

Abstract
Linoleate diol synthases (LDS) are heme enzymes, which oxygenate 18:2n-6 sequentially to (8R)-hydroperoxylinoleic acid ((8R)-HPODE) and to (5S,8R)-dihydroxy-, (7S,8S)-dihydroxy-, or (8R,11S)-dihydroxylinoleic acids (DiHODE). The genome of the rice blast fungus, Magnaporthe oryzae, contains two genes with homology to LDS. M. oryzae oxidized 18:2n-6 to (8R)-HPODE and to (7S,8S)-DiHODE, (6S,8R)-DiHODE, and (8R,11S)-HODE. Small amounts of 10-hydroxy-(8E,12Z)-octadecadienoic acid and traces of 5,8-DiHODE were also detected by liquid chromatography-mass spectrometry. The contribution of the 7,8-LDS gene to M. oryzae pathogenicity was evaluated by replacement of the catalytic domain with hygromycin and green fluorescent protein variant (SGFP) cassettes. This genetically modified strain Δ7,8-LDS infected rice leaves and roots and formed appressoria and conidia as the native fungus. The Δ7,8-LDS mutant had lost the capacity to biosynthesize all the metabolites except small amounts of 8-hydroxylinoleic acid. Studies with stereospecifically deuterated linoleic acids showed that (8R)-HPODE was formed by abstraction of the pro-S hydrogen at C-8 and antarafacial oxygenation, whereas (7S,8S)-DiHODE and (8R,11S)-DiHODE were formed from (8R)-HPODE by suprafacial hydrogen abstraction and oxygenation at C-7 and C-11, respectively. A mac1 suppressor mutant (Δmac1 sum1–99) of M. oryzae, which shows cAMP-independent protein kinase A activity, oxygenated 18:2n-6 to increased amounts of (10R)-HPODE and (5S,8R)-DiHODE. Expression of the 7,8-LDS gene but not of the second homologue was detected in the suppressor mutant. This suggests that PKA-mediated signaling pathway regulates the dioxygenase and hydroperoxide isomerase activities of M. oryzae.
Keywords: Enzymes/Mechanisms, Enzymes/Oxidase, Lipid/Oxidation, Metabolism/Fatty Acid, Methods/HPLC, Methods/Mass Spectrometry, Organisms/Fungi, Oxygen/Peroxidation
Introduction
Magnaporthe oryzae is an ascomycete fungus, which causes rice blast disease (1, 2).2 Rice blast was first described in China and Japan over 300 years ago. The disease is difficult to control, and it remains a serious problem. In Japan, rice blast annually reduces the rice crop by ∼25% (2, 3). M. oryzae has also been found on other continents where wheat, barley, and pearl millet also can be infected. As a consequence of its biological importance, M. oryzae was the first fungal plant pathogen that was sequenced (4).3 The haploid genome (∼40 Mb) is estimated to contain about 11,000 genes in seven chromosomes (5). This study focuses on a linoleate diol synthase (LDS)4 of M. oryzae, which is homologous to LDS of important plant and human fungal pathogens.
LDS was first described in the take-all fungus of wheat, Gaeumannomyces graminis (6). This enzyme (EC 1.13.11.44), oxidizes 18:2n-6 sequentially to (8R)-HPODE and to (7S,8S)-DiHODE (6–8) and was designated 7,8-LDS. (8R)-HPODE is formed by hydrogen abstraction and antarafacial oxygenation at C-8, whereas the diol is formed sequentially by intramolecular suprafacial oxygenation at C-7 by the N-terminal cytochrome P450 domain of LDS, as summarized in Fig. 1 (8, 9). Sequence homology, the reaction mechanism, and results of site-directed mutagenesis suggest that 7,8-LDS and mammalian cyclooxygenases have mechanistic and structural similarities (8, 10–13).
FIGURE 1.
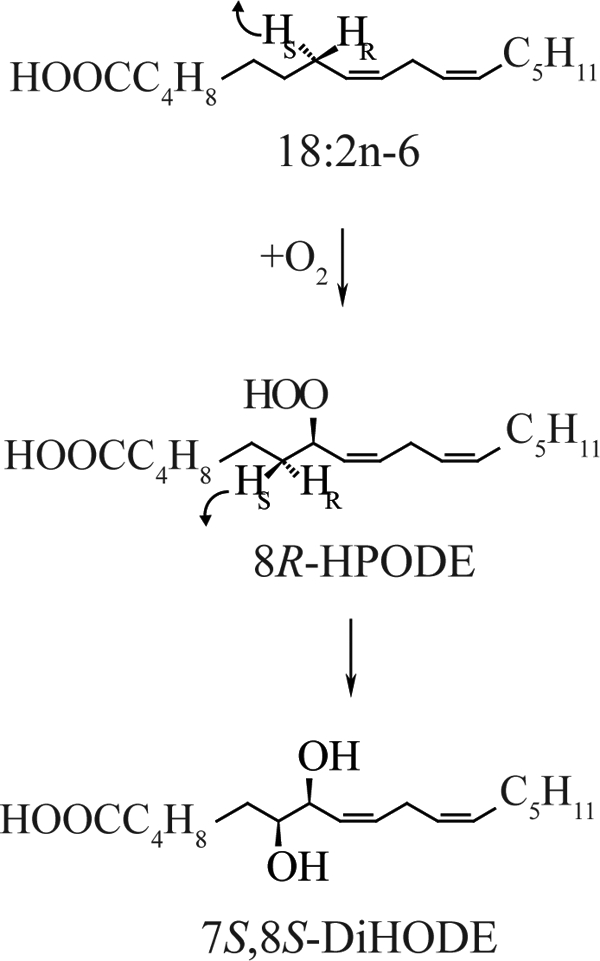
Mechanism of oxidation of linoleic acid to (7S,8S)-DiHODE by 7,8-LDS of G. graminis.
Genes with homology to 7,8-LDS are present in M. oryzae, Magnaporthe aspergilli, and other filamentous fungi (14, 15). Two of them have been studied by gene expression and gene deletion, (10R)-DOX of Aspergillus nidulans and Aspergillus fumigatus (16, 17) and 5,8-LDS of A. nidulans and A. fumigatus (16, 18).5 These enzymes oxygenated 18:2n-6 to (8R)-HPODE or (10R)-HPODE by abstraction of the pro-S hydrogen at C-8 followed by antarafacial oxygen insertion at C-8 by LDS or at C-10 with double bond migration in the case of (10R)-DOX (16). (8R)-HPODE is isomerized to (5S,8R)-DiHODE by 5,8-LDS and to (8R,11S)-DiHODE by A. fumigatus. 8,11-LDS of A. fumigatus has not been identified by gene deletion nor by gene expression. (5S,8R)-DiHODE and (8R,11S)-DiHODE appear to be formed from (8R)-HPODE by the same mechanism as (7S,8S)-DiHODE (7, 16, 19), viz. suprafacial hydrogen abstraction and hydroxylation.
