

Abstract
Rapid molecular collapse mediated by nonlocal interactions is believed to be a crucial event for protein folding. To investigate the role of nonlocal interactions in tertiary structure formation, we performed a nonlocal interaction substitution mutation analysis on staphylococcal nuclease (SNase). Y54 and I139 of wild-type (WT) SNase and Δ140-149 were substituted by cysteine to form intramolecular disulfide bonds, respectively called WT-SS and Δ140-149-SS. Under physiological conditions, the reduced form of Δ140-149-SS appears to assume a denatured structure; in contrast, the oxidized form of Δ140-149-SS forms a native-like structure. From this result, we conclude that the C-terminal region participates in a nonlocal interaction that is indispensable for the native structure. Although the oxidized form of WT-SS assumes a more compact denatured structure under acidic conditions than the WT, the kinetic measurements reveal that the refolding reactions of both the reduced and oxidized forms of WT-SS are similar to those of the WT, suggesting that an intact nonlocal interaction is established within the dead time (22 ms). On the basis of these results, we propose that the native nonlocal contact established at the early stage of the folding process facilitates further secondary structure formation.
Introduction
A protein's tertiary structure contains numerous interatomic contacts. During the folding reaction, free energy is rapidly reduced to attain the native tertiary structure. The formation of one native contact itself facilitates the formation of other native contacts (1,2). It is known that, among the possible native interatomic contacts, nonlocal contacts among several residues separated on a primary sequence can most effectively reduce the molecular size, resulting in a decrease in the size of the conformational search space. It has been revealed that the contact order is closely related to the folding rate (3,4). The contact order is defined by the average separation along the sequence of residues in physical contact within the folded protein. More nonlocal contacts result in a higher contact order. This higher contact order prolongs the folding time, suggesting that the formation of native nonlocal contacts is a crucial step in protein folding. Therefore, to understand the mechanism of tertiary structure formation, we must clarify the role of native nonlocal contacts.
A commonly used technique to investigate protein folding is point mutation analysis, in which the influence of point mutations on protein folding reactions and the stability of thermodynamic states is examined to elucidate the role of local structure formation (5). However, point mutation analysis cannot necessarily be applied to investigate the formation of nonlocal contacts. Nonlocal contacts are the consequence of nonlocal interactions, which in turn are the consequence of several nonpolar amino acid residues forming clusters buried in the molecule, as a result of segregation of the nonpolar side chains from surrounding water molecules (6). Consequently, the site-specific point mutations cannot sufficiently influence nonlocal interactions. In this work, we introduced an artificial nonlocal interaction into staphylococcal nuclease (SNase) via a double-cysteine mutation, and examined the influence of this artificial nonlocal interaction by a disulfide bond on tertiary structure formation and protein folding.
SNase has been extensively studied as a model protein in attempts to elucidate the principles of protein stability and folding (7–14). SNase consists of two subdomains (N-terminal and C-terminal (15,16)) and the substrate-binding site is located in the cleft between the two subdomains (17). Trypsin digestion of SNase generates two long fragments, each containing one of the subdomains. Early studies of trypsin-digested SNase revealed that two separated fragments assume a native-like structure with the aid of an inhibitor binding (15), suggesting that a nonlocal interaction between the two subdomains promotes tertiary structure formation.
The C-terminal 13 amino acid residues truncated mutant (Δ137-149) assumes a denatured structure even under nondenatured conditions (18,19), suggesting that the truncated region contains crucial information specifying the native tertiary structure formation. W140 of SNase is one of the key residues that stabilize the native conformation (10,20). A systematic mutation analysis of W140 revealed that the aromatic ring is essential at this position; the ring gathers the surrounding residues of E129, A132, K133, and I139 to form a small hydrophobic cluster (21). The hydrophobic cluster tightly contacts several residues in the N-terminal subdomain through hydrophobic and/or van der Waals interactions, which are expected to be involved in nonlocal contacts between the N-terminal and C-terminal subdomains, and to stabilize the tertiary structure of SNase (21).
In this work, to confirm the nonlocal interaction between the N-terminal and C-terminal subdomains and elucidate its role in tertiary structure formation, we introduced a disulfide bond between the two subdomains of the intact SNase and the W140-lacking mutant Δ140-149, respectively named WT-SS and Δ140-149-SS, and examined their structures and stabilities. Although the reduced form of Δ140-149-SS lost its tertiary structure, the oxidized forms exhibited a native-like tertiary structure, clearly indicating that the C-terminal cluster around W140 participates in an intact nonlocal interaction to collapse the two subdomains. Furthermore, the nonlocal interaction between the N-terminal and C-terminal subdomains is indispensable for the native-like tertiary structure. To investigate the time region of the nonlocal interaction formation during the folding process, we measured the folding reactions of the disulfide bond-containing mutants using a stopped-flow circular dichroism (CD) apparatus. These mutants exhibit a folding reaction similar to that of the wild-type (WT) within the observed time region from 22 ms to 900 s, and the apparent folding rates are almost identical between the WT and mutants. These observations suggest that the nonlocal interaction forms within the stopped-flow dead time (22 ms). From these observations, we propose that the native nonlocal interaction between the N-terminal and C-terminal subdomains is established at an early stage of the protein-folding reaction, and that this nonlocal interaction promotes subsequent structure formation in the later stages of folding.
