

Abstract
Thiazole synthase in Escherichia coli is an αβ heterodimer of ThiG and ThiH. ThiH is a tyrosine lyase that cleaves the Cα–Cβ bond of tyrosine, generating p-cresol as a by-product, to form dehydroglycine. This reactive intermediate acts as one of three substrates for the thiazole cyclization reaction catalyzed by ThiG. ThiH is a radical S-adenosylmethionine (AdoMet) enzyme that utilizes a [4Fe-4S]+ cluster to reductively cleave AdoMet, forming methionine and a 5′-deoxyadenosyl radical. Analysis of the time-dependent formation of the reaction products 5′-deoxyadenosine (DOA) and p-cresol has demonstrated catalytic behavior of the tyrosine lyase. The kinetics of product formation showed a pre-steady state burst phase, and the involvement of DOA in product inhibition was identified by the addition of 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase to activity assays. This hydrolyzed the DOA and changed the rate-determining step but, in addition, substantially increased the uncoupled turnover of AdoMet. Addition of glyoxylate and ammonium inhibited the tyrosine cleavage reaction, but the reductive cleavage of AdoMet continued in an uncoupled manner. Tyrosine analogues were incubated with ThiGH, which showed a strong preference for phenolic substrates. 4-Hydroxyphenylpropionic acid analogues allowed uncoupled AdoMet cleavage but did not result in further reaction (Cα–Cβ bond cleavage). The results of the substrate analogue studies and the product inhibition can be explained by a mechanistic hypothesis involving two reaction pathways, a product-forming pathway and a futile cycle.
Keywords: Enzymes/Inhibitors, Enzymes/Kinetics, Enzymes/Mechanisms, Protein/Iron-Sulfur, Radicals, Vitamins and Cofactors/Vitamin B, Product Inhibition
Introduction
Thiamine pyrophosphate (TPP)2 is an essential cofactor for several enzymes, including pyruvate decarboxylase, pyruvate dehydrogenase, and transketolase (1). Prokaryotes and some eukaryotes, such as yeast and plants, can biosynthesize TPP (2). In prokaryotes, ThiE catalyzes the formation of thiamine monophosphate by covalently linking two independently formed heterocyclic precursors (3), 4-amino-5-hydroxymethylpyrimidine pyrophosphate 6 and the recently characterized (4) 2-carboxy-4-methyl-5-(β-hydroxyethyl)-thiazole phosphate (Fig. 1A, Thz-P carboxylate 4). Phosphorylation to TPP 7 by ThiL completes the biosynthesis of the catalytically active vitamin (5). The 4-amino-5-hydroxymethylpyrimidine moiety is formed in a complex radical mediated rearrangement of 5-aminoimidazole ribotide 5, in a reaction that requires ThiC, a recent addition to the rapidly expanding radical S-adenosylmethionine (AdoMet) superfamily (6, 7). The formation of 4-methyl-5-(β-hydroxyethyl) thiazole phosphate carboxylate (Fig. 1B) requires the precursor 1-deoxyxyulose 5-phosphate 3, a sulfur atom from cysteine, which is transferred via a thiocarboxyl terminus of the small protein, ThiS 12 (8, 9), and dehydroglycine 10, which serves as the first common intermediate between aerobic and anaerobic thiazole biosynthetic pathways. The aerobe Bacillus subtilis has been the model bacterium for probing the mechanism of the thiazole ring-forming reaction, catalyzed by ThiG (8, 10, 11). B. subtilis, like all other aerobic prokaryotes, uses dioxygen to oxidize glycine to dehydroglycine in a reaction catalyzed by ThiO (12). The pathway to this electron-deficient intermediate differs significantly in anaerobes such as E. coli, which cannot oxidize glycine. As an alternative, anaerobes use ThiH to produce dehydroglycine by the cleavage of the Cα–Cβ bond of tyrosine. ThiH has been shown to belong to the “radical AdoMet” family, with the characteristic iron-sulfur cluster-binding motif (13–15), and in vitro studies on Escherichia coli ThiGH have demonstrated the requirement for AdoMet and a reductant for activity (16, 17).
FIGURE 1.

Biosynthesis of thiamine pyrophosphate in prokaryotes. A, biosynthetic enzymes and precursors of TPP in E. coli. B, proposed mechanism for formation of the thiazole moiety. The formation of 13 has been characterized using ThiG from B. subtilis (8, 10, 11) and is proposed to be, along with dehydroglycine, a common intermediate in the biosynthetic pathway in all prokaryotes. The generation of dehydroglycine varies between anaerobic and aerobic organisms. The thiazole moiety is coupled with 4-amino-5-hydroxymethylpyprimidine (HMP) pyrophosphate and phosphorylated to TPP in the later steps of the biosynthesis of TPP.
The products of tyrosine cleavage in vitro have been characterized as glyoxylate and p-cresol. The glyoxylate is proposed to form as a result of the hydrolysis of dehydroglycine (18). The reactivity of the intermediate dehydroglycine presents a potential problem, especially during its transfer from ThiH to ThiG as the release of dehydroglycine into aqueous solution would result in hydrolysis. The consequence of this wasteful outcome is the net loss of 1 eq of AdoMet, tyrosine, and NADPH, with no overall benefit. Product inhibition of ThiGH has recently been observed with the products of AdoMet cleavage, DOA and methionine. This product inhibition can be overcome by the addition of 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase (MTAN), which catalyzes the hydrolysis of DOA to adenine and 5′-deoxyribose (19).
Here, we report the application of an in vitro activity assay for ThiH-mediated tyrosine cleavage to demonstrate that ThiH can undergo more than a single turnover when provided with sufficient substrates and reductant. Tyrosine lyase activity of ThiGH was found to exhibit a “burst phase” kinetic profile due to release of the products becoming rate-limiting when undergoing multiple turnovers. The addition of MTAN was found to alter this rate-limiting step in subsequent turnovers and changed the profile of the time course. Addition of MTAN also resulted in a significant increase in the amount of uncoupled AdoMet cleavage. Experiments with tyrosine analogues or combinations of products indicated that AdoMet cleavage is coupled to the generation of a substrate radical, most likely by abstraction of the phenolic hydrogen atom. The experiments also suggest a potential mechanism for uncoupled turnover of AdoMet. These results clarify the likely site of substrate radical formation and indicate the complex regulation required to efficiently form and successfully utilize dehydroglycine during thiamine biosynthesis.