Studies on the biological function of fungal oxylipins started 20 years ago in A. nidulans. (8R)-HODE and (5S,8R)-DiHODE were found to induce premature sexual sporulation (20). Gene deletion of 5,8-LDS and (10R)-DOX in A. nidulans and A. fumigatus was later found to influence sporulation, development, mycotoxin production, and pathogenicity (16, 21, 22). In contrast, little is known about the biological function of 7,8-LDS of G. graminis. The hyphae of G. graminis penetrate and destroy wheat roots, and the take-all disease causes significant economical losses (23). The genome of G. graminis is not yet available. Transformation and genetic manipulation of G. graminis are difficult and preclude gene deletion studies of 7,8-LDS. We therefore turned our attention to the genetically tractable M. oryzae, another fungal grass pathogen of the Magnaporthaceae family (24), to better understand the biological roles of the 7,8-LDS. Amino acid identities between deduced oxygenases of the two genes of M. oryzae with homology to 7,8-LDS of G. graminis and fatty acid oxygenases of aspergilli are summarized in Table 1. M. oryzae has previously been found to oxygenate 18:2n-6 to 7,8-DiHODE and 8-HODE, and the transcript of the tentative 7,8-LDS gene was sequenced (14).6
TABLE 1.
Oxygenases of M. oryzae with homology to 7,8-LDS of G. graminis and (10R)-DOX, 5,8-, and 8,11-LDS of A. fumigatus
| Locus, GenBankTM (hypothetical proteins) | Percent identitya |
|||
|---|---|---|---|---|
| 7,8-LDSgg | 5,8-LDSaf | 8,11-LDSaf | (10R)-DOXaf | |
| % | ||||
| MGG_13239 | 63 | 48 | 39 | 40 |
| MGG_10859 | 41 | 49 | 48 | 48 |
a Alignments were performed with the ClustalW algorithm. 7,8-LDSgg is 7,8-LDS of G. graminis var. triticie; 5,8-LDSaf and 8,11-LDSaf designate 5,8-LDS and the tentatively identified 8,11-LDS of A. fumigatus (16), respectively; (10R)-DOXaf designates the (10R)-DOX enzyme (GenBankTM accession number XP_75170, XP_754409, and XP_746438).
M. oryzae is a model organism for studies of fungal diseases of blasts (25), and it also infects rice roots (26). The spores of M. oryzae attach to the hydrophobic surface of leaves and contain all that is needed to develop an injection apparatus for cuticle penetration, designated an appressorium with penetrating hyphae. In this process, the conidium undergoes apoptosis with transfer of lipid bodies, trehalose, and glycerol to the appressorium. The subsequent mobilization and oxidation of fatty acids of lipid bodies are controlled by PKA, MAPK, triacylglycerol lipases, and β-oxidation (27–32). Glycerol accumulates and generates the unprecedented internal turgor pressure of the appressorium (27). These processes are deranged in mutants of the cAMP-protein kinase cascade (29). Whether LDS enzymes are involved in lipid mobilization and pathogenicity and can be regulated by kinases are unknown.
The first goal of this study was to characterize the oxygenation of 18:2n-6 by M. oryzae with respect to all products, oxygenation mechanisms, and stereochemistry. Our second goal was to use gene targeting to identify and study the biological importance of 7,8-LDS during rice infection and sporulation. The third goal was to determine whether LDS activity is influenced by the cAMP-mediated signaling pathway. Finally, we also assayed oxygenation of unsaturated fatty acids by mycelia to identify the major products formed by cytochrome P450.
EXPERIMENTAL PROCEDURES
Materials
Fatty acids were dissolved in ethanol and stored in stock solutions (50–100 mm) at −20 °C. 18:2n-6 (99%), 18:3n-6 (99%), and 18:3n-3 (99%) were from VWR, all other fatty acids (98–99%) were from Larodan (Malmö, Sweden). (7R)-[2H]18:2n-6, (8R)-[2H]18:2n-6, and (11S)-[2H]18:2n-6 were prepared as described previously (7, 16). (13R)-[2H4]HODE, (8R)-HPODE, and stereoisomers of 8,11-DiHODE were prepared and purified as described previously (19). Chemically competent Escherichia coli (Top10), DNA ladders, reverse transcriptase (Superscript III), and pCR2.1 were from Invitrogen. pGEM5Zf+ and DNase I were obtained from Promega, and pCAMBIA0380 was from CAMBIA (Canberra, Australia). Phusion DNA polymerase was from Finnzymes, and GeneJET cloning kit was from Fermentas. Taq DNA polymerase, hexadecyltrimethylammonium bromide, cefotaxime, hygromycin B, kanamycin, 3,5-dimethoxy-4-hydroxyacetophenone, and tetranitromethane were from Sigma. Restriction enzymes were from New England Biolabs and Fermentas. Plasmid Midi kits, and QIAquick gel extraction kits were from Qiagen. SYBR Green Supermix and RNA StdSens analysis kit were from Bio-Rad. Agrobacterium tumefaciens (strain AGL-1) were obtained as described previously (26).
Fungal Strains and Growth
M. oryzae Guy11 was used in the LDS gene deletion study. M. oryzae was grown in liquid culture for 7–14 days in complete medium, minimal medium (33), minimal medium without glucose, or without nitrate salts (34). Conidia were prepared from mycelia grown for 8–12 days on complete medium agar at 22 °C under 16-h light and 8-h dark cycles of fluorescent light (True-light). The agar plates were flooded with distilled water and harvested with a glass spreader, and the conidial suspensions were filtered through one layer of Miracloth.
The following genetically modified strains of M. oryzae were studied: deletion of adenylyl cyclase MAC1 gene (Δmac1) (35); deletion of the adenylyl cyclase MAC1 gene with a suppressor mutation in the cAMP binding domain of the regulatory subunit of PKA (Δmac1 sum1–99 (29)); deletion of the catalytic subunit of protein kinase A (Δcpka) (36); deletion of PMK1 (PMK1, pathogenicity MAPK 1) (31); and deletion of the MAPK substrate and transcription factor MST12 (32). The mycelium was ground up in liquid nitrogen and homogenized, and the supernatant was assayed for enzyme activity with 18:2n-6 as substrate (see below). The experiment was performed in triplicate.
Enzyme Assays
Mycelia (0.5–20 g wet weight) of M. oryzae were incubated in 0.1 m sodium borate buffer (pH 8.0) with 0.5–1 mg of fatty acids ml−1 (4–5 h, 21 °C), as described previously (14). Mycelia were also ground up in liquid nitrogen, and the fine powder was stored at −80 °C. The nitrogen powder was homogenized (glass-Teflon, 10 passes; 4 °C) in 10 volumes (w/v) of 0.1 mm KHPO4 buffer (pH 7.3), 2 mm EDTA, 0.04% Tween 20, centrifuged at 13,000 × g (10 min, 4 °C), and used immediately for enzyme assay. Microsomal fractions and high speed supernatants were prepared by centrifugation (100,000 × g, 60 min; 4 °C) of low speed supernatant and assayed for enzyme activity. An aliquot (0.5–1 ml) was incubated with 100 μm of fatty acids for 30–40 min on ice. In some experiments, 50 pmol of (13R)-[2H4]HODE was added as an internal standard. The products were extracted with ethyl acetate or on SepPak/C18 (19). Triphenylphosphine or NaBH4 was used to reduce hydroperoxides to alcohols to simplify analysis. The products from incubations with mycelia were purified by TLC (Kieselgel 60, Merck) or by preparative RP-HPLC.