Materials and Methods
Materials
Construction and purification of SNase mutants were carried out as described previously (21). The genes for WT-SS and Δ140-149-SS were prepared by polymerase chain reaction using oligonucleotide mutation primers, and cloned into the pET-16b vector. WT and mutant SNases were expressed in Escherichia coli BL-21 (DE3) as inclusion bodies, and purified by a series of urea extraction, ethanol precipitation, and ion-exchange chromatography as described previously (21). The double-cysteine mutants were purified using buffer solutions containing 2 mM dithiothreitol (DTT) to avoid oxidation of the sulfhydryl group. The disulfide bond was formed in an air-purged buffer without DTT. Disulfide bond formation was verified by means of nonreducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). All samples were further purified by a Superdex-75 prepacked column (GE Healthcare UK Ltd., Buckinghamshire, UK). The protein concentration was determined by absorbance at 280 nm, using an excitation coefficient at 280 nm (E1% 1cm of 9.3 for WT, 6.7 for Δ140-149, and 8.5 for WT-SS) (5,22).
Far-UV CD spectrum measurements
CD spectra were measured using a Jasco (Jasco, Tokyo, Japan) model J-820 spectropolarimeter with a quartz cell of 1 mm path length. The proteins were prepared at 0.2 mg/mL in the buffer solutions containing 50 mM MOPS, 50 mM NaCl, 1 mM EDTA, pH 6.6 (physiological condition), and 50 mM HCl/NaCl, pH 2.0 (acidic condition). The temperature of the sample was kept at 20°C by circulating temperature-controlled water. Thermal denaturation experiments were performed by monitoring molar residue ellipticity at 222 nm as the temperature increased. All solutions for the reduced forms of the double-cysteine mutants contain 2 mM DTT. The thermodynamic parameters were obtained as described previously (21).
Small-angle x-ray scattering experiments
Small-angle x-ray scattering (SAXS) measurements were performed using a solution x-ray scattering apparatus installed at Photon Factory BL-10C in Tsukuba, Japan (23). The measurements were carried out at 20°C as described previously (21). The buffer condition was the same as for the far-UV CD spectrum measurements.
Size-exclusion chromatography
Size-exclusion chromatography was carried out on an ÄKTAexplorer system (GE Healthcare) on a Superdex 75 prepacked column. The running buffer was 50 mM HCl/NaCl, pH 3.0. The buffer solution for the reduced forms of the mutants also contained 2 mM DTT.
Stopped-flow CD experiments
Kinetic far-UV CD measurements of refolding reactions were carried out using the stopped-flow apparatus (UNISOKU, Osaka, Japan) installed in the cell compartment of a JASCO J-725 spectropolarimeter (Jasco). The measurements were carried out at 20°C. The proteins were initially dissolved in hydrochloride buffer, 50 mM HCl/NaCl, pH 2.2, at a protein concentration of 0.8 mg/mL. The reaction was initiated by mixing the protein solution with a MOPS buffer with a mixing ratio of 1:3. The final solution condition was 50 mM MOPS, 50 mM NaCl, 1 mM EDTA, pH 6.6, with a protein concentration of 0.2 mg/mL. The dead time of the stopped-flow apparatus was 22 ms. Each reaction curve was observed by the ellipticity changes at 222 nm, using the cell with 1 mm optical path length, at 20 ms intervals. To improve the signal/noise ratio, the measurements were repeated 50 times and averaged. The kinetic data were fitted by the following equation:
where θ(t) and θ(∞) are ellipticity values at time t and infinite time, respectively. Δθi and ki are the ellipticity amplitude and the apparent rate of the ith phase, respectively. Decay-associated difference (DAD) far-UV CD spectra were obtained by fitting the refolding curves measured at various wavelengths ranging from 212 nm to 238 nm using the global fitting procedure included in the IGOR software package (WaveMetrics, SW Nimbus, Portland, OR).
Results and Discussion
Preparation of nonnative disulfide bond-introduced mutants
The crystal structure of SNase shows that the C-terminal cluster around W140 is composed of G107, A132, K133, and I139, which are located on the C-terminal region on the amino acid sequence (10,17) (Fig. 1). The C-terminal cluster comes into contact with P42 and Y54 on the N-terminal subdomain. The distances between the Cβ and Cα atoms of I139 on the C-terminal cluster and those of Y54 on the N-terminal subdomain are 7.1 and 5.6 Å, respectively. Although the Cβ-Cβ distance is longer than the moderate distance for disulfide bond formation (3.5–4.2 Å), the Cα-Cα distance is relatively close to the suitable distance range (3.8–7.0 Å) (24). The disulfide bond is thought to form spontaneously when both Y54 and I139 are simultaneously substituted with cysteine. The introduced artificial disulfide bond provides an effective means to examine the role of nonlocal interaction. For our purpose, we prepared the double-cysteine mutant, Y54C/I139C, for WT-SS and Δ140-149-SS. The reduced forms of the double-cysteine mutants were prepared in the buffer containing 5 mM DTT. The proteins were oxidized in the air-purged buffer in the absence of DTT to form the disulfide bond. Fig. 2 shows SDS-PAGE patterns of the reduced [SS(−)] and oxidized [SS(+)] forms of the mutants under nonreducing conditions. Each sample exhibits a single band, excluding the possibility of dimer formation due to an intermolecular disulfide bond. Although the band positions of WT and the reduced form of the WT-SS mutant, WT-SS(−), are almost identical, the oxidized form, WT-SS(+), is slightly shifted downward. The oxidized form of Δ140-149-SS also exhibits a downward shift compared to its reduced form. The downward band shift that occurs upon the oxidization of these mutants is interpreted as a decrease in the molecular dimension under the SDS-denaturation condition. We conclude that the intramolecular disulfide bond between C54-C139 is established under the oxidized condition.