EXPERIMENTAL PROCEDURES
Materials
Reagents and materials were obtained from the following suppliers: far-UV HPLC-grade acetonitrile from Fisher; protein chromatography media from GE Healthcare; arabinose from Alfa-Aesar (Heysham, UK); dithiothreitol, NADPH, and antibiotics from Melford Laboratories (Ipswich, UK). All other chemicals used were of the highest quality available and were purchased from Sigma or Fluka. AdoMet was purchased from Sigma and used without further purification.
Methods
All proteins were purified and reconstituted, and activities were assayed in an anaerobic glove box maintained at less than 1 ppm O2 (Belle Technology, Portesham, UK). HPLC analysis and UV spectra were recorded as described previously (17). Proteins were expressed and purified as reported previously (17, 20). ThiGH and ThiH were further purified by Superdex 200 gel filtration chromatography as follows; the protein was chemically reconstituted as described previously (17) and concentrated to a volume of less than 5 ml using a 10-kDa molecular mass cutoff filter (Millipore). The concentrated protein was then applied to a Superdex 200 gel filtration column (2.6 × 60 cm) pre-equilibrated in anaerobic buffer A (50 mm MOPS (pH 7.7), 100 mm NaCl, 12.5% (w/v) glycerol, 5 mm dithiothreitol). The column was eluted with buffer A (∼100 ml) until the A280 was observed to rise, at which point 7.5-ml fractions were collected. The ThiGH complex typically eluted in the first six fractions and the monomeric ThiH in the following six fractions (supplemental Fig. S1). An aliquot of each fraction (150 μl) was retained for SDS-PAGE and iron analysis, and the remainder was stored at −80 °C in sealed 15-ml Falcon tubes for further investigation. Iron content of protein samples was determined by the method of Fish (21).
In Vitro Assay for Tyrosine Lyase Activity
The two most concentrated fractions of either the ThiGH complex or monomeric ThiH (1–3 mg/ml, 15 ml) were thawed inside an anaerobic glove box. To ensure a full complement of 4Fe-4S cluster, the protein was carefully reconstituted, first by the addition of aqueous dithiothreitol solution (to a final concentration of 5 mm). After 20 min of gentle mixing, 2.5 mol eq of FeCl3 (from a freshly prepared anaerobic 10 mm aqueous stock solution) were added dropwise, and after a further 15 min, 2.5 mol eq of Na2S was added. The resulting solution was stirred at room temperature for 1 h before concentrating the protein to a volume of 1 ml. The protein was then exchanged into buffer B (50 mm MOPS (pH 7.5), 100 mm NaCl, and 5% (w/v) glycerol) via a Sephadex G-25 pre-packed column. Assays (150 μl) were prepared in 1.6-ml microcentrifuge tubes by addition of components to the following final concentrations: ThiGH or ThiH (35–95 μm; the exact concentration of ThiH or ThiGH for time course experiments is given in the figure legends), AdoMet (1 mm), and tyrosine (1 mm). The assay solutions were equilibrated at 37 °C by incubating in a water bath contained within an anaerobic glove box for 5 min. A stock solution of the reductant system was prepared containing flavodoxin 1 (280 μm), flavoprotein:NADPH oxidoreductase (70 μm), and NADPH (15 mm) and incubated at room temperature for 15 min to permit the formation of the blue-colored semiquinone (22). The assays were initiated by the addition (20 μl) of the reductant system (final concentrations of flavodoxin 1 (37 μm), flavoprotein:NADPH oxidoreductase (9 μm), and NADPH (2 mm)). Each time point (1–60 min) was stopped by protein precipitation with 20% perchloric acid (10 μl) and then cleared by centrifugation (Eppendorf 5415D microcentrifuge, maximum speed). Supernatants were analyzed by HPLC using a Gemini C18, 5-μm, 110-Å reverse phase column (Phenomenex). The mobile phase solvents were 0.1% AcOH in water (solvent A) or 0.1% AcOH in acetonitrile (solvent B), and the flow rate was 0.8 ml/min. An initial isocratic phase of 100% solvent A for 8 min was followed by a linear gradient to 50% solvent B over 32 min, followed by an increase to 100% solvent B over 3 min, which was held for 5 min before returning to 100% solvent A over 2 min and re-equilibration for 10 min (total time of 60 min). To investigate the effect of removing DOA formed in situ during the experiment, the reaction assays were supplemented with MTAN (10 μm) that had been previously exchanged into anaerobic buffer B. To investigate inhibition by glyoxylate and ammonia, these compounds (10–2000 μm) were added to the assay.
Experiments with Substrate Analogues
Stock solutions (33 mm) of substrate analogues were prepared in 100 mm ammonium bicarbonate and deoxygenated in an anaerobic glove box overnight. ThiGH/ThiH isolated by nickel-affinity chromatography was chemically reconstituted as described previously (17), concentrated, and exchanged into buffer B. Assays were prepared with chemically reconstituted ThiGH/ThiH mixture (60–90 μm), AdoMet (1 mm), substrate (1 mm) and analyzed as described above.
