

Abstract
We recently identified a missense single nucleotide polymorphism (SNP) in DDX5 (rs1140409, p.S480A) that enhances the risk of developing cirrhosis. DDX5 is an ATP-dependent RNA helicase and transcriptional modulator. We hypothesized that the activity of DDX5 in regulating fibrogenic gene transcription in hepatic stellate cells (HSCs) is altered by the S480A SNP. To test this, we employed two approaches: 1) transient overexpression of DDX5 cDNA or siRNA knockdown of endogenous DDX5, with replacement by either DDX5 wild type (WT) or SNP cDNA, or 2) stable expression of exogenous DDX5 WT and SNP in HSC lines. WT DDX5 mRNA in HSCs was inversely correlated with gene expression for α2(I) collagen, tissue inhibitor of metalloproteinase-1, and transforming growth factor-β1. Stable DDX5 SNP-expressing cells had higher basal and transforming growth factor-β1-stimulated expression and enhanced promoter activities of fibrogenic genes. DDX5 variant-expressing cells also had higher Smad3 and AP-1-responsive reporter activities. In a one-hybrid GAL4 system, co-expression of the DDX5 SNP variant with chimeras of GAL4 DNA binding domain linked to JunD or Sp1 displayed higher transactivation of a GAL4-responsive reporter than that of DDX5 WT. Increased fibrogenic gene expression in DDX5 SNP-expressing cells was associated with reduced recruitment of DDX5 homodimers to responsive promoters, but there was no difference in the recruitment of the co-repressor HDAC1 (histone deacetylase 1). These data suggest that DDX5 is a repressor of fibrogenic genes in HSCs through interaction with transcriptional complexes. The enhanced fibrogenic activity of the DDX5 risk variant is linked to a reduced repressive function toward these target genes.
Keywords: Genetics/Polymorphism, DNA-Protein Interaction, Gene Regulation, Promoters, Transcription Factors, DDX5, Fibrogenic Genes, Hepatic Stellate Cells, Homodimers, Single Nucleotide Polymorphism
Introduction
Hepatic fibrosis reflects the orchestrated response to liver injury, culminating in deposition of extracellular matrix within the hepatic parenchyma (1). Estimated to affect hundreds of millions of patients worldwide, the disease occurs in patients with chronic hepatitis B or hepatitis C, non-alcoholic steatohepatitis, alcohol abuse, toxic drug injury, and a number of other etiologies. Of these, hepatitis C virus (HCV)3 infection is a major cause of chronic liver disease in North America and Western Europe. The patients usually harbor the infection for 2–3 decades on average, and begin to display an accelerating risk of decompensation and hepatocellular carcinoma (2). Although up to 40% of patients with chronic hepatitis C will die of complications directly related to the infection, the remainder do not, indicating not only the pressure of competing morbidities but, more importantly, the remarkably variable rate of fibrosis progression in these patients. A number of well defined external risk factors contribute to this variability, including alcohol abuse, gender, age, body mass index, co-infection with human immunodeficiency virus, and cannabis use, yet these risks in aggregate do not sufficiently explain the variable progression. Instead, a growing number of studies highlight the impact of genetic polymorphisms that modulate the vigor of the fibrogenic response (3–11). Typically, these have been uncovered by seeking single nucleotide polymorphisms (SNPs) within genes mechanistically linked to pathways of fibrosis and inflammation (3, 12).
Recognizing, however, that there might be other genetic determinants whose link to fibrosis pathways might not yet be appreciated, we undertook an unbiased whole genome functional, gene-centric scanning approach to reveal SNPs correlating with fibrosis progression in a well characterized cohort of patients with chronic HCV infection who displayed variable fibrosis progression (4, 7). Among the genes identified using this approach was DDX5 (DEAD box polypeptide 5) (7), a gene that is located on chromosome 17q21 and not previously connected to liver injury or fibrosis. The DDX5 risk allele that confers accelerated fibrosis progression encodes an amino acid change from serine to alanine in exon XIII (S480A) (4). The potential mechanism underlying the impact of a DDX5 SNP variant on fibrosis is unknown.
DDX5 is a prototypical member of the DEAD box helicase family of proteins, which are characterized by the conserved motif Asp-Glu-Ala-Asp (DEAD). DEAD box proteins have vital roles in almost all aspects of RNA synthesis, processing, and activity, including pre-mRNA processing; ribosome biogenesis; RNA turnover, export, and translation; the multistep association and dissociation of large RNP complexes; and the modulation of complex RNA structures (13, 14). DDX5 is also a transcriptional co-regulator (15) that interacts with many transcription factors, including p53 (16), estrogen receptor α, Smad3 (17), Runx2 (18), MyoD (14), and several transcriptional co-activators and co-repressors, including p300, CREB-binding protein, RNA polymerase II (19), and HDAC1 (histone deacetylase 1) (20). DDX5 is a nucleocytoplasmic shuttling protein (21) that exists in heterodimeric and homodimeric forms (22).