Figure 1.
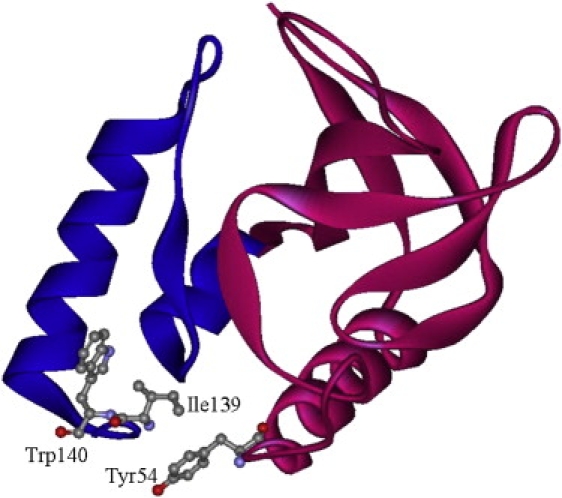
X-ray crystal structure of WT SNase (Protein Data Bank code: 1EY0). Trp-140, Ile-139, and Tyr-54 are shown in a ball and stick model.
Figure 2.
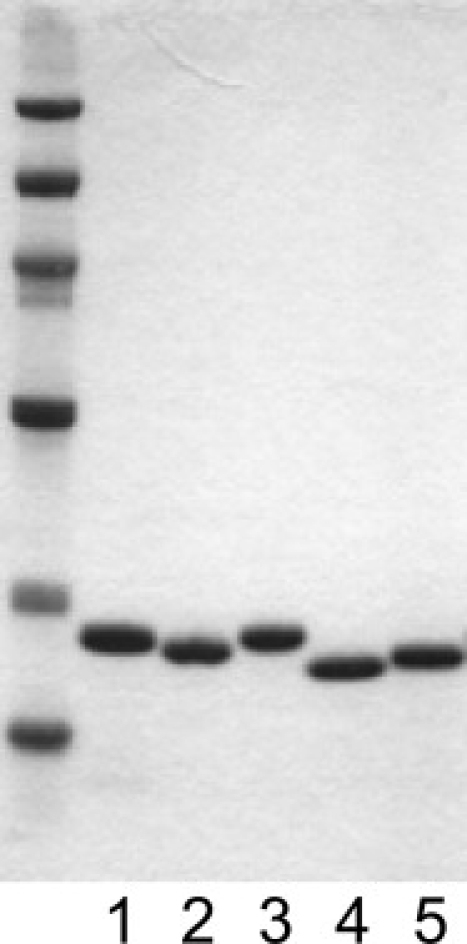
Nonreducing SDS-PAGE. Lane 1, WT; lane 2, WT-SS(+); lane 3, WT-SS(−); lane 4, Δ140-149-SS(+); lane 5, Δ140-149-SS(−). SS(+) and SS(−) represent the thiol-oxidized form and the reduced form, respectively.
Solution structures of the SS mutants under the physiological condition
The solution structures of the oxidized and reduced forms of the double-cysteine mutants were examined by means of far-UV CD and SAXS measurements. Fig. 3 A shows the far-UV CD spectra of WT and the double-cysteine mutants under physiological conditions. The far-UV CD spectra of Δ140-149-SS(−) indicates that the mutants lose their secondary structure, in agreement with previous results obtained with Δ140-149 (10,21). On the other hand, the spectra of the oxidized form, Δ140-149-SS(+), is close to that of WT. The far-UV CD spectra of WT-SS(+) and WT-SS(−) exhibit the native-like CD spectra regardless of the presence or absence of the disulfide bond. Although the ellipticities at 222 nm are slightly smaller than that of WT, Δ140-149-SS(+) and WT-SS(+/−) are expected to assume native tertiary structures. Fig. 3 B shows the scattering profiles of the reduced and oxidized forms of the double-cysteine mutants and WT in the form of a Kratky plot. The Kratky plot of Δ140-149-SS(−) does not show any clear peaks; instead, it exhibits a broad bump with a monotonously increasing tail, indicating that the reduced forms assume a denatured structure (10). The Rg values of Δ140-149-SS(+) and WT-SS(+/−) were almost identical to that of WT (Table 1). In addition, their Kratky plots are superimposed on that of WT. Their tertiary structures are almost identical despite the slight difference in the far-UV CD spectra between WT and the mutants. Therefore, we conclude that the slight difference in the far-UV CD spectra reflects the structural perturbation localized around the mutated amino acid residues.
Figure 3.
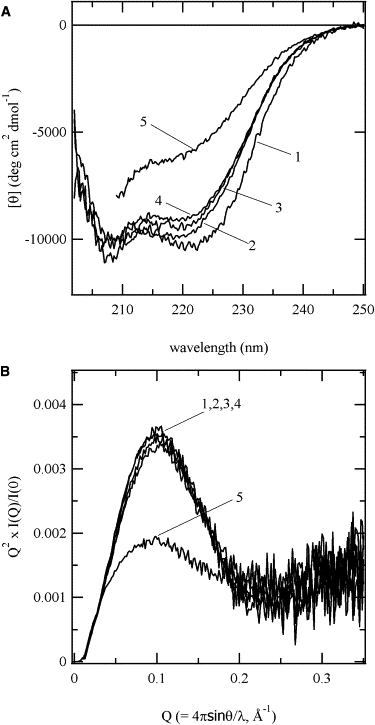
Far-UV CD spectra (A) and SAXS profiles in the form of Kratky plots (B) of the SNase mutants under the physiological condition. Curve 1, WT; 2, WT-SS(+); 3, WT-SS(−); 4, Δ140-149-SS(+); 5, Δ140-149-SS(−).