Gene Replacement Construct
The gene replacement construct of 7,8-LDS is outlined in Fig. 2. Standard procedures of molecular biology methods were used (37). SGFP was excised from pCAMBgfp (26) with NcoI and NotI and cloned into pGEM5Zf+, yielding pGEMgfp. A 1424-bp SalI-fragment from pCAMBgfp containing a hygromycin B resistance cassette (hph, which encodes hygromycin phosphotransferase, controlled by the TrpC promoter) was ligated downstream of SGFP in pGEMgfp (yielding pGEMgfp/hph). The flanking regions of 7,8-LDS (MGG_13239) were obtained by PCR. Genomic DNA was prepared from M. oryzae Guy11 (38). The primers MG_F-5′-UTR (5′-gggcccGAATTCTTCTAGCCTGAAGAA-3′) and MG_R-5′-UTR (5′-atgggcccCGAGCTTTTGGAAGAATG-3) with restriction sites for ApaI (underlined) amplified 1375 bp of 5′-UTR and the first 87 bp of the coding region of 7,8-LDS. This fragment was cloned, excised (ApaI), and ligated in-frame and upstream of SGFP in pGEMgfp/hph (under control of the 7,8-LDS promotor). Primers MG_F-3′-UTR (5′-cagagctcTCAAGACGGAGAATGGAG) and MG_R-3′-UTR (5′-gagctcCCGGGAACATCAAAAAGTTCAG) with restriction sites for SacI (underlined) amplified the last 1206 bp of 7,8-LDS coding sequence and 55 bp of its 3′-UTR. This fragment was cloned, excised (SacI), and ligated into pGEM-5′-UTR-gfp/hph, resulting in the gene deletion construct (5′-UTR-gfp-hph-3′-UTR; Fig. 2). The construct (4917 bp) was restricted (EcoRI and SmaI) and ligated into pCAMB0380, and the plasmid was designated pCAMBΔ7,8-LDS. This plasmid was introduced into chemically competent A. tumefaciens AGL-1 by heat shock.
FIGURE 2.

Mutant Δ7,8-LDS was produced by homologous recombination using A. tumefaciens-mediated transformation. 2.5 kb of the LDS gene, including putative heme ligands of 7,8-LDS, was replaced with SGFP and TrpC/hph cassettes. The modified strains were characterized by PCR analysis, Southern hybridization, and by analysis of enzymatic activity.
A. tumefaciens-mediated Transformation
Competent A. tumefaciens containing pCAMBΔ7,8-LDS was used to transform M. oryzae Guy11 conidia (39). Selection medium agar consisted of complete medium supplemented with 250 μg/ml hygromycin B and with 200 μm cefotaxime to eliminate A. tumefaciens. Transformants were grown individually on selection medium in 24-well plates before analysis.
Analysis of Δ7,8-LDS Transformants
Transformants were analyzed by PCR, Southern blot, and for expression of LDS activity. PCR was used as a first screening to distinguish between homologous and nonhomologous recombination. Primers F-MG-43646 (5′-GGTTGTTTGTAGTACTGCAGCAGC) and R-SGFP-149 (5′-GCAGATGAACTTCAGGTGCAGCTT) amplified a fragment of 1736 bp in transformants resulting from homologous recombination. This primer pair would generate amplicons of different size, or no amplicon at all, in ectopic transformants. Primers F-Δlds (5′-AACGGCAACGGTATACATCAGAAC-3) and R-Δlds (5′-TATTGCCAAATGTTTGAACGATCGG-3′) generated amplicons of 1105 bp in ectopic transformants, because the R-Δlds primer binds to a sequence in the T-DNA of pCAMBΔ7,8-LDS outside of the gene replacement construct. Confirmation of the integration pattern was determined by Southern hybridization (see supplemental material). Finally, the deletion mutants of 7,8-LDS were assayed for enzyme activity, as above.
PCR Analysis of Gene Transcripts in M. oryza, the Δmac1 Mutant, and the Suppressor Mutant (Δmac1 sum1–99)
Total RNA was prepared from nitrogen powder of mycelia (grown for 7–14 days) using LiCl extraction (17) and treated with DNase I. Quality was assessed on the Experion Automated Electrophoresis System (Bio-Rad). First-strand cDNA was synthesized using Superscript III. Real time PCR was performed in an iCycler (Bio-Rad) with SYBR Green Supermix. 500 nm forward and reverse primers and ∼200 ng of cDNA were used in 50 μl. Primers used for the detection of MGG_13239 were 5′-AGCCTTCAACACGTTGATGAAG (forward) and 5′-GGAGGAACGTCGAGTCCTTG (reverse) and for MGG_10859 were 5′-ACCGCGTCTTTGTATCCTTTG (forward) and 5′-CATCTCGGTGATGGCAATCTG (reverse). Both primers were designed to bridge an intron to distinguish amplification of cDNA from any contaminating genomic DNA. Primers for detection of actin as housekeeping gene (40) were 5′-CCTGGCACCGTCGTCGATGAAGG and 5′-GCGAGGCGAGAATGGAACCACCG. Cycling conditions were as follows: 95 °C, 3 min followed by 50 cycles (95 °C, 30 s; 58 °C, 30 s; 72 °C, 30 s). Melting point curve analysis and agarose gel electrophoresis verified amplification of one single product of the expected size. Cycle threshold (CT) values were obtained from the iCycler software (Bio-Rad).
Infection Assays
Rice (Oryzae sativa spp. indica var. CO39) seedlings were grown at 85% relative humidity, 25 °C, and 16-h light/8-h dark photoperiod. Two-week-old plants with second and third expanded leaves were used for leaf infection assays. Plants were spray-inoculated with 2 ml of a suspension of 105 conidia/ml with 0.25% gelatin per pot. Symptoms were scored after 5 days.
Root infection assays were carried out using thick and moist vermiculite. Wet vermiculite was prepared by immersing in distilled water (2 h), and excess water was then removed by a sieve. We filled a 50-ml Falcon tube with 30 cm of wet vermiculite, followed by a mycelial plug with the same diameter as the Falcon tube, a further layer of 5 cm of wet vermiculite, and five rice seeds of the same strain covered with the other 5-cm layer of wet vermiculite. The tube was sealed with parafilm to prevent loss of humidity. Lesions were scored and compared with wild type strain Guy11 after 15 days.
Appressorium Formation
Aliquots (150 μl) of conidia suspensions (104 spores/ml) were placed on plastic microscope coverslips (BDH, Germany) and incubated under humid conditions at 25 °C in 16-h light/8-h dark photoperiod. At least 200 conidia per strain were examined microscopically for germination and formation of appressoria in each experiment.
Radial Growth and Asexual Sporulation
Mycelial plugs of M. oryzae Guy11 and Δ7,8-LDS were placed in triplicates on agar plates with complete and minimal medium. The fungal growth area, and the number of conidia were determined after 10 days (22 °C, fluorescent light as above). The conidia were harvested as above, and the conidia were counted in a hemocytometer and corrected for the fungal growth area.
LC- and GC-MS Analysis
RP-HPLC with MS/MS analysis was performed with a Surveyor MS pump (ThermoFisher) and an analytical octadecyl silica column (5 μm; 2.0 × 150 mm; Phenomenex), which was usually eluted with methanol/water/acetic acid, 800:200:0.05, at 0.3 ml/min. The effluent was subject to electrospray ionization in an ion trap mass spectrometer (LTQ, ThermoFisher). The heated transfer capillary was set at 315 °C, the ion isolation width at 1.5, and the collision energy at 25–35 (arbitrary scale). Prostaglandin F1α (100 ng/min) was infused for tuning. Products formed from stereospecifically deuterated 18:2n-6 were analyzed as described previously (16), and the deuterium content of the recovered 18:2n-6 was determined (LC-MS/MS analysis; m/z 276–282→ full scan). For preparative use, we employed a large column (20 × 150-mm Reprosil 100 ODS-A, 5 μ; Dr. Maisch, Ammerbuch, Germany), eluted with 85–90% methanol in water with 0.1% acetic acid.
