation of relative free energy is not as good as that given by PMF.
Although the relative folding free energy is quantitatively reproduced by our PMF calculation using the AMBER force field, the absolute folding free energies are overestimated for both hairpins. This error is likely to originate from the sampling limitation, but it may also come from the AMBER force field. The limitations in sampling can cause the conformational entropy of the unfolded states to be underestimated, which leads to the overstabilization of the folded state. Explorations aimed at enhancing the sampling are being investigated currently.
The results of CHARMM27 are qualitatively different from those obtained with AMBER, in terms of both the free energy profile and the conformational property. The RNA hairpins modeled by CHARMM27 are significantly less rigid, which probably led to more complete sampling of the conformational space and the more accurate estimate of the folding free energy for UUUU. Although the structural stability of the UUUU hairpin is maintained in CHARMM27, the global stability of the UUCG hairpin is not reproduced by the PMF calculated. Separate simulations using CHARMM27 show that the force field related instability of the UUCG hairpin originates from the loop region, and propagates to the stem region after several nanoseconds. Consequently, the problem may not be as pronounced in shorter simulations and/or with longer stem regions.
Supporting Material
Details of umbrella sampling, MD protocol, three tables, and 16 figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01682-8.
Supporting Material
Acknowledgments
We thank Prof. Yunyu Shi for her support of this work; Prof. C. L. Brooks III for very helpful discussions; Prof. Haiyan Liu for providing the computing facility; and Mr. Jian Zhan for help maintaining the Linux clusters on which the calculations are carried out.
References
- 1.Tinoco I., Bustamante C. How RNA folds. J. Mol. Biol. 1999;293:271–281. doi: 10.1006/jmbi.1999.3001. [DOI] [PubMed] [Google Scholar]
- 2.Proctor D.J., Ma H., Bevilacqua P.C. Folding thermodynamics and kinetics of YNMG RNA hairpins: specific incorporation of 8-bromoguanosine leads to stabilization by enhancement of the folding rate. Biochemistry. 2004;43:14004–14014. doi: 10.1021/bi048213e. [DOI] [PubMed] [Google Scholar]
- 3.Uhlenbeck O.C. Tetraloops and RNA folding. Nature. 1990;346:613–614. doi: 10.1038/346613a0. [DOI] [PubMed] [Google Scholar]
- 4.Varani G. Exceptionally stable nucleic acid hairpins. Annu. Rev. Biophys. Biomol. Struct. 1995;24:379–404. doi: 10.1146/annurev.bb.24.060195.002115. [DOI] [PubMed] [Google Scholar]
- 5.Akke M., Fiala R., Palmer A.G. Base dynamics in a UUCG tetraloop RNA hairpin characterized by 15N spin relaxation: correlations with structure and stability. RNA. 1997;3:702–709. [PMC free article] [PubMed] [Google Scholar]
- 6.MacKerell A.D., Nilsson L. Nucleic acid simulations. In: Becker O., MacKerell A.D., Roux B., Watanabe M., editors. Computational Biochemistry and Biophysics. Marcel Dekker; New York: 1999. pp. 441–463. [Google Scholar]
- 7.Orozco M., Pérez A., Luque F.J. Theoretical methods for the simulation of nucleic acids. Chem. Soc. Rev. 2003;32:350–364. doi: 10.1039/b207226m. [DOI] [PubMed] [Google Scholar]
- 8.Cheatham T.E. Simulation and modeling of nucleic acid structure, dynamics and interactions. Curr. Opin. Struct. Biol. 2004;14:360–367. doi: 10.1016/j.sbi.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Sorin E.J., Engelhardt M.A., Pande V.S. RNA simulations: probing hairpin unfolding and the dynamics of a GNRA tetraloop. J. Mol. Biol. 2002;317:493–506. doi: 10.1006/jmbi.2002.5447. [DOI] [PubMed] [Google Scholar]
- 10.Sorin E.J., Rhee Y.M., Pande V.S. Insights into nucleic acid conformational dynamics from massively parallel stochastic simulations. Biophys. J. 2003;85:790–803. doi: 10.1016/S0006-3495(03)74520-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sorin E.J., Rhee Y.M., Pande V.S. Does water play a structural role in the folding of small nucleic acids? Biophys. J. 2005;88:2516–2524. doi: 10.1529/biophysj.104.055087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarzynska J., Nilsson L., Kulinski T. Effects of base substitutions in an RNA hairpin from molecular dynamics and free energy simulations. Biophys. J. 2003;85:3445–3459. doi: 10.1016/S0006-3495(03)74766-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams D.J., Hall K.B. Experimental and computational studies of the G[UUCG]C RNA tetraloop. J. Mol. Biol. 2000;297:1045–1061. doi: 10.1006/jmbi.2000.3623. [DOI] [PubMed] [Google Scholar]
- 14.Deng N., Cieplak P. A molecular dynamics and free energy study of the conformational equilibria in RNA hairpin. J. Chem. Theory Comput. 2007;3:1345–1360. doi: 10.1021/ct6003388. [DOI] [PubMed] [Google Scholar]
- 15.Garcia A.E., Paschek D. Simulation of the pressure and temperature folding/unfolding equilibrium of a small RNA hairpin. J. Am. Chem. Soc. 2008;130:815–817. doi: 10.1021/ja074191i. [DOI] [PubMed] [Google Scholar]
- 16.Villa A., Widjajakusuma E., Stock G. Molecular dynamics simulation of the structure, dynamics, and thermostability of the RNA hairpins uCACGg and cUUCGg. J. Phys. Chem. B. 2008;112:134–142. doi: 10.1021/jp0764337. [DOI] [PubMed] [Google Scholar]
- 17.Srinivasan J., Cheatham T., Case D. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate−DNA helices. J. Am. Chem. Soc. 1998;120:9401–9409. [Google Scholar]
- 18.Torrie G., Valleau J. Nonphysical sampling distribution in Monte Carlo free-energy estimation: umbrella sampling. J. Comput. Phys. 1977;23:187–199. [Google Scholar]
- 19.Chen S.J., Dill K.A. RNA folding energy landscapes. Proc. Natl. Acad. Sci. USA. 2000;97:646–651. doi: 10.1073/pnas.97.2.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang W., Chen S.J. RNA hairpin-folding kinetics. Proc. Natl. Acad. Sci. USA. 2002;99:1931–1936. doi: 10.1073/pnas.032443099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma H., Proctor D.J., Gruebele M. Exploring the energy landscape of a small RNA hairpin. J. Am. Chem. Soc. 2006;128:1523–1530. doi: 10.1021/ja0553856. [DOI] [PubMed] [Google Scholar]
- 22.Cheatham T.E., Cieplak P., Kollman P.A. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn. 1999;16:845–862. doi: 10.1080/07391102.1999.10508297. [DOI] [PubMed] [Google Scholar]
- 23.Foloppe N., MacKerell A. All atom empirical force field for nucleic acids: 1) parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000;21:86–104. [Google Scholar]
- 24.Fürtig B., Richter C., Schwalbe H. NMR spectroscopy of RNA. ChemBioChem. 2003;4:936–962. doi: 10.1002/cbic.200300700. [DOI] [PubMed] [Google Scholar]
- 25.Molinaro M., Tinoco I. Use of ultra stable UNCG tetraloop hairpins to fold RNA structures: thermodynamic and spectroscopic applications. Nucleic Acids Res. 1995;23:3056–3063. doi: 10.1093/nar/23.15.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sitkoff D., Sharp K., Honig B. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 1994;98:1978–1988. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.