Table 1.
SAXS parameters of SNase mutants
| Rg (Å) | I(0)/conc | |
|---|---|---|
| WT | 18.0 ± 0.2 | 4900 ± 85 |
| WT-SS(+) | 18.4 ± 0.2 | 4980 ± 85 |
| WT-SS(−) | 17.8 ± 0.3 | 4770 ± 94 |
| Δ-SS(+) | 18.8 ± 0.3 | 4430 ± 87 |
| Δ−SS(−) | 23.1 ± 1.2 | 4600 ± 290 |
Thermal denaturation experiments of the SS mutants
The equilibrium states of the mutants with the native-like structure at 20°C were determined by the thermal denaturation experiments. The ellipticity change at 222 nm with increasing temperature was monitored. The fraction change of the denatured state was calculated assuming a two-state transition between the native and denatured states (Fig. 4) (21). The curves of Δ140-149-SS(+), WT-SS(+), and WT-SS(−) clearly show plateaus at the lower and higher temperatures, indicating that the equilibrium between the folded and unfolded states is sufficiently shifted toward the folded state under the neutral pH condition at 20°C. The thermodynamic parameters, ΔH and mid-temperature, Tm, are summarized in Table 2. These parameters decrease in the following order: WT, WT-SS(+), WT-SS(−), and Δ140-149-SS(+). It is well known that a disulfide bond without steric strain stabilizes the protein tertiary structure (25–27). It was previously reported that the propensity for disulfide bond formation is influenced by various stereochemical effects when a pair of residues are replaced by Cys (24,28). In those studies, the propensity for disulfide bond formation was examined with the use of MODIP, which assumes empirical criteria for disulfide bond formation propensities and provides recommended interresidue pairs based on a crystal structure (24,28). However, the pair we consider here, Y54 and I139, were not listed, mainly due to the longer interatomic distance between their Cβ atoms. It is possible that structural perturbation is accompanied by the formation of the artificial disulfide bond between Cys-54 and Cys-139. The decrease in the thermostability may be partly due to such a perturbation, brought about by the disulfide bond. However, both the native-like CD spectrum and SAXS profile lead to the conclusion that WT-SS(+) and Δ140-149-SS(+) assume a native-like structure in solution. The spontaneous formation of the disulfide bond also suggests a native-like conformation.
Figure 4.
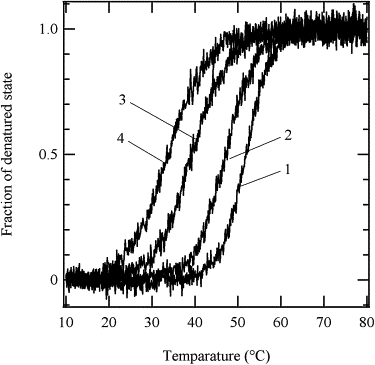
Thermal denaturation curves of the SNase mutants monitored by far-UV CD at 222 nm. Curve 1, WT; 2, WT-SS(+); 3, WT-SS(−); 4, Δ140-149-SS(+).
Table 2.
Thermodynamic parameters
| ΔH(kcal/mol) | ΔS(kcal/mol/K) | Tm (°C) | |
|---|---|---|---|
| WT | 71.0 ± 1.2 | 0.219 ± 0.004 | 51.2 ± 0.5 |
| WT-SS(+) | 55.8 ± 0.6 | 0.175 ± 0.002 | 46.4 ± 0.4 |
| WT-SS(−) | 46.5 ± 0.5 | 0.149 ± 0.002 | 39.7 ± 0.4 |
| Δ-SS(+) | 42.8 ± 0.6 | 0.139 ± 0.002 | 34.9 ± 0.5 |
Role of the nonlocal interaction in the tertiary structure formation
In previous work, we showed that the W140-lacking mutants lose their tertiary structure under physiological conditions (10,21). G107, A132, K133, and I139 on the C-terminal subdomains closely contact the indole ring of W140 to form the C-terminal cluster. Because the C-terminal cluster is in contact with two amino acid residues, P42 and Y54, on the N-terminal subdomain, we speculated that the C-terminal cluster formation is responsible for the nonlocal interaction between the N-terminal and C-terminal subdomains. In this study, we found that an artificial nonlocal interaction brought about by a disulfide bond recovers the foldability, even in the mutant lacking W140. This observation clearly indicates that the nonlocal interaction between the N-terminal and C-terminal subdomains, which can be replaced by the disulfide bond, is essential for tertiary structure formation. Furthermore, it shows that the presumed C-terminal cluster around W140 is indeed responsible for generating the nonlocal interaction.
C-terminal deletion mutants lacking W140 are known to attain a native-like structure with the aid of an inhibitor (8,14). We recently described the induced folding mechanism in detail (14). During the induced folding reaction of Δ140-149, a defined intermediate state can be observed within a few tens of milliseconds. The intermediate state exhibits a partially denatured structure but is bound to an inhibitor, prAp. The induced folding event was considered to be an equilibrium shift toward the native state, brought about by ligand binding. The existence of the induced folding intermediate indicates that the binding occurs before formation of the native tertiary structure. In other words, the intermediate formation is an indispensable step in the formation of the native tertiary structure. This observation implies that the free form of Δ140-149 loses its foldability due to the lack of the C-terminal region. The binding site lies on the cleft between the N-terminal and C-terminal subdomains. Based on these observations, in combination with our findings, we hypothesize that inhibitor binding acts as an alternative nonlocal interaction, replacing the nonlocal interaction that is missing due to the lack of W140.