Normal phase HPLC with MS/MS analysis was performed in a silicic acid column (5 μm; Kromasil 100SI, 250 × 2 mm, Dalco Chromtech) using 1% isopropyl alcohol in hexane for separation of hydroxy fatty acids and 7% isopropyl alcohol in hexane for separation of DiHODE (0.3–0.5 ml/min; Constametric 3200 pump, LDC/MiltonRoy). The effluent was combined with isopropyl alcohol/water (3:2; 0.2–0.3 ml/min) from a second pump (Surveyor MS pump (16)). The combined effluents were introduced by electrospray ionization into the ion trap mass spectrometer (LTQ, ThermoFisher). Steric analysis of 8-HODE and 10-HODE was performed by chiral phase HPLC-MS/MS (19).
GC-MS analysis, hydrogenation, and synthesis of methyl ester and TMS ether derivatives were performed as described previously (41, 42). Carbon values were estimated from the retention times of saturated fatty acid methyl esters (41).
RESULTS
7,8-LDS Activity
Mycelia of M. oryzae Guy11 and nitrogen powder preparations of mycelia transformed 18:2n-6 to 8-HPODE, 8-HODE, and 7,8-DiHODE as major metabolites (Fig. 3A) in agreement with previous reports (14, 43) and to 6,8-DiHODE. 6,8-DiHODE eluted on the left shoulder of 7,8-DiHODE but could be separated from the latter by preparative LC-MS (Fig. 3B). The relative amounts of these metabolites were ∼5 and ∼95%, respectively. The MS/MS spectrum of 6,8-DiHODE (Fig. 3C) showed structurally important signals at m/z 173 (−OOC-(CH2)4-CHOH-CH2-COH), m/z 155 (173–18), and m/z 129 (−OOC-(CH2)4-COH). The MS/MS spectrum of 6,8-[U-13C]DiHODE was consistent with this fragmentation mechanism.
FIGURE 3.

RP-HPLC separation with MS/MS analysis of products formed from 18:2n-6 by M. oryzae Guy11. A, nitrogen powder of M. oryzae was incubated with (8S)-[2H]18:2n-6, and the major metabolites and unchanged substrate were analyzed by LC-MS (total ion current). The deuterium content of the metabolites and recovered 18:2n-6 was determined by MS/MS analysis (see Table 2). Small amounts of 6,8-DiHODE eluted on the left shoulder of 7,8-DiHODE. B, mycelia were incubated with 18:2n-6, and 6,8- and 7,8-DiHODE were separated by preparative RP-HPLC and analyzed by MS/MS. Top, total ion current (m/z 311→full scan); middle, reconstructed ion chromatogram for a characteristic ion of 7,8-DiHODE (m/z 173; −OOC-(CH2)5-CHOH-CHO), and bottom, a characteristic ion of 6,8-DiHODE (m/z 127; −OOC-(CH2)4-CHO). C, MS/MS spectrum of 6,8-DiHODE. Inset shows formation of characteristic fragments.
Steric analysis was performed after chemical reduction of HPODE to alcohols. 8-HODE consisted of over 95% of the 8R stereoisomer (supplemental material). We confirmed that (8R)-HPODE was a precursor of 7,8- and 6,8-DiHODE. The absolute configuration of (7,8S)-DiHODE was determined by GC-MS analysis after hydrogenation (Fig. 4A); the designation 8S in (7S,8S)-DiHODE is due to the hydroxyl at C-7 and the Cahn-Ingold-Prelog nomenclature rules. The retention time of the biological product was the same as the threo stereoisomer of 7,8-octadecanoic acid (methyl ester TMS ether derivative, as described in the supplemental material), and only a few percent of the erythro isomer was detected. The configuration at C-6 of 6, (8R)-DiHODE was also determined by GC-MS analysis after hydrogenation. The hydrogenated product had the same retention time as the anti stereoisomer of 6,8-DiHODE (TMS ether methyl ester derivative, supplemental material), as shown in Fig. 4B. This suggested that (6S,8R)-DiHODE was formed. We conclude that both dihydroxymetabolites were formed with high stereo selectivity.
FIGURE 4.

Steric analysis of (7,8S)-DiHODE and (6,8R)-DiHODE by capillary GC-MS. A, top, chromatogram of (7,8S)-DiHODE methyl ester after hydrogenation and silylation with selective ion monitoring of m/z 231. Bottom, chromatogram of the methyl esters and TMS ether derivatives of erythro and threo 7,8-hydroxyoctadecanoic acid. As the precursor 8-HPODE has an R configuration, the results show that (7S,8S)-DiHODE was formed. B, top, chromatogram shows selective ion chromatogram of 6,8-DiHODE methyl ester after hydrogenation and silylation with selective ion monitoring of m/z 217. Bottom, selective ion chromatogram of the syn and anti stereoisomers of methyl 6,8-hydroxyoctadecanoate (TMS ether derivative). As the metabolite was formed from (8R)-HPODE, the results show that (6S,8R)-DiHODE was produced. The ion intensities were normalized to 100%, as indicated.
7,8-LDS activity was present without noticeable differences in mycelia grown in all studied media as follows: complete and minimal media and minimal medium minus glucose or minus nitrate salts. This suggest that the 7,8-LDS activity is not altered under nutrient-rich or starvation conditions.
10-DOX, 8,11-LDS, and 5,8-LDS Activities
In addition to the metabolites discussed above, we also noticed that M. oryzae formed detectable amounts of 10-HODE, 8,11-DiHODE, and 5,8-DiHODE. 10-HODE was identified by its MS/MS spectrum, selective ion monitoring of characteristic MS/MS ions (m/z 295 → m/z 155; m/z 295 → m/z 183), the retention time, and by comparison with authentic 10-HODE (19). Selective ion monitoring of 8-HODE (m/z 295 → m/z 157) and 10-HODE suggested 10-HODE only constituted a few percent of (8R)-HODE.
Significant amounts of 8,11-DiHODE were mainly formed by nitrogen powder of M. oryzae Guy11, and it was a minor product (2–3%) compared with (7S,8S)-DiHODE. 8,11-DiHODE was identified by LC-MS/MS analysis (supplemental material). Nitrogen powder preparations of mycelia also formed 8,11- DiHODE from exogenous (8R)-HPODE and from [U-13C]18:2n-6. The configuration of the hydroxyl at C-11 was S (supplemental material). We conclude that (8R,11S)-DiHODE was formed. Finally, traces of 5,8-DiHODE were detected (<0.5% of (7S,8S)-DiHODE) in most experiments (MS/MS spectrum with the characteristic signals at m/z 115 and m/z 173 at the retention time of authentic 5,8-DiHODE during RP-HPLC).
Oxygenation Mechanism
The mechanism of biosynthesis of (8R)-HPODE, (7S,8S)-DiHODE, and (8R,11S)-DiHODE was studied with (8R)-[2H]18:2n-6, (7R)-[2H]18:2n-6, and (11S)-[2H]18:2n-6 as substrates with nitrogen powder preparations of mycelia. The results are summarized in Table 2. The deuterium labels were retained in both (8R)-HODE and in (7S,8S)- DiHODE, respectively. This suggested antarafacial hydrogen abstraction and oxygen insertion at C-8 and suprafacial hydrogen abstraction and oxygenation at C-7. (11S)-[2H]18:2n-6 (>95%) was transformed to (8R,11S)-DiHODE with loss of the deuterium label, suggesting suprafacial oxygenation at C-11 (Table 2).