Data Analysis
Data were analyzed and graphs prepared using SigmaPlot (Systat Software Inc., London). Data from experiments where a pre-steady state burst phase was observed (Fig. 2 and Fig. 6, A and B) were fitted to a function that contains a single exponential and a linear component (23) as shown in Equation 1,
where [P] is the observed concentration of product; [E] is the amplitude of the burst phase and is equal to the concentration of enzyme active sites in the reaction; kburst is the observed single exponential rate constant for the burst phase; L is the observed linear rate; and t is time. The rate constant for the steady state phase (kss) was derived by dividing the observed linear rate by the burst amplitude. For experiments where the reaction displayed first order kinetics (Fig. 3), data were fitted to Equation 2,
where [P] is the observed concentration of product; [P]max is the maximum observed product concentration, and k is the observed first order rate constant. To derive the initial turnover number (kcat0), Equation 3 was used,
 |
where [E] is the ThiGH concentration as estimated by the Bradford assay (24). For the product inhibition experiment (Fig. 4), data were fitted to a 4-parameter logistic sigmoid as shown in Equation 4,
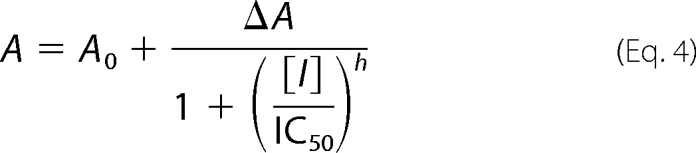 |
where A is the observed activity; A0 is the minimum activity; ΔA is the difference between the maximum and minimum observed activity; [I] is the concentration of inhibitor (glyoxylate and ammonium), and h is the Hill coefficient.
FIGURE 2.

In vitro time course of tyrosine lyase activity by ThiGH. Formation of DOA (○) and p-cresol (●) in a ThiGH activity assay (containing 80 μm ThiGH) was monitored by HPLC. Data are the average of experiments carried out in duplicate, shown with the standard error, and were fitted to a pre-steady state burst phase function (Equation 1) to give the results shown in Table 1.
FIGURE 6.

In vitro time course of monomeric ThiH activity. A, formation of DOA (○) and p-cresol (●) in a monomeric ThiH assay (containing 35 μm ThiH). B, formation of adenine (♢) and p-cresol (●) in a monomeric ThiH assay (containing 90 μm of ThiH) coupled with MTAN-mediated hydrolysis of DOA. To facilitate a comparison, the data are presented in number of equivalents of product formed per enzyme. Data are the average of experiments carried out in duplicate, shown with the standard error, and were fitted to a pre-steady state burst phase function (Equation 1) to give the results shown in Table 1.
FIGURE 3.

In vitro time course of tyrosine lyase activity by ThiGH coupled with MTAN-mediated hydrolysis of DOA. Formation of adenine (○) and p-cresol (●) in a ThiGH activity assay (containing 95 μm of ThiGH), coupled with MTAN-mediated hydrolysis of DOA, was monitored by HPLC. Data are the average of experiments carried out in duplicate, shown with standard errors, and were fitted to a first order exponential function (Equation 2) to give the results shown in Table 2.
FIGURE 4.

Inhibition of tyrosine lyase activity by glyoxylate and ammonium. Assays were coupled with MTAN-mediated hydrolysis of DOA to preclude the possibility of inhibition by DOA and methionine. A, tyrosine Cα–Cβ bond cleavage was measured by the formation of p-cresol and was fitted to Equation 4. B, AdoMet cleavage was measured by the formation of adenine. Assays were incubated for 1 h and analyzed by HPLC, and values are relative to standard assays with no additions. Data are the average of experiments carried out in duplicate, shown with the standard errors, and were fitted to Equation 4.
RESULTS
Isolation of Monomeric ThiH and ThiGH Complex
Experiments with tyrosine lyase purified by nickel-affinity chromatography have permitted much initial characterization of the protein (17, 18). In addition, analytical gel filtration chromatography (14) has shown that the protein isolated from the initial nickel-charged chelating Sepharose column contained a mixture of two forms of ThiH as follows: as part of a large multimeric complex with at least six ThiGH heterodimers (apparent mass ∼440 kDa) or as a monomer (apparent mass ∼42 kDa). To simplify the interpretation of subsequent experiments, it was preferable to isolate these species so that their properties could be compared. Resolution of these two species was achieved by preparative gel filtration chromatography using Superdex 200 resin. The protein isolated after nickel-charged chelating Sepharose chromatography (ThiGH/ThiH mixture) was chemically reconstituted to improve the stability of ThiH, allowing it to be concentrated to ∼25 mg/ml for application to the preparative gel filtration column. Typically, a 1:1 ratio of ThiGH: ThiH, with ∼1 μmol of each, was successfully isolated (supplemental Fig. S1). Despite working under anaerobic conditions (less than 1 ppm O2), the labile 4Fe-4S cluster did not remain intact during this purification step, and the iron content of purified protein fractions was reduced to 3.1 ± 0.2 mol eq of iron per ThiGH and 2.5 ± 0.3 mol eq of iron per ThiH (supplemental Table S1). The 4Fe-4S cluster of ThiH shows a broad absorbance at 400 nm (17). UV-visible spectroscopy was used to monitor the loss of the labile 4Fe-4S cluster during chromatography and its efficient reconstitution (supplemental Fig. S2). To ensure optimal activity of protein samples, the 4Fe-4S cluster was reconstituted immediately before activity measurements.
Activity of the ThiGH Complex
The time dependence of product formation was studied with ThiGH complex isolated by gel filtration chromatography. In previous experiments, tyrosine lyase activity had been limited to less than one turnover (18), but this may have resulted from insufficient reductant or substrate in the assay. ThiGH assays were therefore prepared with greater than 10 mol eq of tyrosine and AdoMet and a large excess of reductant (2 mm NADPH) to ensure the [4Fe-4S] cluster could repeatedly access the +1 oxidation state during catalytic turnover. Analytical HPLC was used to quantify the formation of the products DOA and p-cresol. Reactions were stopped at a range of time points up to 1 h, and the time course showed a pre-steady state burst phase for product formation (Fig. 2), followed by a slower reaction once steady state was reached. In a typical experiment, after 1 h ThiGH had generated 1.8 mol eq of p-cresol and 2.3 mol eq of DOA, with an overall DOA:p-cresol ratio of 1.3:1, indicating that there was some uncoupled turnover of AdoMet to DOA. The time-dependent product formation could be fitted to Equation 1. This analysis gave the rate constant for p-cresol formation in the pre-steady state exponential phase of 53 ± 6 × 10−4 s−1, and during steady state phase, kcat was calculated as 1.6 ± 0.2 × 10−4 s−1. The rate constant for formation of 5′-deoxyadenosine was 63 ± 10 × 10−4 s−1 during the burst phase, but this slowed to 2.9 ± 0.2 × 10−4 s−1 during the steady state phase. The fitting of the data gave values of 89 ± 4 and 90 ± 4 μm for burst phase amplitude, which corresponds to the concentration of ThiH active sites and is in good agreement with the ThiGH concentration estimated by the Bradford assay (24) of 80 μm.