In summary, the nonlocal interaction in the native SNase requires the C-terminal region. However, even if the C-terminal region is truncated, the inhibitor binding and the artificial disulfide bond can act as an alternative nonlocal interaction responsible for the tertiary formation. The inhibitor-binding site is distant from the nonlocal interaction ascribable to the C-terminal region, suggesting that there may be alternative global patterns of nonlocal interactions that facilitate the formation of the tertiary structure.
Solution structures of the SS mutants under the acidic condition
Fig. 5 A shows the far-UV CD spectra of the acid-denatured states of the mutants and WT. All of these species lost their secondary structures. The far-UV CD spectra of the denatured mutants are identical to that of WT in the acid-denatured state, regardless of the reduced and oxidized forms, indicating that the disulfide bonds do not influence the secondary structures in the acid-denatured states. However, the molecular dimensions of the oxidized forms are more compact than those of the reduced form under the SDS-denatured state, as shown in Fig. 2. The molecular dimensions of the acid-denatured states were examined by size-exclusion chromatography (Fig. 5 B). Each sample showed a single elution peak. Although the retention time of WT-SS(−) is identical to that of WT, oxidation delays its elution peak, indicating that WT-SS(+) assumes a more compact form than WT-SS(−). Although Δ140-149 lacks the C-terminal 10 residues, the retention time of Δ140-149 is faster than that of WT, suggesting that the acid-denatured state of Δ140-149 is slightly swollen compared to that of WT. However, the oxidized form of Δ140-149 also showed a prolongation of the retention time. These results suggest that the disulfide bond of the unfolded states of WT-SS and Δ140-149 reduced their molecular dimension even under the acidic condition.
Figure 5.
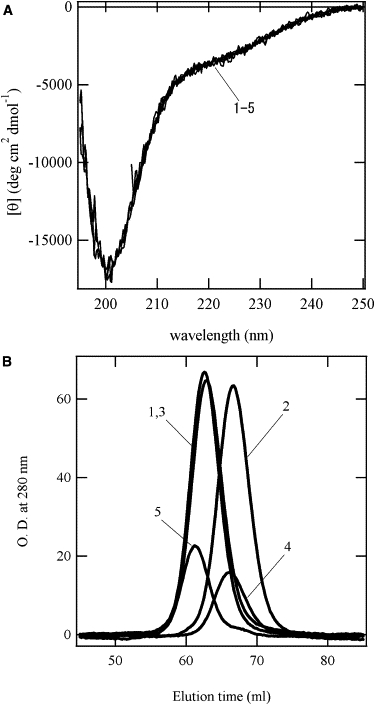
(A) Far-UV CD spectra under acidic conditions. (B) Elution profiles of the size-exclusion chromatography. Curve 1, WT; 2, WT-SS(+); 3, WT-SS(−); 4, Δ140-149-SS(+); 5, Δ140-149-SS(−).
Folding reactions of the SS mutants
We investigated the folding kinetics of the double-cysteine mutants using stopped-flow CD. The reaction was initiated by a pH jump from pH 2.2 to pH 6.6. Fig. 6 shows the ellipticity change at 222 nm. The refolding reactions of the mutants and WT reached their equilibrium states within 900 s. These proteins exhibited a partial ellipticity increase within the dead time of the stopped-flow apparatus (22 ms), indicating that folding intermediates are accumulated in the early stage of the folding reaction. Table 3 shows the kinetic parameters obtained from the reaction curves. The refolding reaction curves can be fit by using triple-exponential functions, except for Δ140-149-SS(+). The apparent rates of WT were 15 s−1, 1.6 s−1, and 0.03 s−1, in agreement with those reported previously for WT (29). Although the reaction curves of WT, WT-SS(−), and WT-SS(+) appear to be substantially different from each other (Fig. 6), the apparent rates of the phases are almost identical, suggesting that the refolding reactions of WT-SS(+) and WT-SS(−) obey a mechanism similar to that of WT within the observed time region. The major difference among the refolding reactions was found in the amplitude of each phase. The total ellipticity changes were 5200 for WT, 4600 for WT-SS(+), and 4200 for WT-SS(−). The difference in the amplitude is mainly due to the difference in the ellipticities of the native states (Fig. 3 A). Previous kinetic studies of SNase suggested that the third phase is attributed to rate-limiting proline isomerization (9), and the second phase is also partly influenced by proline isomerization (29). The effective prolines in the refolding reaction were assigned to P11, P31, and P56 (29). The cysteine mutations perturb the local structure around the substituted positions, I139 and Y54, as mentioned above. Therefore, we assume that the alteration of the fractional amplitudes is probably due to local structural perturbation near P56. It should be stressed that neither the double mutations of Y54 and I139 nor the nonnative disulfide bond formation influences the folding rates of SNase in the observed time region (>22 ms). If the nonlocal interaction forms in WT during the observed time region, the preexistence of the artificial nonlocal interaction, the disulfide bond, inevitably affects the folding reaction. Together with the fact that the unfolded state of WT-SS(+) is more compact than that of WT-SS(−), these findings suggest that the intact nonlocal interaction between the N-terminal and C-terminal subdomains is established in the early stage of the refolding reaction (<22 ms).
Figure 6.