TABLE 2.
Transformation of stereospecifically deuterated linoleic acids by nitrogen powder preparation of mycelia of M. oryzae Guy11
| [2H]18:2n-6 | Percent 2H | Percent Incorporation of 2H |
|
|---|---|---|---|
| 18:2n-6a | Oxygenated metabolites | ||
| % | % | ||
| (8R)-[2H]18:2n-6 | 64 | 57 | 59% (8R)-8-[2H]HODE |
| 59% (8R)-7,8-[2H]DiHODE | |||
| (7R)-[2H]18:2n-6 | 41 | 39 | 40% (7R)-8-[2H]HODE |
| 41% (7R)-7,8-[2H]DiHODE | |||
| (11S)-[2H]18:2n-6 | >95 | >95 | <1% (11S)-8,11-[2H]DiHODE |
a The deuterium content was determined in recovered 18:2n-6 from the experiments to correct for dilution of [2H]18:2n-6 by endogenous 18:2n-6 in the preparations (cf. Fig. 3A).
Tetranitromethane (30 and 100 μm) inhibited the biosynthesis of (8R)-HODE and (7S,8S)-DiHODE dose-dependently (supplemental material). This finding is in agreement with a key role of tyrosine radicals for hydrogen abstraction at C-8 (8), as this reagent will nitrate tyrosine residues.
Nitrogen powder preparations of M. oryzae Guy11 oxidized 18:1n-9 (cis) and 18:3n-3 in analogy with 18:2n-6 to 8-hydroxy- and 7,8-dihydroxymetabolites (supplemental material). 20:2n-6 was only oxidized at C-10, suggesting that 20:2n-6 entered the catalytic site for dioxygenation “tail first.” 18:1n-9 (trans), 18:3n-6, and 20:4n-6 were not oxidized.
Gene Targeting of 7,8-LDS and Biosynthesis of Oxylipins
Gene replacement of the 7,8-LDS gene at the MGG_13239 locus (supplemental material) resulted in complete loss of biosynthesis of (7S,8S)-DiHODE, which clearly links the M. oryzae 7,8-LDS activity to this gene. The Δ7,8-LDS mutant incubated with 18:2n-6 appeared to form trace amounts of 8-HODE (Fig. 5). 8-HPODE can be formed during autoxidation as a minor product (∼1%) in comparison with 9- and 13-HPODE (44). 8-HODE was present only in somewhat larger amounts (∼3%), and we cannot exclude minor enzymatic contribution. Nitrogen powder preparations of M. oryzae Guy11 transformed exogenous (8R)-HPODE to 7,8-, 6,8-, 8,11-, and 5,8-DiHODE, whereas nitrogen powder preparations of Δ7,8-LDS did not form detectable amounts of these products.
FIGURE 5.

LC-MS/MS analysis of oxylipins produced by M. oryzae Guy11 (wild type) and the genetically modified strain, Δ7,8-LDS. The top two chromatograms show MS/MS analysis of m/z 311→ m/z 173, a characteristic fragment of 7,8-DiHODE (−OOC-(CH2)5-CHOH-CHO). This metabolite could not be detected in the mutant Δ7,8-LDS (ion chromatogram magnified 100 times). The bottom two chromatograms show MS/MS analysis of m/z 295 → m/z 157, the characteristic fragment of 8-HODE (−OOC-(CH2)6-CHO). This metabolite could be detected only in trace amounts in mutant Δ7,8-LDS (ion chromatogram magnified 100 times), and it could possibly be formed by autoxidation.
Infection Assays
The spray inoculation assays with the Δ7,8-LDS mutant did not show any differences in frequency or severity of lesions as compared wild type M. oryzae Guy11. The Δ7,8-LDS mutant also infected rice roots as the wild type (Fig. 6). Appressoria formation also appeared to occur without abnormality in the Δ7,8-LDS mutant (supplemental material). The expression of SGFP inserted within the 7,8-LDS coding sequence was readily detected in mycelia of the Δ7,8-LDS mutant by epifluorescence microscopy (supplemental material).
FIGURE 6.
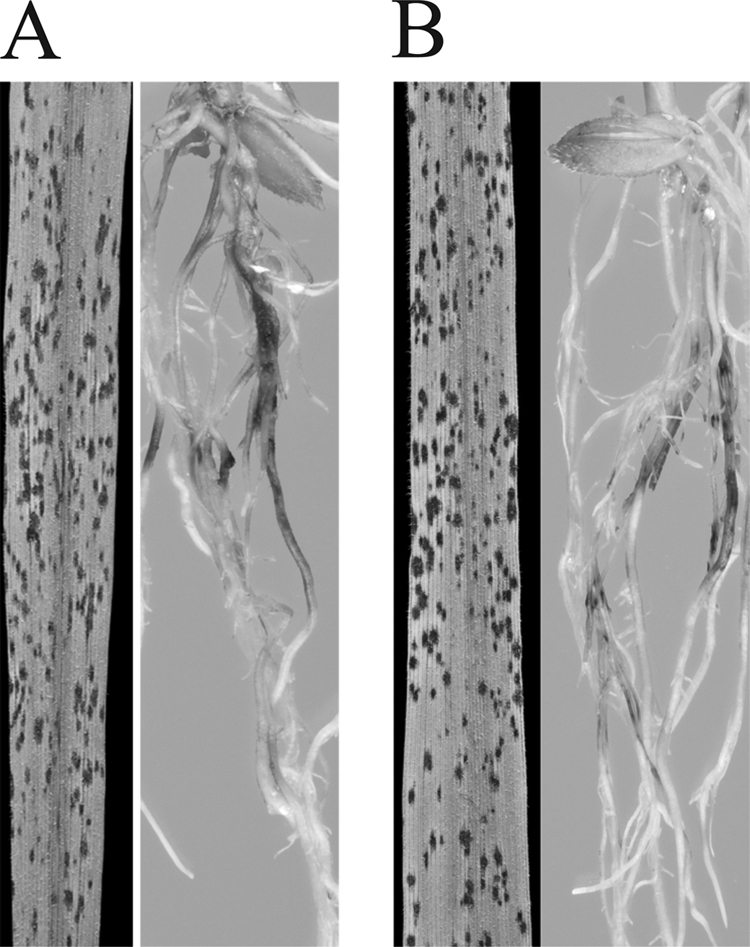
Infection of rice leaves and roots. A, leaves and roots infected by M. oryzae. B, leaves and roots infected by Δ7,8-LDS.
Conidia production and spore morphology of the Δ7,8-LDS mutant was not affected. Radial growth and colony morphology were also unchanged both on complete or minimal media.
Oxylipins Formed by Mutants Implicated in the cAMP-dependent and Mitogen-activated Protein Kinase Cascades
There are two important signaling pathways required for M. oryzae appressorium development and leaf penetration as follows: the PMK1 pathway (31), and the cAMP-activated PKA pathway (36). Nitrogen powder preparations of M. oryzae mutants lacking the adenylyl cyclase (Δmac1), a catalytic subunit of the PKA (Δcpka), the PMK1, or the transcription factor MST12 oxidized 18:2n-6 as did the native M. oryzae Guy11. The biosynthesis of 6,8-DiHODE and 8,11-DiHODE also appeared to be unchanged. These results suggest that both PMK1 and PKA-dependent signaling pathways have no influence in the 7,8-LDS activity or that their effect was not detectable under our experimental conditions.