The pre-steady state burst phase (up to ∼10 min, Fig. 2 and Table 1) reflects the catalytic rate for the chemical reaction, and during this phase there is efficient coupling of DOA formation to tyrosine cleavage; for example, after 5 min, the concentration of DOA was 79 ± 6 μm, and the concentration of p-cresol was 71 ± 2 μm, giving a DOA to p-cresol ratio of 1.1:1. However, after the first turnover, when the release of products may become rate-limiting, the reaction slows down, and uncoupled turnover of AdoMet becomes more significant. In the steady state phase (after about 15 min, Fig. 2), the ratio of the rate of formation of DOA to the rate of p-cresol formation increases to 1.7:1
TABLE 1.
Kinetic analysis of tyrosine lyase activity showing a burst phase of product formation (Figs. 2 and 6)
These results were obtained by fitting data to Equation 1. [P] is the observed concentration of product; [E] is the observed burst amplitude; kburst is the observed single exponential rate constant for the burst phase, and kss is the steady state rate constant, derived by dividing the observed linear rate by the burst amplitude. Data are presented with standard errors, and R2 is a measure of the goodness of fit.
| Form of ThiH | [P] | [E] | kburst | kss | R2 |
|---|---|---|---|---|---|
| μm | ×10−4s−1 | ×10−4s−1 | |||
| ThiGH complex | p-Cresol | 89 ± 4 | 53 ± 6 | 1.6 ± 0.2 | 0.99 |
| DOA | 90 ± 4 | 63 ± 10 | 2.9 ± 0.2 | 0.99 | |
| Monomeric ThiH | p-Cresol | 38 ± 4 | 32 ± 8 | 5.9 ± 0.2 | 0.99 |
| DOA | 42 ± 9 | 27 ± 11 | 10.7 ± 1.0 | 0.99 | |
| Monomeric ThiH with MTAN | p-Cresol | 92 ± 16 | 32 ± 10 | 4.3 ± 0.1 | 0.98 |
| Adenine | 180 ± 75 | 32 ± 23 | 16.0 ± 3.3 | 0.95 |
Addition of MTAN to in Vitro Assays Changes the Rate-limiting Step
The addition of MTAN to time course experiments with ThiGH resulted in the rapid hydrolysis of DOA to 5′-deoxyribose and adenine (19). Adenine could be readily quantified using the same analytical HPLC protocol (Rt = 6.0 min, see supplemental Fig. S2). During the quantification of adenine produced in these time course experiments, particular care was required with negative control samples (lacking reductant), as commercial supplies of AdoMet contain a small proportion of 5′-methylthioadenosine. 5′-Methylthioadenosine also yields adenine upon hydrolysis by MTAN (20), and this background was subtracted from activity assays.
The addition of MTAN changed the profile of the ThiGH activity time course (Fig. 3). In situ hydrolysis of the DOA eliminated the burst phase and extended the period of relatively rapid turnover (where kcat for the formation of cresol was in the range 30–50 × 10−4 s−1) beyond a single turnover; however, the rate of product formation slowly declined and could be fitted as a first order process (Equation 2) with a final p-cresol concentration of 220 ± 5 μm (∼2.3 turnovers). Subsequent experiments with lower ThiGH concentrations demonstrated that after 1 h, ThiGH could produce up to 7 eq of p-cresol (supplemental Table S2). The initial turnover numbers (kcat0) for ThiGH calculated from the initial rates are 37 ± 3 × 10−4 and 51 ± 6 × 10−4 s−1 for the formation of p-cresol and adenine, respectively. These initial rates are similar to those observed in the absence of MTAN (Fig. 2 and Tables 1 and 2). At time points beyond 5 min, a further increase in uncoupled turnover of AdoMet was observed, and the final concentration of adenine reached 590 ± 24 μm, corresponding to a ratio of adenine:p-cresol of 2.7:1.
TABLE 2.
Kinetic analysis of tyrosine lyase activity showing an exponential profile of product formation (Fig. 3)
These results were obtained by fitting data to Equation 2. [P] is the observed concentration of product; [P]max is the maximum observed product concentration; k is the observed first order rate constant, and kcat0 is the initial turnover number obtained from Equation 3. Data are presented with standard errors, and R2 is a measure of the goodness of fit.
| Form of ThiH | [P] | [P]max | k | kcat0 | R2 |
|---|---|---|---|---|---|
| μm | ×10−4s−1 | ×10−4s−1 | |||
| ThiGH complex with MTAN | p-Cresol | 220 ± 5 | 16 ± 1 | 37 ± 3 | 0.99 |
| Adenine | 590 ± 24 | 8 ± 1 | 51 ± 6 | 0.99 |
Glyoxylate and Ammonia Inhibit the Tyrosine Cleavage Reaction
A different strategy was required for investigating the potential of dehydroglycine to inhibit tyrosine lyase. The hydrolysis of dehydroglycine is a rapidly reversible equilibrium, and the addition of relatively high concentrations of glyoxylate and ammonium yields a low concentration of dehydroglycine in the solution. This method was used advantageously by Begley and co-workers (11) during studies in which they demonstrated that the addition of glyoxylate and ammonium provided sufficient dehydroglycine to reconstitute the ThiG-dependent cyclization reaction.