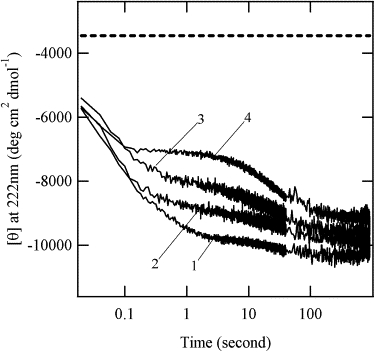
Kinetic refolding reaction curves immediately after the pH jump from pH 2.2 to pH 6.6, monitored by far-UV CD at 222 nm. Curve 1, WT; 2, WT-SS(+); 3, WT-SS(−); 4, Δ140-149-SS(+). Dashed line indicates the average ellipticity (−3456) of the initial state for these proteins at 222 nm.
Table 3.
Kinetic parameters of the refolding reaction
| k1 (s−1) | k2 (s−1) | k3 (s−1) | |
|---|---|---|---|
| WT | 14.7 ± 0.6 | 1.62 ± 0.06 | 0.0325 ± 0.003 |
| WT-SS(+) | 13.2 ± 1.8 | 0.925 ± 0.09 | 0.0545 ± 0.003 |
| WT-SS(−) | 11.8 ± 0.7 | 1.10 ± 0.2 | 0.0609 ± 0.003 |
| Δ-SS(+) | 25.0 ± 1.0 | N.D. | 0.0371 ± 0.0006 |
| Δθ1 | Δθ2 | Δθ3 | Δθtotal | |
|---|---|---|---|---|
| WT | 3140 ± 65 (60%) | 1530 ± 50 (29%) | 570 ± 21 (11%) | 5200 ± 140 |
| WT-SS(+) | 3530 ± 94 (77%) | 490± 33 (11%) | 580 ± 13 (13%) | 4600 ± 140 |
| WT-SS(−) | 2700 ± 160 (65%) | 420 ± 62 (10%) | 1050 ± 22 (25%) | 4200 ± 240 |
| Δ−SS(+) | 2170 ± 48 (54%) | N.D. | 1880 ± 13 (46%) | 4050 ± 60 |
Although WT and the WT-SS(+/−) mutants exhibit the three phases, the refolding reaction of Δ140-149-SS(+) can be fitted by using a double-exponential function. The kinetic parameters are summarized in Table 3. The apparent rates of the first and second phases were similar to the apparent rates of the first and third phases of WT and the WT-SS(+/−) mutants, respectively. The fractional amplitude of the first phase of Δ140-149-SS(+) is comparable to those of WT and the WT-SS(+/−) mutants. The fractional amplitude of the second phase of Δ140-149-SS(+) is close to the summation of the fractional amplitudes of the second and third phases of WT.
To find the reason for the differences in the second phase between Δ140-149-SS(+) and the WT-SS(+/−) mutants, we measured the ellipticity changes at various wavelengths ranging from 212 nm to 238 nm to obtain DAD CD spectra (Fig. 7). The DAD CD spectra of the second and third phases of WT resemble each other, but that of the first phase is slightly shifted toward the shorter wavelength. It was previously reported that SNase folds into the native structure through two parallel reaction paths: the first phase lies on one path, and the second and third phases lie on the other (12,29,30). The similarity of the DAD CD spectrum between the second and third phases may indicate that these phases lie on the sequential reaction path. The DAD CD spectrum of the first phase of Δ140-149-SS(+) is shifted toward the shorter wavelength as compared with the second phase, in similarity to WT. Therefore, we consider that the second phase of Δ140-149-SS(+) is the same phase as the third phase of WT, i.e., the phase corresponding to the second phase of WT disappears in Δ140-149-SS(+). It is unclear why the second phase disappears in Δ140-149-SS(+), but we assume that the decay rate of the phase of Δ140-149-SS(+) decreases due to the lack of the C-terminal region, and the second and third phases are degenerated into one observed phase. The similarity in the folding rates of the first and third phases, along with the DAD CD spectral shift, suggests that the folding mechanism of Δ140-149-SS(+) is also similar to that of WT.
Figure 7.
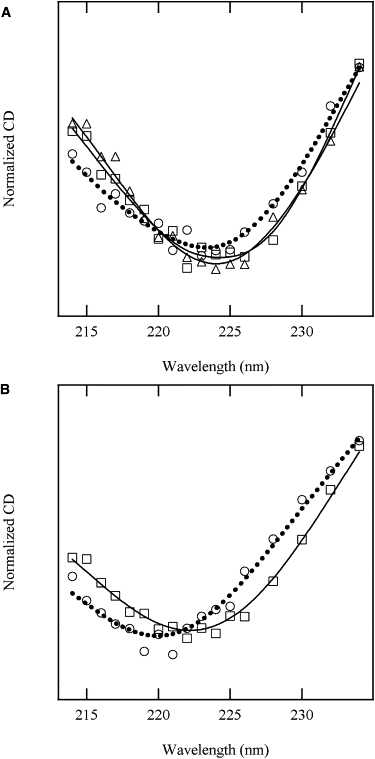
DAD CD spectra of the refolding of WT (A) and Δ140-149-SS(+) (B). The DAD CD spectra of the first, second, and third phases are represented by open circles, open squares, and open triangles, respectively. These spectra were normalized by the integrated ellipticity ranging from 212 nm to 238 nm. The broken and solid lines represent the interpolated lines among the data points.