The correlation of a DDX5 gene variant with HCV-associated fibrosis progression was especially intriguing because it reportedly interacts with the HCV NS5B protein and might thereby regulate HCV RNA replication (23). However, the SNP has subsequently been associated with fibrosis progression in non-alcoholic steatohepatitis (24), where HCV is not present.
Therefore, we undertook this study to identify which regulatory pathways might be affected by the DDX5 variant in activated hepatic stellate cells (HSCs), the principal fibrogenic cell in injured liver (25). Specifically, we reasoned that given its potential role in gene regulation and repression, DDX5 might regulate transcription of fibrogenic genes.
EXPERIMENTAL PROCEDURES
Isolation of Primary Human HSCs
Primary HSCs were isolated from normal liver margins in selected patients undergoing hepatic resection for primary benign tumors or a single metastasis from colon cancer, as described previously (26). Briefly, immediately posthepatectomy, an isolated liver section was washed, and the portal vessels were cannulated for in situ digestion with collagenase (Roche Applied Science) and Pronase (Roche Applied Science), followed by density gradient centrifugation (27). HSC populations were consistently found between 95 and 99% purity with viability of 95%. HSCs were plated on plastic in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum in a 5% CO2 humidified atmosphere. HSCs were activated by culturing on plastic for 7–10 days and subcultured to passage 4 for the following experiments. In addition to primary HSCs, we also utilized a well validated immortalized human stellate cell line, LX-2, whose features closely resemble those of primary activated HSCs (28).
Expression Plasmids and Reporter Constructs
Full-length DDX5 cDNA was PCR-cloned from a human liver cDNA library and TOPO-ligated into pcDNA3.1 vector (Invitrogen). DDX5 S480A SNP was generated by site-directed mutagenesis (see primer sequences in Table 1) and the QuikChange®II-E site-directed mutagenesis kit (Stratagene, La Jolla, CA). The DDX5 WT and SNP cDNAs were then PCR-amplified using Pfx50TM DNA polymerase (Invitrogen) and a pair of cloning primers (Table 1) that allowed incorporation of FLAG epitope coding sequence to be added at the C terminus of the cDNAs. The PCR products were then TOPO-cloned into pCR®8/GW/TOPO® Entrez vector (Invitrogen) and transferred into destination vectors via LR recombination reactions. The destination vectors selected were pcDNA-DEST40 GatewayTM vector (Invitrogen) for transient transduction and Plenti4/V5-DEST GatewayTM vector (Invitrogen) for lentivirus-mediated stable transduction of hu-DDX5 cDNAs into LX-2 cells. Constructs in which the N-terminal GAL4 DNA-binding domain (GAL4-DBD) was linked to DDX5 cDNA were generated by recombinant ligation of DDX5 WT and SNP cDNA into a GAL4-DBD pcDNA3-expressing vector. The vector sequences were validated by commercial sequencing (GENEWIZ, Inc., South Plainfield, NJ).
TABLE 1.
Sequences used for mutagenesis, cloning, siRNA, and real-time quantitative PCR
Boldface type indicates mutated oligonucleotides.
| Sequence | |
|---|---|
| Site-directed mutagenesis | |
| DDX5 | Sense: 5′-GGTCGAAGACAGAGGTGCAGGTCGTTCCAGGGGTAGA-3′ |
| Antisense: 5′-TCTACCCCTGGAACGACCTGCACCTCTGTCTTCGACC-3′ | |
| α2(I) collagen promoter | |
| Sp1(1) | Sense: 5′-GGCGGGAGGATGCGGAGGGATGAGGTATGCAGACAACGAGTC-3′ |
| Antisense: 5′-GACTCGTTGTCTGCATACCTCATCCCTCCGCATCCTCCCGCC-3′ | |
| Sp1(2, 3) | Sense: 5′-CCAAACTTGGAAAGGGATGGGGAGGGATGGAGGATGCGGAGGGCG-3′ |
| Antisense: 5′-CGCCCTCCGCATCCTCCATCCCTCCCCATCCCTTTCCAAGTTTGG-3′ | |
| Smad3 | Sense: 5′-GAGGGCGGAGGTATGCACCCAACGAGTCAGAGTTTCCCCTTG-3′ |
| Antisense: 3′-CAAGGGGAAACTCTGACTCGTTGGGTGCATACCTCCGCCCTC-3′ | |
| AP-1 | Sense: 5′-GCGGAGGTATGCAGACAACGCCTCAGAGTTTCCCCTTGAAAGC-3′ |
| Antisense: 5′-GCTTTCAAGGGGAAACTCTGAGGCGTTGTCTGCATACCTCCGC-3′ | |
| Cloning primers | |