However, the suppressor mutant (Δmac1 sum1–99), which shows cAMP-independent PKA activity due to a mutation in the regulatory subunit of PKA leading to a partially constitutive PKA activity (27), formed increased amounts of 10-HPODE, 10-HODE, and 5,8-DiHODE (Fig. 7). Compared with the Δmac1 mutant (and to wild type M. oryzae Guy11), the biosynthesis of 10-HODE and 5,8-DiHODE appeared to be augmented ∼8- and 100-fold, respectively, in the experiment illustrated in Fig. 7. LC-MS/MS analysis suggested that 10-HPODE amounted to ∼30% of (8R)-HPODE and 5,8-DiHODE to ∼20% of (7S,8S)-DiHODE. In other experiments, 5,8-DiHODE and 10-HODE were formed in even larger amounts than (7S,8S)-DiHODE and (8R)-HODE, respectively. This seems to be related to an increased growth time of the mycelia but was not further investigated.
FIGURE 7.

LC-MS analysis of products formed from 18:2n-6 by two mutants of M. oryzae, Δmac1 with disruption of adenylate cyclase and Δmac1 sum1–99 with augmented PKA activity. A, RP-HPLC-MS/MS analysis of DiHODE formed by Δmac1 sum1–99. The top chromatogram shows total ion current (TIC) during MS/MS analysis (m/z 311→ full scan), the middle m/z 311→ m/z 173 (a characteristic ion in the MS/MS spectra of both 7,8-DiHODE and 5,8-DiHODE), and the bottom chromatogram m/z 311→ m/z 115 (−OOC-(CH2)3-COH; a characteristic ion in the MS/MS spectrum of 5,8-DiHODE). Only traces of 5,8-DiHODE were formed by Δmac1 mutant (data not shown). B, RP-HPLC-MS/MS analysis of 8- and 10-HODE. The reconstructed ion chromatograms for detection of for 8-HODE (m/z 295→157) and 10-HODE (m/z 295→155 (183–18)) in the Δmac1 strain. The bottom chromatograms showed an 8-fold relative increase in formation of 10-HODE by the Δmac1sum1–99 strain. The ion chromatograms of 10-HODE were magnified 10 times, as indicated.
Chiral phase HPLC-MS/MS analysis showed that 10-HODE mainly consisted of the R stereoisomer (∼85% R; supplemental material). Normal phase HPLC-MS/MS analysis showed that 5,8-DiHODE eluted with the same retention time as authentic (5S,8R)-DiHODE (supplemental material). Finally, we determined that 5,8- and 7,8-LDS activities were present in high speed supernatant of nitrogen powder prepared from mycelia of the Δmac1 sum1–99 mutant.
Real Time PCR Analysis
Real time PCR was performed to investigate whether the increased formation of (5S,8R)- DiHODE and (10R)-HPODE in Δmac1 sum1–99 strain could be due to relative up-regulation of the second gene with homology to 7,8-LDS, MGG_10859. We therefore compared the expression of the transcripts of MGG_10859, MGG_13239, and actin in three strains (Guy11 and two mutants, Δmac1 and Δmac1 sum1–99).
In Guy11 and Δmac1 mycelia, transcripts of MGG_13239 (7,8-LDS) were more (ΔCT ∼ 1.9) and less abundant (ΔCT ∼ 1.9) compared with transcripts of actin. This suggested that 7,8-LDS could be down-regulated in Δmac1 (ΔCT ∼ 3.8). The transcripts of MGG_10859 were less abundant than actin in Guy11 (ΔCT ∼ 10.6) and Δmac1 (ΔCT ∼ 5.7) and thus up-regulated in the latter (ΔCT ∼ 4.9). We confirmed that the nitrogen powder preparations used for RNA extraction oxidized linoleic acid to 7,8-DiHODE with insignificant formation of 5,8-DiHODE (<1%), although MGG_10859 mRNA was detected.
In the Δmac1 sum1–99 mutant, transcripts of MGG_13239 (7,8-LDS) were detected by real time PCR analysis, but we could not detect transcripts of MGG_10859 (n = 7; 50 cycles). We confirmed that the PCR efficiencies for both primer pairs of MGG_10859 and MGG_13239 were comparable with genomic DNA as template. We also confirmed that the nitrogen powder preparation of the Δmac1 sum1–99 mutant used for RNA extraction formed 5,8-DiHODE and 10-HODE as major metabolites.
Hydroxylation and Epoxidation of Unsaturated Fatty Acids
Mycelia of M. oryzae catalyzed ω2- and/or ω3-hydroxylation of all investigated unsaturated fatty acids and oxidation to vicinal diols also occurred. ω-Hydroxymetabolites could not be detected. The diols were likely formed by epoxidation of the terminal double bonds of the fatty acids followed by enzymatic hydrolysis. Over 50% of the fatty acids were typically consumed in 4–5 h, and these hydroxy- and dihydroxymetabolites were obtained in significant yields, as judged from TLC analysis. Experimental details of GC-MS analysis and a summary of the major metabolites of unsaturated C18-C22 fatty acids are given in the supplemental material.
DISCUSSION
We have studied oxygenation of 18:2n-6 by M. oryzae Guy11 and report four major findings. First, gene deletion allowed us to identify the 7,8-LDS gene (MGG_13239). Second, we could also determine the metabolites of 18:2n-6 associated with the 7,8-LDS activity, their structures, and mechanism of formation with the aid of stereospecifically deuterated 18:2n-6 and by chiral analysis. Third, gene deletion showed that 7,8-LDS activity was not critical for sporulation or rice infection. Fourth, a suppressor mutant of M. oryzae with cAMP-independent PKA activity (Δmac1 sum1–99) transformed 18:2n-6 to two other major metabolites, (10R)-hydroxy-(8E,12Z)-octadecadienoic acid and (5S,8R)-DiHODE, which were barely detectable in M. oryzae Guy11 and a mutant of the adenylate cyclase MAC1 gene (Δmac1). These results suggest that (10R)-DOX and 5,8-hydroperoxide isomerase activities could be up-regulated by PKA.
The genome of M. oryzae contains two genes with homology to the dioxygenase and P450 domains of 7,8-LDS of G. graminis. Disruption of the gene (MGG_13239) with a deduced protein of 63% amino acid identity to 7,8-LDS of G. graminis abolished formation of all major DiHODE both from 18:2n-6 and from exogenous (8R)-HPODE. It seems likely that the P450 domain of 7,8-LDS catalyzes isomerization of (8R)-HPODE to (7S,8S)-DiHODE in analogy with 7,8-LDS of G. graminis. This hydroperoxide isomerase may not be specific for hydroxylation of C-7, and (6S,8R)-DiHODE is likely formed in this way. (8R,11S)-DiHODE biosynthesis was detected from 18:2n-6 and from (8R)-HPODE in nitrogen powder preparations of M. oryzae Guy11 but not in preparations of Δ7,8-LDS. (8R,11S)-DiHODE could conceivably be formed by the hydroperoxide isomerase of 7,8-LDS as a minor by-product. (8R,11S)-DiHODE was formed by antarafacial dioxygenation at C-8 and suprafacial hydroxylation at C-7 and C-11. This mechanism of biosynthesis appears to be a characteristic feature of the family of LDS enzymes (7, 16, 42). We conclude that the metabolites discussed above can be formed by 7,8-LDS, as summarized in Fig. 8.