Role of the nonlocal interaction in the folding process
A number of NMR studies have investigated the folding core in the early stage of the SNase folding process (31,32). The stable core structure forms in a part of the N-terminal β-barrel at the early stage of the folding process (31). Furthermore, in a pulsed hydrogen-deuterium exchange NMR experiment revealed that ∼10 ms after initiation of the folding reaction, a region from residues 134–141 in the C-terminal domain is protected against hydrogen-deuterium exchange, as is the region in the N-terminal β-barrel (32). The protected C-terminal region is superimposed on the hydrophobic cluster around W140 (21). Subsequently, at 10–100 ms, the amide protons of other residues in helices 2 and 3, and in strands 4 and 5 are protected against pulse labeling, indicating that additional secondary structure formation is followed by cluster formation (32). The core region in the β-barrel that forms in the early stage of folding (<10 ms) may serve as a nucleation site for further structure formation (32), although this has not been confirmed. NMR studies of Δ136-149 revealed a residual structure in the N-terminal subdomain, although the mutant assumes a denatured structure under a physiological condition (33). The partially folded region in the deletion mutant is well superimposed on the presumed folding core in the β-barrel. This suggests that under physiological conditions, the core region cannot work as a folding core for mutants that lack a C-terminus. Although the artificial nonlocal interaction introduced by the disulfide bond exists before initiation of the folding reaction, the folding kinetics are essentially identical to those of WT. We therefore propose that, in addition to the core region in the N-terminal β-barrel, the formation of the nonlocal interaction between the N-terminal and C-terminal subdomains is also an essential step of SNase folding, and that this step occurs in the early stage of folding.
Summary
This study clearly shows that the structure of the W140-lacking mutant can recuperate from the denatured structure with the aid of an artificial nonlocal interaction between the N-terminal and C-terminal subdomains. These results support our model, which proposes that the C-terminal hydrophobic cluster plays an essential role in a nonlocal interaction that is indispensable for tertiary structure formation. The unfolded structures of the disulfide bond-introduced mutants take a more compact conformation than WT and the reduced form of the double-cysteine mutants. By comparing the refolding reactions, one can see that the disulfide bond-introduced mutant exhibits a native-like refolding reaction, regardless of the difference in the molecular dimension of the unfolded structure. Therefore, the nonlocal interaction between the N-terminal and C-terminal subdomains forms before the formation of the burst intermediates, within the dead time of the stopped-flow apparatus. From the observation that the major ellipticity change occurs at the late stage of the reaction, we conclude that nonlocal interactions in the early stage of the folding reaction can promote subsequent secondary structure formation.
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (15076208, 20370062, and 20107006) to M.K. The experiments at Photon Factory BL-10C were performed under the approval of the Photon Factory Advisory Committee (proposal Nos. 2006G209, 2004G186, and 2002G169).
Contributor Information
Hironari Kamikubo, Email: kamikubo@ms.naist.jp.
Mikio Kataoka, Email: kataoka@ms.naist.jp.
References
- 1.Brooks C.L., Gruebele M., Wolynes P.G. Chemical physics of protein folding. Proc. Natl. Acad. Sci. USA. 1998;95:11037–11038. doi: 10.1073/pnas.95.19.11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker D. A surprising simplicity to protein folding. Nature. 2000;405:39–42. doi: 10.1038/35011000. [DOI] [PubMed] [Google Scholar]
- 3.Plaxco K.W., Simons K.T., Baker D. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 4.Ivankov D.N., Garbuzynskiy S.O., Finkelstein A.V. Contact order revisited: influence of protein size on the folding rate. Protein Sci. 2003;12:2057–2062. doi: 10.1110/ps.0302503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shortle D., Stites W.E., Meeker A.K. Contributions of the large hydrophobic amino acids to the stability of staphylococcal nuclease. Biochemistry. 1990;29:8033–8041. doi: 10.1021/bi00487a007. [DOI] [PubMed] [Google Scholar]
- 6.Dill K.A., Bromberg S., Chan H.S. Principles of protein folding—a perspective from simple exact models. Protein Sci. 1995;4:561–602. doi: 10.1002/pro.5560040401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shortle D., Meeker A.K., Freire E. Stability mutants of staphylococcal nuclease: large compensating enthalpy-entropy changes for the reversible denaturation reaction. Biochemistry. 1988;27:4761–4768. doi: 10.1021/bi00413a027. [DOI] [PubMed] [Google Scholar]
- 8.Flanagan J.M., Kataoka M., Engelman D.M. Truncated staphylococcal nuclease is compact but disordered. Proc. Natl. Acad. Sci. USA. 1992;89:748–752. doi: 10.1073/pnas.89.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walkenhorst W.F., Green S.M., Roder H. Kinetic evidence for folding and unfolding intermediates in staphylococcal nuclease. Biochemistry. 1997;36:5795–5805. doi: 10.1021/bi9700476. [DOI] [PubMed] [Google Scholar]