Using this approach to examine the effect of dehydroglycine on tyrosine lyase activity, increasing concentrations of glyoxylate and ammonium ions were added to activity assays. To ensure product inhibition by DOA did not disguise any effects of glyoxylate and ammonium, these assays also contained sufficient MTAN to hydrolyze the DOA produced in the assay to adenine and 5′-deoxyribose. The effects of glyoxylate and ammonium were measured using HPLC analysis, measuring tyrosine cleavage by detecting the production of p-cresol and reductive cleavage of AdoMet by detecting the production of adenine. The combination of ammonium and glyoxylate inhibited the cleavage of tyrosine (Fig. 4A) with an apparent IC50 of 440 ± 55 μm and a Hill coefficient of 1.1 ± 0.1, suggesting a 1:1 complex of the inhibitor and the enzyme. Subsequent experiments in which 1 mm of the individual species was added to activity assays showed this inhibition was due solely to glyoxylate and that ammonium had no effect (supplemental Fig. S4).
In contrast, no inhibition of AdoMet cleavage was observed over the whole range of glyoxylate and ammonium concentrations (Fig. 4B). At high glyoxylate and ammonium concentrations (2 mm), greater than 90% of the reductive cleavage was uncoupled, resulting in the accumulation of up to 600 μm adenine but only 40 μm p-cresol. Similar results were obtained in experiments using either ThiGH complex or monomeric ThiH, suggesting the presence of ThiG alone does not influence this inhibition.
Activity Assays with Tyrosine Analogues
To obtain further insight into the mechanism of tyrosine cleavage, the reactivity of structural analogues of tyrosine in ThiGH activity assays was investigated. Analysis of these analogue assays by HPLC allowed two steps in the mechanism to be monitored; the formation of DOA indicated the extent of the reductive cleavage of AdoMet, and the formation of p-cresol or other aromatic products allowed the measurement of the Cα–Cβ bond cleavage step. The negative control samples (which contained ThiGH, AdoMet, and reductant but no tyrosine) were particularly important in this experiment, as very little uncoupled AdoMet cleavage occurred in the absence of tyrosine (less than 10% relative to a positive control sample containing tyrosine). Several tyrosine analogues were investigated using this strategy (Fig. 5 and Table 3). Tyramine 17 and l-phenylalanine 18 were not substrates and did not show an increase in the cleavage of AdoMet relative to the negative control. However, it was found that 4-hydroxyphenylpropionic acid (4-HPPA, 16) and 4-hydroxyphenyl-α-hydroxypropionic acid (4-HPHPA, 15) lead to the formation of ∼50% of the amount of DOA formed compared with the positive control (with tyrosine as the substrate). Careful examination of the HPLC trace confirmed that little or no p-cresol was formed (less than 5% relative to positive control samples). These results suggest 4-HPPA and 4-HPHPA did not undergo Cα–Cβ bond cleavage but are able to support uncoupled AdoMet cleavage.
FIGURE 5.

HPLC analysis of ThiH assays with substrate analogues. A, structure of substrate analogues. B, analytical HPLC traces of tyrosine lyase activity assays with tyrosine analogues, displaced upwards for clarity. The detector measures the absorbance at 280 nm. The traces are for assays with the following (from top to bottom): l-tyrosine 1; 4-HPHPA 15; 4-HPPA 16; tyramine 17; l-phenylalanine 18 and with no substrate (negative control). Comparison with authentic samples identified the following peaks: a, tyrosine; b, DOA; c, 5′-methylthioadenosine (MTA); d, 4-HPHPA; e, 4-HPPA; f, p-cresol.
TABLE 3.
Amount of turnover from substrate analogues
AdoMet cleavage was assessed by monitoring the amount of DOA produced. Values are given as a percentage with respect to the amount of activity observed in a standard assay (including tyrosine as the substrate) that was measured in parallel. Standard errors are given where the results have been replicated.
| Compound | AdoMet cleavage |
|---|---|
| % | |
| l-Tyrosine 1 | 100 |
| 4-Hydroxyphenylpropionic acid 16 | 50.9 ± 3.0 |
| 4-Hydroxyphenyl-α-hydroxypropionic acid 15 | 50.9 ± 6.8 |
| d-Tyrosine | 14.3 |
| 4-Hydroxyphenylcinnamic acid | 7.2 ± 2.2 |
| Tyramine 17 | 5.9 ± 3.0 |
| l-Phenylalanine 18 | 5.4 ± 1.3 |
| 4-Amino-l-phenylalanine | 4.8 ± 0.6 |
| 4-Methyl-l-phenylalanine | 3.0 |
| 4-Fluoro-l-phenylalanine | 2.6 |
| No substrate (negative control) | 6.6 ± 1.7 |
This strategy of analyzing for DOA formation was used to screen further tyrosine analogues (see Table 3). Analogues with different functional groups at the phenol position were investigated, specifically 4-amino-, 4-methyl-, and 4-fluoro-l-phenylalanines. None of these tyrosine analogues increased AdoMet cleavage or yielded detectable aromatic products. Assays with d-tyrosine showed relatively slow uncoupled AdoMet cleavage, but no p-cresol formation was observed. Further experiments in which reaction products or potential substrate mimics were added to activity assays are shown in Table 4. They show that combinations of reaction products do not increase uncoupled AdoMet cleavage unless l-tyrosine is present in the mixture.
TABLE 4.
Amount of turnover measured from combinations of in vitro products or potential substrate mimics
AdoMet cleavage was assessed by monitoring the amount of DOA generated, and Cα—Cβ bond cleavage was assessed by monitoring the amount of p-cresol formed. Values are given as a percentage with respect to the amount of activity observed in a standard assay (including tyrosine as the substrate) that was measured in parallel. Standard errors are given where the results have been replicated. NA means not applicable.