FIGURE 8.

Overview of products formed by 7,8-LDS of M. oryzae Guy11 and the mechanism of biosynthesis. (8R,11S)-DiHODE might be formed by the hydroperoxide isomerase (P450) of 7,8-LDS, as indicated.
Δ7,8-LDS infected rice leaves and roots to the same extent as native M. oryzae. We found no apparent defects in conidia and appressoria formation or in radial growth. This might due to alternative compensatory pathways, which seem to be common in rice blast (45). Recent genome analysis suggests that LDS homologues occur in many filamentous fungi (46), and it may be linked to housekeeping functions. A protein with homology to LDS is abundant in teliospores of Ustilago maydis and is thought to participate in the mobilization of stored lipids, although gene deletion of this protein did not produce a distinct phenotype (47). In contrast, gene deletion of 5,8-LDS and (10R)-DOX affected biological processes of A. fumigatus and A. nidulans (15, 16).
Degradation of lipid bodies, activation of lipases, and generation of glycerol are important steps in formation of the turgor pressure of appressoria of M. oryzae (27). Protein kinases are important regulators of this process (29–32). Glycogen formation and lipid degradation occur rapidly in the Δmac1 sum1–99 mutant (27). Interestingly, mycelia of Δmac1 sum1–99 formed much larger amounts of (10R)-HPODE and (5S,8R)-DiHODE in comparison with the Δmac1 mutant and M. oryzae Guy11. Could this be due to up-regulation of the second gene product (MGG_10859) with homology to 7,8-LDS?
Transcripts of the two LDS homologues were detected by PCR analysis in Guy11 and the Δmac1 mutant, and MGG_10859 mRNA appeared to be up-regulated in the Δmac1 mutant. In contrast, MGG_10859 mRNA was not detectable in the mutant with cAMP-independent PKA activity (Δmac1 sum1–99). MGG_10859 could therefore not be linked to biosynthesis of (10R)-HPODE nor (5S,8R)-DiHODE. As shown in Table 1, MGG_10859 did not show specific sequence homology to any of the listed enzymes, and the homology did not suggest a specific catalytic activity. Further studies will be needed to determine its function.
The genome of M. oryzae contains over 120 CYP genes, grouped with only a few members per family (5). It is conceivable that 5,8-DiHODE could be formed by any of these P450s of the suppressor mutant. We found, however, that 5,8-LDS (and 7,8-LDS) activities were present in the high speed supernatant and negligible in the microsomal fraction, which seems to exclude microsomal enzymes.
The P450 domain of 7,8-LDS contains several sequences with homology to the PKA motif, Arg-Arg-Xaa-(Ser/Thr). Phosphorylation of P450 at this motif by PKA has so far only been found to reduce P450 activity without influence on product specificity (48). Whether the oxylipin biosynthesis of the Δmac1 sum1–99 mutant can be attributed to post-translational modification of 7,8-LDS merits further investigation.
Mycelia of M. oryzae also oxidized medium and long chain polyunsaturated fatty acids by hydroxylation and epoxidation, as summarized in the supplemental material. CYP102 of Bacillus megaterium is the prototype microbiological fatty acid ω2- and ω3-hydroxylase and fatty acid epoxygenase (49). Blast search with CYP102 revealed three homologous hypothetical enzymes (numbers 3–5) of the CYP505A family in M. oryzae. Further studies will be needed to link CYP505 of M. oryzae to fatty acid oxygenation in the same way as the terminal oxygenases of Fusarium oxysporum were linked to CYP404 (50). Recent studies of Talbot and co-workers (28) showed that peroxisomal β-oxidation is essential for M. oryzae during appressorium-mediated rice infection, and the lipid bodies of spores may provide the fuel for the β-oxidation.
In summary, we report the first studies on the biological function of 7,8-LDS in M. oryzae, an important fungal grass pathogen and model organism. We identified the 7,8-LDS gene (MGG_13239) and determined the structure and mechanism of formation of metabolites. Gene deletion showed that 7,8-LDS was not critical for sporulation nor for rice infection. PKA, which has profound effects on these processes, augmented formation of two previously unrecognized oxylipins of M. oryzae, (5S,8R)-DIHODE and (10R)-HPODE. We conclude that M. oryzae can oxidize to 18:2n-6 to a more complex set of oxylipins than previously anticipated.
This work was supported by Vetenskapsrådet Medicin/Grant 03X-06523 (to E. O.), The Knut and Alice Wallenberg Foundation/Grant KAW 2004.0123 and Formas/Grant 222-2005-1733, The Swedish Research Council for Environmental, Agricultural Sciences, and Spatial Planning Project 229-2004-833 (to M. H.), and The Biotechnology and Biological Sciences Research Council Grant BB/C520720/1, United Kingdom (to A. S.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1S–12S and Tables 1S and 2S.
M. oryzae designates the rice-infecting species of Magnaporthe isolates separate from M. grisea (51).
Data are from the Magnaporthe grisea Sequencing Project at Broad Institute of Harvard and Massachusetts Institute of Technology.
Hoffmann, I., Jernerén, F., and Oliw, E. H. (2009) Regulatory Oxylipins, An International Symposium, Lausanne, Switzerland, June 4–6, 2009.
M. Cristea, A. E. Osbourn, and E. H. Oliw, GenBankTM accession number AY372822.
- LDS
- linoleate diol synthase
- DiHODE
- dihydroxy-(9Z,12Z)-octadecadienoic acid
- DOX
- dioxygenase
- (8R)-HODE
- (8R)-hydroxy-(9Z,12Z)-octadecadienoic acid
- (8R)-HPODE
- (8R)-hydroperoxy-(9Z,12Z)-octadecadienoic acid
- (10R)-HPODE
- (10R)-hydroperoxy-(8E,12Z)-octadecadienoic acid
- HPODE
- hydroperoxyoctadecadienoic acid
- MAPK
- mitogen-activated protein kinase
- PKA
- protein kinase A
- RP
- reversed phase
- TMS
- trimethylsilyl ether
- HPLC
- high pressure liquid chromatography
- MS/MS
- tandem mass spectrometry
- GC-MS
- gas chromatography-mass spectrometry
- LC-MS
- liquid chromatography-mass spectrometry
- UTR
- untranslated region.