- 10.Hirano S., Mihara K., Kataoka M. Role of C-terminal region of staphylococcal nuclease for foldability, stability, and activity. Proteins. 2002;49:255–265. doi: 10.1002/prot.10216. [DOI] [PubMed] [Google Scholar]
- 11.Kamagata K., Sawano Y., Kuwajima K. Multiple parallel-pathway folding of proline-free staphylococcal nuclease. J. Mol. Biol. 2003;332:1143–1153. doi: 10.1016/j.jmb.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 12.Maki K., Cheng H., Roder H. Early events during folding of wild-type staphylococcal nuclease and a single-tryptophan variant studied by ultrarapid mixing. J. Mol. Biol. 2004;338:383–400. doi: 10.1016/j.jmb.2004.02.044. [DOI] [PubMed] [Google Scholar]
- 13.Tsong T.Y., Hu C.K., Wu M.C. Hydrophobic condensation and modular assembly model of protein folding. Biosystems. 2008;93:78–89. doi: 10.1016/j.biosystems.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 14.Onitsuka M., Kamikubo H., Kataoka M. Mechanism of induced folding: both folding before binding and binding before folding can be realized in staphylococcal nuclease mutants. Proteins. 2008;72:837–847. doi: 10.1002/prot.21978. [DOI] [PubMed] [Google Scholar]
- 15.Anfinsen C.B. The formation and stabilization of protein structure. Biochem. J. 1972;128:737–749. doi: 10.1042/bj1280737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griko Y.V., Gittis A., Privalov P.L. Residual structure in a staphylococcal nuclease fragment. Is it a molten globule and is its unfolding a first-order phase transition? J. Mol. Biol. 1994;243:93–99. doi: 10.1006/jmbi.1994.1632. [DOI] [PubMed] [Google Scholar]
- 17.Loll P.J., Lattman E.E. The crystal structure of the ternary complex of staphylococcal nuclease, Ca2+, and the inhibitor pdTp, refined at 1.65 A. Proteins. 1989;5:183–201. doi: 10.1002/prot.340050302. [DOI] [PubMed] [Google Scholar]
- 18.Flanagan J.M., Kataoka M., Engelman D.M. Mutations can cause large changes in the conformation of a denatured protein. Biochemistry. 1993;32:10359–10370. doi: 10.1021/bi00090a011. [DOI] [PubMed] [Google Scholar]
- 19.Shortle D., Meeker A.K. Residual structure in large fragments of staphylococcal nuclease: effects of amino acid substitutions. Biochemistry. 1989;28:936–944. doi: 10.1021/bi00429a003. [DOI] [PubMed] [Google Scholar]
- 20.Su Z., Wu J.M., Chen H.M. Local stability identification and the role of a key aromatic amino acid residue in staphylococcal nuclease refolding. FEBS J. 2005;272:3960–3966. doi: 10.1111/j.1742-4658.2005.04814.x. [DOI] [PubMed] [Google Scholar]
- 21.Hirano S., Kamikubo H., Kataoka M. Elucidation of information encoded in tryptophan 140 of staphylococcal nuclease. Proteins. 2005;58:271–277. doi: 10.1002/prot.20333. [DOI] [PubMed] [Google Scholar]
- 22.Pace C.N., Vajdos F., Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ueki T., Hiragi Y., Muroga Y. Aggregation of bovine serum albumin upon cleavage of its disulfide bonds, studied by the time-resolved small-angle X-ray scattering technique with synchrotron radiation. Biophys. Chem. 1985;23:115–124. doi: 10.1016/0301-4622(85)80069-7. [DOI] [PubMed] [Google Scholar]
- 24.Dani V.S., Ramakrishnan C., Varadarajan R. MODIP revisited: re-evaluation and refinement of an automated procedure for modeling of disulfide bonds in proteins. Protein Eng. 2003;16:187–193. doi: 10.1093/proeng/gzg024. [DOI] [PubMed] [Google Scholar]
- 25.Katz B.A., Kossiakoff A. The crystallographically determined structures of atypical strained disulfides engineered into subtilisin. J. Biol. Chem. 1986;261:15480–15485. [PubMed] [Google Scholar]
- 26.Pace C.N., Grimsley G.R., Barnett B.J. Conformational stability and activity of ribonuclease T1 with zero, one, and two intact disulfide bonds. J. Biol. Chem. 1988;263:11820–11825. [PubMed] [Google Scholar]
- 27.Tidor B., Karplus M. The contribution of cross-links to protein stability: a normal mode analysis of the configurational entropy of the native state. Proteins. 1993;15:71–79. doi: 10.1002/prot.340150109. [DOI] [PubMed] [Google Scholar]
- 28.Sowdhamini R., Srinivasan N., Balaram P. Stereochemical modeling of disulfide bridges. Criteria for introduction into proteins by site-directed mutagenesis. Protein Eng. 1989;3:95–103. doi: 10.1093/protein/3.2.95. [DOI] [PubMed] [Google Scholar]
- 29.Maki K., Ikura T., Kuwajima K. Effects of proline mutations on the folding of staphylococcal nuclease. Biochemistry. 1999;38:2213–2223. doi: 10.1021/bi981962+. [DOI] [PubMed] [Google Scholar]
- 30.Maki K., Cheng H., Roder H. Folding kinetics of staphylococcal nuclease studied by tryptophan engineering and rapid mixing methods. J. Mol. Biol. 2007;368:244–255. doi: 10.1016/j.jmb.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacobs M.D., Fox R.O. Staphylococcal nuclease folding intermediate characterized by hydrogen exchange and NMR spectroscopy. Proc. Natl. Acad. Sci. USA. 1994;91:449–453. doi: 10.1073/pnas.91.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walkenhorst W.F., Edwards J.A., Roder H. Early formation of a β hairpin during folding of staphylococcal nuclease H124L as detected by pulsed hydrogen exchange. Protein Sci. 2002;11:82–91. doi: 10.1110/ps.28202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng Y., Liu D., Wang J. Native-like partially folded conformations and folding process revealed in the N-terminal large fragments of staphylococcal nuclease: a study by NMR spectroscopy. J. Mol. Biol. 2003;330:821–837. doi: 10.1016/s0022-2836(03)00660-0. [DOI] [PubMed] [Google Scholar]