| Compound(s) | AdoMet cleavage | Cα–Cβ bond cleavage |
|---|---|---|
| % | % | |
| l-Tyrosine | 100 | 100 |
| NH4+ + glyoxylate + l-tyrosine | 117 ± 7 | 25.2 ± 5.1 |
| Glycine + p-cresol | 8.4 | NA |
| p-Cresol | 7.8 | NA |
| NH4+ + glyoxylate + p-cresol | 6.9 | NA |
| Glycine | 6.8 | NA |
| NH4+ + glyoxylate | 4.8 | NA |
Activity of Monomeric ThiH
Size exclusion chromatography allowed the purification of ThiGH complex and monomeric ThiH. The natural pathway for thiazole biosynthesis includes efficient transfer of dehydroglycine from ThiH to ThiG (Fig. 1B), and it was therefore of interest to determine the kinetic properties of ThiH in the absence of ThiG. The time courses obtained using HPLC analysis of activity assays with monomeric ThiH are shown in Fig. 6A. The kinetics of product formation from ThiH showed burst phase similar to that observed with ThiGH. Fitting of the data from Fig. 6A to Equation 1 gave a burst phase rate constant (32 ± 8 × 10−4 s−1 for p-cresol) that is marginally slower than for ThiGH but a substantially faster steady state rate (5.9 ± 0.2 × 10−4 s−1 for p-cresol), which is consistent with a faster (but still rate-limiting) product release step. The addition of MTAN to the ThiH assays did not abolish the burst phase but did increase the degree of uncoupled turnover (Table 1). This suggests that for monomeric ThiH, a step other than release of DOA is rate-limiting.
DISCUSSION
Dehydroglycine is a precursor of the vitamin thiamine pyrophosphate in a wide range of prokaryotes (2). It is derived from (at least) two sources. Aerobes use dioxygen as an electron acceptor to facilitate the oxidation of glycine in a reaction catalyzed by ThiO (11, 12). Anaerobes cannot use this pathway, but instead they make use of a radical AdoMet enzyme, tyrosine lyase (ThiH), to break the Cα–Cβ bond of tyrosine to yield p-cresol and dehydroglycine (17, 18). The energetic challenge of using AdoMet to generate and control the deoxyadenosyl radical has been highlighted previously (25, 26). Not only does the reaction need to be isolated in the active site to permit the radical reaction to proceed, the reaction product dehydroglycine must also be protected from the aqueous environment as it is hydrolytically unstable. To use dehydroglycine for the formation of thiazole carboxylate 4, the protection of dehydroglycine must extend to its transfer from ThiH to ThiG. This requirement may explain the functional role of complex formation between ThiG and ThiH.
The time course of product formation (Fig. 2) showed that ThiGH is capable of more than one cycle of tyrosine cleavage, but the enzyme exhibited burst phase kinetics. The observation of burst phase kinetics indicates a rate-limiting step that occurs after chemical catalysis and can be due to product release or a conformational change (27). ThiGH yields four reaction products, methionine, DOA, p-cresol, and dehydroglycine, providing potential for a complex pattern of product inhibition. Earlier in vitro studies have determined that tyrosine lyase activity was inhibited by accumulation of DOA and methionine (19) and that this could be overcome by the addition of MTAN, a nucleosidase that hydrolyzes DOA. The observed changes in the time course of product formation in the presence of MTAN (Fig. 3) indicates a change in the rate-determining step during subsequent turnovers and suggests an ordered sequence of product release from ThiGH. The addition of MTAN gave a profile of product formation that could be fitted to a single rate constant across the whole time course. The turnover number derived from this rate constant was similar to that of the burst phase when MTAN was not present (Tables 1 and 2). The rate-limiting step for ThiH reaction catalysis is unknown, but for anaerobic sulfatase-maturating enzyme, another member of the radical AdoMet family, the observed deuterium isotope effect indicates that hydrogen atom abstraction by the 5′-deoxyadenosyl radical is the rate-limiting step (28). The burst phase rate constant obtained for ThiH reaction catalysis (53 ± 6 × 10−4 s−1) is comparable with other members of the radical AdoMet family; for example, apparent rate constants have been reported as 60 × 10−4 and 12 × 10−4 s−1 for the AtsB (formylglycine-generating enzyme) (29) and biotin synthase (30), respectively.
The hydrolytic instability of dehydroglycine implies that a tight interaction with ThiGH would be advantageous, helping to sequester the imine away from the aqueous medium. The observed inhibition of tyrosine cleavage by glyoxylate may indicate competitive binding to the active site, but further rationalization of this observation is difficult in the absence of structural data. A more surprising observation was the almost complete (>90%) uncoupling of AdoMet turnover from the tyrosine cleavage reaction in the presence of relatively high concentrations of glyoxylate and ammonium ions (2 mm each). This apparently wasteful process is unlikely to occur in a cellular context, and the effect was maximized under specific in vitro conditions with the addition of MTAN plus high concentrations of glyoxylate and ammonium. However, the experiment does indicate that in cells where ThiGH is not actively synthesizing the thiazole carboxylate 4, one of the functions of product inhibition by DOA may be to reduce the uncoupled turnover of AdoMet. The release of dehydroglycine observed in vitro is unlikely to occur during cellular thiamine biosynthesis, when transfer of the dehydroglycine probably occurs directly between ThiH and ThiG. The full biosynthetic reaction pathway for ThiGH (which additionally requires ThiS thiocarboxylate 12 and 1-deoxyxyulose 5-phosphate 3, see Fig. 1) may be faster than the steady state rate observed in vitro for the partial reaction, making it difficult to assess the physiological relevance of the slow steady state rate. Although evidence has been found in these studies for product inhibition of tyrosine lyase by both DOA and dehydroglycine, the complexity of the reaction catalyzed by ThiGH does not allow the description of a definitive kinetic model for product release at this stage.
The problem of uncoupled AdoMet cleavage in radical AdoMet proteins is well documented (26). The reactivity of the primary 5′-deoxyadenosyl radical makes its uncontrolled formation potentially hazardous for the cell. There are several mechanisms by which this family of enzymes regulate uncoupled AdoMet cleavage, including modifying the redox potential of the 4Fe-4S cluster in response to substrate binding (25) or by cooperative substrate binding (31). The nature of the reductant has been proposed to have a role in the degree of uncoupled AdoMet cleavage with less uncoupling being observed when using the natural electron donor systems. Our studies made use of the NADPH, flavodoxin, and flavoprotein:NADPH oxidoreductase system, which is assumed to be the natural intracellular reductant for E. coli ThiGH.