REFERENCES
- 1.Wilson R. A., Talbot N. J. (2009) Nat. Rev. Microbiol. 7, 185–195 [DOI] [PubMed] [Google Scholar]
- 2.Skamnioti P., Gurr S. J. (2009) Trends Biotechnol. 27, 141–150 [DOI] [PubMed] [Google Scholar]
- 3.Ribot C., Hirsch J., Balzergue S., Tharreau D., Nottéghem J. L., Lebrun M. H., Morel J. B. (2008) J. Plant Physiol. 165, 114–124 [DOI] [PubMed] [Google Scholar]
- 4.Dean R. A., Talbot N. J., Ebbole D. J., Farman M. L., Mitchell T. K., Orbach M. J., Thon M., Kulkarni R., Xu J. R., Pan H., Read N. D., Lee Y. H., Carbone I., Brown D., Oh Y. Y., Donofrio N., Jeong J. S., Soanes D. M., Djonovic S., Kolomiets E., Rehmeyer C., Li W., Harding M., Kim S., Lebrun M. H., Bohnert H., Coughlan S., Butler J., Calvo S., Ma L. J., Nicol R., Purcell S., Nusbaum C., Galagan J. E., Birren B. W. (2005) Nature 434, 980–986 [DOI] [PubMed] [Google Scholar]
- 5.Deng J., Carbone I., Dean R. A. (2007) BMC Evol. Biol. 7, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brodowsky I. D., Hamberg M., Oliw E. H. (1992) J. Biol. Chem. 267, 14738–14745 [PubMed] [Google Scholar]
- 7.Hamberg M., Zhang L. Y., Brodowsky I. D., Oliw E. H. (1994) Arch. Biochem. Biophys. 309, 77–80 [DOI] [PubMed] [Google Scholar]
- 8.Su C., Sahlin M., Oliw E. H. (1998) J. Biol. Chem. 273, 20744–20751 [DOI] [PubMed] [Google Scholar]
- 9.Lee D. S., Nioche P., Hamberg M., Raman C. S. (2008) Nature 455, 363–368 [DOI] [PubMed] [Google Scholar]
- 10.Jernerén F., Garscha U., Hoffmann I. M., Oliw E. H. (2010) Biochim. Biophys. Acta, in press [DOI] [PubMed] [Google Scholar]
- 11.Su C., Oliw E. H. (1996) J. Biol. Chem. 271, 14112–14118 [DOI] [PubMed] [Google Scholar]
- 12.Hörnsten L., Su C., Osbourn A. E., Garosi P., Hellman U., Wernstedt C., Oliw E. H. (1999) J. Biol. Chem. 274, 28219–28224 [DOI] [PubMed] [Google Scholar]
- 13.Garscha U., Oliw E. H. (2008) FEBS Lett. 582, 3547–3551 [DOI] [PubMed] [Google Scholar]
- 14.Cristea M., Osbourn A. E., Oliw E. H. (2003) Lipids 38, 1275–1280 [DOI] [PubMed] [Google Scholar]
- 15.Tsitsigiannis D. I., Kowieski T. M., Zarnowski R., Keller N. P. (2005) Microbiology 151, 1809–1821 [DOI] [PubMed] [Google Scholar]
- 16.Garscha U., Jernerén F., Chung D., Keller N. P., Hamberg M., Oliw E. H. (2007) J. Biol. Chem. 282, 34707–34718 [DOI] [PubMed] [Google Scholar]
- 17.Garscha U., Oliw E. H. (2009) J. Biol. Chem. 284, 13755–13765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brodhun F., Göbel C., Hornung E., Feussner I. (2009) J. Biol. Chem. 284, 11792–11805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garscha U., Oliw E. H. (2007) Anal. Biochem. 367, 238–246 [DOI] [PubMed] [Google Scholar]
- 20.Champe S. P., el-Zayat A. A. E. (1989) J. Bacteriol. 171, 3982–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dagenais T. R., Chung D., Giles S. S., Hull C. M., Andes D., Keller N. P. (2008) Infect. Immun. 76, 3214–3220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsitsigiannis D. I., Keller N. P. (2006) Mol. Microbiol. 59, 882–892 [DOI] [PubMed] [Google Scholar]
- 23.Cook R. J. (2003) Physiol. Mol. Plant Pathol. 62, 73–86 [Google Scholar]
- 24.Cannon P. F. (1994) Systema Ascomycetum 13, 25–42 [Google Scholar]
- 25.Talbot N. J., Foster A. J. (2001) Adv. Bot. Res. 34, 263–287 [Google Scholar]
- 26.Sesma A., Osbourn A. E. (2004) Nature 431, 582–586 [DOI] [PubMed] [Google Scholar]
- 27.Thines E., Weber R. W., Talbot N. J. (2000) Plant Cell 12, 1703–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z. Y., Soanes D. M., Kershaw M. J., Talbot N. J. (2007) Mol. Plant Microbe Interact. 20, 475–491 [DOI] [PubMed] [Google Scholar]
- 29.Adachi K., Hamer J. E. (1998) Plant Cell 10, 1361–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mitchell T. K., Dean R. A. (1995) Plant Cell 7, 1869–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu J. R., Hamer J. E. (1996) Genes Dev. 10, 2696–2706 [DOI] [PubMed] [Google Scholar]
- 32.Park G., Xue C., Zheng L., Lam S., Xu J. R. (2002) Mol. Plant Microbe Interact. 15, 183–192 [DOI] [PubMed] [Google Scholar]
- 33.Foster A. J., Jenkinson J. M., Talbot N. J. (2003) EMBO J. 22, 225–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ou S. (1985) Rice Diseases, CAB International, Kew, UK [Google Scholar]
- 35.Choi W., Dean R. A. (1997) Plant Cell 9, 1973–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu J. R., Urban M., Sweigard J. A., Hamer J. E. (1997) Mol. Plant Microbe Interact. 10, 187–194 [Google Scholar]
- 37.Davis L., Kuehl M., Battey J. (1994) Basic Methods in Molecular Biology, pp. 1–304, Appleton and Lange, Norwalk [Google Scholar]
- 38.Talbot N. J. (2001) in Molecular and Cellular Biology of Filamentous Fungi (Talbot N., ed) pp. 25–26, Oxford University Press, Bath [Google Scholar]
- 39.Rho H. S., Kang S., Lee Y. H. (2001) Mol. Cells 12, 407–411 [PubMed] [Google Scholar]
- 40.Carbone I., Kohn L. M. (1999) Mycologia 91, 553–556 [Google Scholar]
- 41.Oliw E. H., Stark K., Bylund J. (2001) Biochem. Pharmacol. 62, 407–415 [DOI] [PubMed] [Google Scholar]
- 42.Wadman M. W., van Zadelhoff G., Hamberg M., Visser T., Veldink G. A., Vliegenthart J. F. (2005) Lipids 40, 1163–1170 [DOI] [PubMed] [Google Scholar]
- 43.Oliw E. H., Su C., Skogström T., Benthin G. (1998) Lipids 33, 843–852 [DOI] [PubMed] [Google Scholar]
- 44.Haslbeck F., Grosch W., Firl J. (1983) Biochim. Biophys. Acta 750, 185–193 [DOI] [PubMed] [Google Scholar]
- 45.Oh Y., Donofrio N., Pan H., Coughlan S., Brown D. E., Meng S., Mitchell T., Dean R. A. (2008) Genome Biol. 9, R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soanes D. M., Alam I., Cornell M., Wong H. M., Hedeler C., Paton N. W., Rattray M., Hubbard S. J., Oliver S. G., Talbot N. J. (2008) PLoS ONE 3, e2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huber S. M., Lottspeich F., Kämper J. (2002) Mol. Genet. Genomics 267, 757–771 [DOI] [PubMed] [Google Scholar]
- 48.Oesch-Bartlomowicz B., Oesch F. (2003) Arch. Biochem. Biophys. 409, 228–234 [DOI] [PubMed] [Google Scholar]
- 49.Capdevila J. H., Wei S., Helvig C., Falck J. R., Belosludtsev Y., Truan G., Graham-Lorence S. E., Peterson J. A. (1996) J. Biol. Chem. 271, 22663–22671 [DOI] [PubMed] [Google Scholar]
- 50.Kitazume T., Takaya N., Nakayama N., Shoun H. (2000) J. Biol. Chem. 275, 39734–39740 [DOI] [PubMed] [Google Scholar]
- 51.Couch B. C., Kohn L. M. (2002) Mycologia 94, 683–693 [DOI] [PubMed] [Google Scholar]