The experiments with tyrosine analogues and reaction products (Fig. 5 and Tables 3 and 4) had two related objectives as follows: to identify the structural features that were essential for the turnover of tyrosine and to elucidate the factors that promoted uncoupled turnover of AdoMet. With regard to understanding the uncoupled turnover, several negative controls were important as follows: incubation of ThiGH with the reductant and AdoMet in the absence of tyrosine resulted in very little uncoupled turnover (less than 10% relative to assays with tyrosine present), and the addition of reaction products, again in the absence of tyrosine, did not increase the uncoupled cleavage of AdoMet (Table 4). Comparing the tyrosine structural analogues, all of the analogues that replaced the phenol functional group were inactive, and it appears that ThiH is very sensitive to modifications at this position. Fluorine has 97% of the van der Waals radius of oxygen, but 4-fluoro-l-phenylalanine was not sufficiently similar to l-tyrosine to increase AdoMet cleavage above the background level. Although the ArO–H bond is weak in phenols such as tyrosine, a comparison of bond dissociation energy of phenol (ArO–H, 360 kJ mol−1), aniline (ArNH–H, 388 kJ mol−1), and toluene (ArCH2–H, 355 kJ mol−1) (32) indicate that 4-amino- and 4-methyl-l-phenylalanine ought to be susceptible to hydrogen atom abstraction. As neither of these analogues resulted in AdoMet cleavage, the subtle substrate selectivity of tyrosine lyase cannot be based purely on the bond strength. The substrates that permitted AdoMet cleavage, l- and d-tyrosine, 4-HPPA 16, and 4-HPHPA 15 (Fig. 5 and Table 3) share a common structural motif of being 4-hydroxyphenylpropionic acids, and the phenol appears to be required for AdoMet cleavage.
The observed product inhibition, uncoupled turnover, and studies with these tyrosine analogues can be integrated with a mechanistic model (Fig. 7). The strong preference shown by tyrosine lyase for a phenolic substrate does not provide evidence for concerted AdoMet cleavage and phenolic hydrogen atom abstraction, but it does highlight the importance of a phenolic O–H bond in the active site as a prerequisite for AdoMet cleavage. The formation of a tyrosine radical is thermodynamically favorable as a result of the reactivity of the primary deoxyadenosyl radical, the strength of the C–H bond formed in 5′-deoxyadenosine and relative weakness of the phenolic O–H bond. The model proposes two possible fates for the phenolic radical as follows: either turnover leading to product formation or completion of a futile cycle leading back to tyrosine. During turnover leading to product formation, the electron-deficient phenolic radical can undergo Cα–Cβ bond cleavage with the assistance of the lone pair of electrons on the α-amine of tyrosine. Such a mechanism provides a direct route to dehydroglycine and the resonance stabilized radical anion 21a↔ 21b that requires reduction and protonation to form p-cresol 8. To achieve uncoupled turnover, the phenolic radical 19 is proposed to undergo immediate reduction followed by protonation of the phenoxide 20, giving a direct route back to tyrosine. In this model, the difference between the uncoupled and product-forming pathways depends on the timing of the reduction step (reduction of radical 19 or 21). In the case of d-tyrosine and the analogues 4-HPPA and 4-HPHPA, the Cα–Cβ bond does not undergo cleavage, and these substrates are limited to futile cycling with all of the AdoMet cleavage being uncoupled. A precise stereoelectronic explanation for the observed substrate selectivity will require a structural model of the ThiH-active site. The formation of p-cresol from l-tyrosine requires a reduction step, in addition to the reductive cleavage of AdoMet, which is unusual for a radical AdoMet protein (33). The immediate source of the additional reducing equivalent is unknown, but the [4Fe-4S] cluster that is used to accelerate the reductive cleavage of AdoMet is a possible candidate.
FIGURE 7.

Proposed mechanism for tyrosine lyase. The reductive cleavage of AdoMet yields the 5′-deoxyadenosyl radical that can abstract a hydrogen atom from the phenolic O–H bond. The resultant phenolic radical 19 can follow two possible reaction pathways, and the nature of the group R can affect which of these two pathways is favored. Immediate reduction, followed by protonation, completes an unproductive futile cycle back to the substrate. For l-tyrosine (R = NH2), cleavage of the Cα–Cβ bond yields dehydroglycine and the resonance-stabilized radical anion 21a↔ 21b, which requires the addition of two protons and an electron to give p-cresol 8.
During the time course with l-tyrosine as a substrate, the burst phase of the reaction is efficiently coupled (up to 90%), but during the steady state phase, the ratio of coupled to uncoupled reaction pathways is 1.7:1. Uncoupled turnover is further increased by the addition of MTAN and becomes dominant when glyoxylate and ammonia are added to the assay. One interpretation of these data is that dehydroglycine is able to bind to the ThiGH complex, and this suppresses the Cα–Cβ bond cleavage step. The mechanism by which dehydroglycine and/or DOA might modulate the ratio of coupled to uncoupled activities is not clear but may result from dehydroglycine remaining bound at the interface between ThiG and ThiH. This provides a convenient mechanism to ensure tyrosine cleavage catalyzed by ThiH is synchronized with the incorporation of dehydroglycine by ThiG into the thiazole carboxylate 14.
In conclusion, catalytic turnover of the tyrosine lyase activity of E. coli ThiGH complex has been demonstrated. However, tyrosine cleavage is strictly controlled by the accumulation of the products, which results in a burst phase kinetic profile. The function of this product inhibition in cells actively synthesizing thiamine may be to coordinate the rate of formation of dehydroglycine by ThiH with its utilization by ThiG for thiazole formation. Structural analogues of tyrosine have been used to define the substrate requirements of the enzyme and in particular the need for a phenolic O–H bond. Furthermore, phenolic tyrosine analogues that lacked a correctly positioned amine functional group were able to undergo a partial reaction leading to uncoupled AdoMet cleavage but with no evidence for Cα–Cβ bond cleavage.
Acknowledgments
We thank R. J. Wood and J. C. McKelvie for helpful discussions.
This work was supported by the Biotechnology and Biological Sciences Research Council, the Royal Society, and the University of Southampton.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4 and Tables S1 and S2.
- TPP
- thiamine pyrophosphate
- AdoMet
- S-adenosylmethionine
- HPLC
- high pressure liquid chromatography
- DOA
- 5′-deoxyadenosine
- MTAN
- 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase
- 4-HPPA
- hydroxyphenylpropionic acid
- 4-HPHPA
- 4-hydroxyphenyl-α-hydroxypropionic acid
- MOPS
- 4-morpholinepropanesulfonic acid.
REFERENCES
- 1.Pohl M., Sprenger G. A., Müller M. (2004) Curr. Opin. Biotechnol. 15, 335–342 [DOI] [PubMed] [Google Scholar]
- 2.Jurgenson C. T., Begley T. P., Ealick S. E. (2009) Annu. Rev. Biochem. 78, 569–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanes J. W., Ealick S. E., Begley T. P. (2007) J. Am. Chem. Soc. 129, 4860–4861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hazra A., Chatterjee A., Begley T. P. (2009) J. Am. Chem. Soc. 131, 3225–3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCulloch K. M., Kinsland C., Begley T. P., Ealick S. E. (2008) Biochemistry 47, 3810–3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez-Gomez N. C., Downs D. M. (2008) Biochemistry 47, 9054–9056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chatterjee A., Li Y., Zhang Y., Grove T. L., Lee M., Krebs C., Booker S. J., Begley T. P., Ealick S. E. (2008) Nat. Chem. Biol. 4, 758–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dorrestein P. C., Huili, Zhai H., Taylor S. V., McLafferty F. W., Begley T. P. (2004) J. Am. Chem. Soc. 126, 3091–3096 [DOI] [PubMed] [Google Scholar]
- 9.Settembre E. C., Dorrestein P. C., Zhai H., Chatterjee A., McLafferty F. W., Begley T. P., Ealick S. E. (2004) Biochemistry 43, 11647–11657 [DOI] [PubMed] [Google Scholar]
- 10.Chatterjee A., Han X., McLafferty F. W., Begley T. P. (2006) Angew. Chem. Int. Ed. Engl. 45, 3507–3510 [DOI] [PubMed] [Google Scholar]
- 11.Park J. H., Dorrestein P. C., Zhai H., Kinsland C., McLafferty F. W., Begley T. P. (2003) Biochemistry 42, 12430–12438 [DOI] [PubMed] [Google Scholar]
- 12.Settembre E. C., Dorrestein P. C., Park J. H., Augustine A. M., Begley T. P., Ealick S. E. (2003) Biochemistry 42, 2971–2981 [DOI] [PubMed] [Google Scholar]
- 13.Sofia H. J., Chen G., Hetzler B. G., Reyes-Spindola J. F., Miller N. E. (2001) Nucleic Acids Res. 29, 1097–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leonardi R., Fairhurst S. A., Kriek M., Lowe D. J., Roach P. L. (2003) FEBS Lett. 539, 95–99 [DOI] [PubMed] [Google Scholar]
- 15.Martinez-Gomez N. C., Robers M., Downs D. M. (2004) J. Biol. Chem. 279, 40505–40510 [DOI] [PubMed] [Google Scholar]
- 16.Leonardi R., Roach P. L. (2004) J. Biol. Chem. 279, 17054–17062 [DOI] [PubMed] [Google Scholar]
- 17.Kriek M., Martins F., Leonardi R., Fairhurst S. A., Lowe D. J., Roach P. L. (2007) J. Biol. Chem. 282, 17413–17423 [DOI] [PubMed] [Google Scholar]
- 18.Kriek M., Martins F., Challand M. R., Croft A., Roach P. L. (2007) Angew. Chem. Int. Ed. Engl. 46, 9223–9226 [DOI] [PubMed] [Google Scholar]
- 19.Challand M. R., Ziegert T., Douglas P., Wood R. J., Kriek M., Shaw N. M., Roach P. L. (2009) FEBS Lett. 583, 1358–1362 [DOI] [PubMed] [Google Scholar]
- 20.Cornell K. A., Riscoe M. K. (1998) Biochim. Biophys. Acta 1396, 8–14 [DOI] [PubMed] [Google Scholar]
- 21.Fish W. W. (1988) Methods Enzymol. 158, 357–364 [DOI] [PubMed] [Google Scholar]
- 22.McIver L., Leadbeater C., Campopiano D. J., Baxter R. L., Daff S. N., Chapman S. K., Munro A. W. (1998) Eur. J. Biochem. 257, 577–585 [DOI] [PubMed] [Google Scholar]
- 23.Grant B. D., Adams J. A. (1996) Biochemistry 35, 2022–2029 [DOI] [PubMed] [Google Scholar]
- 24.Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 25.Wang S. C., Frey P. A. (2007) Biochemistry 46, 12889–12895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duschene K. S., Veneziano S. E., Silver S. C., Broderick J. B. (2009) Curr. Opin. Chem. Biol. 13, 74–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Segel I. H. (1993) Enzyme Kinetics, John Wiley & Sons Ltd., Chichester, UK [Google Scholar]
- 28.Benjdia A., Leprince J., Sandström C., Vaudry H., Berteau O. (2009) J. Am. Chem. Soc. 131, 8348–8349 [DOI] [PubMed] [Google Scholar]
- 29.Grove T. L., Lee K. H., St Clair J., Krebs C., Booker S. J. (2008) Biochemistry 47, 7523–7538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor A. M., Farrar C. E., Jarrett J. T. (2008) Biochemistry 47, 9309–9317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ugulava N. B., Frederick K. K., Jarrett J. T. (2003) Biochemistry 42, 2708–2719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fossey J., Lefort D., Sorba J. (1995) Free Radicals in Organic Chemistry, John Wiley & Sons Ltd., Chichester, UK [Google Scholar]
- 33.Wang S. C., Frey P. A. (2007) Trends Biochem. Sci. 32, 101–110 [DOI] [PubMed] [Google Scholar]