| DDX5 | Sense: 5′-CGGATCCACCGCAACCATTGACGCC-3′ |
| Antisense: 5′-GGGATCCTTACTTATCATCGTCGTCCTTGTAGTCTTGGGAATATCCTGTTGGC-3′ | |
| siRNA | |
| DDX5 5′-untranslated region | Sense: 5′-GATCCCCATGAAGGCAATGCATGGCCTTCAAGAGAGGCCATGCATTGCCTTCATTTTTTA-3′ |
| Antisense: 5′-AGCTTAAAAAATGAAGGCAATGCATGGCCTCTCTTGAAGGCCATGCATTGCCTTCATGGG-3′ | |
| ChIP primers | |
| α2(I) collagen promoter (162 bp) | Sense: 5′-ACTCCGACGTGTCCCATAGTGTTT-3′ |
| Antisense: 5′-GGGCTGGCTTCTTAAATTGGTTCC-3′ | |
| TIMP-1 promoter (153 bp) | Sense: 5′-AGGAATAGTGACTGACGTGGAGGT-3′ |
| Antisense: 5′-GCATTACTCATCCACCCACCATCA-3′ | |
| TIMP-1 promoter (240 bp) | Sense: 5′-CGGCTTGGAAGGAATAGTGACTGA-3′ |
| Antisense: 5′-CGCTAGAGGATAAATGTCCACGCT-3′ | |
| TGF-β1 promoter (264 bp) | Sense: 5′-AGGAGGCAGCACCCTGTTT-3′ |
| Antisense: 5′-AGGGAGGTGGGAGGGAGAT-3′ | |
| TGF-β1 promoter (152 bp) | Sense: 5′-TACCTTGTTTCCCAGCCTGACTCT-3′ |
| Antisense: 5′-TGATCCAGATGCGCTGTGGCTTT-3′ | |
| Real-time quantitative PCR primers | |
| Human DDX5 (131 bp) | Sense: 5′-ACAGAATTTCACTGAACCCACTGC -3′ |
| Antisense: 5′-GACAATGGCAGGAAGCAAATAAGA-3′ | |
| Human collagen type 1 (118 bp) | Sense: 5′-GGCTTCCCTGGTCTTCCTGG-3′ |
| Antisense: 5′-CCAGGGGGTCCAGCCAAT-3′ | |
| Human TGF-β1 (101 bp) | Sense: 5′-CCCAGCATCTGCAAAGCTC-3′ |
| Antisense: 5′-GTCAATGTACAGCTGCCGCA-3′ | |
| Human glyceraldehyde-3-phosphate dehydrogenase (104 bp) | Sense: 5′-GGTGAAGGTCGGAGTCAACGG-3′ |
| Antisense: 5′-TGAAGGGGTCATTGATGGCAACA-3′ | |
The promoter constructs of the α2(I) collagen gene containing mutations of either Smad3, AP-1 (activating protein-1), or three Sp1 binding sites were generated by site-directed mutagenesis (see primer sequences in Table 1) using a luciferase reporter construct of −378/+1 α2(I) collagen gene promoter (29) as template. The GAL4-Sp1 and GAL4-JunD vectors expressing fusion proteins of Sp1 or JunD linked to GAL4-DBD were generated by recombinant ligation of a GAL4-DBD insert from a pcDNA3 vector, with Sp1 and JunD cDNAs expressing pCMV plasmids. The mutations and the recombinants were confirmed by DNA sequencing. The TIMP-1 promoter and the mutant promoter constructs for AP-1 and Sp1 binding sites (30, 31); TGF-β1 promoter constructs (32, 33); GAL4-responsive TATA luciferase reporter construct; and plasmids that express JunD, c-Fos, Runx2, Smad3, or Sp1 have been described elsewhere (34–37).
Knockdown and Reconstitution of DDX5 in Cultured Cells
LX-2 and primary human HSCs were grown in 6-well culture plates to 60–70% confluence. Either a duplex of DDX5 StealthTM siRNA (Invitrogen) or a Stealth RNA interference negative control (Invitrogen) was transfected into LX-2 or primary human HSCs using TransIT-LT1 reagent (Mirus Bio, Madison, WI). After 6 h, the medium was replaced with normal growth medium, and the cells were harvested at 12, 24, and 48 h. In order to reconstitute the DDX5 SNP in LX-2 cells that contain endogenous DDX5, the cells were transiently co-transfected with pSUPERshRNA DDX5 (Table 1), a pSUPER plasmid containing short hairpin RNA targeting the 5′-untranslated region of DDX5, then transfected with DDX5 WT or SNP-containing cDNA expression plasmids 24 h later. Samples for RNA and protein analysis were harvested at 12, 24, and 48 h thereafter.
Generation of Stable HSC Lines Expressing DDX5 WT and SNP
Replication-incompetent lentiviral vectors were prepared by co-transfection of a ViraPowerTM packaging mix (Invitrogen), along with FLAG-tagged DDX5 WT- or SNP-expressing constructs or a pLenti4/TO/V5-GW/LacZ control plasmid, into the 293FT producer cell line mediated by LipofectamineTM 2000 transfection reagent. The medium was changed with Dulbecco's modified Eagle's medium containing 10% fetal bovine serum at 12 h thereafter. The virus-containing supernatants were harvested at 48 h post-transfection by collecting medium and centrifuging at 3,000 rpm for 5 min at 4 °C to pellet and remove cell debris. The viral supernatants were aliquoted and stored at −80 °C.
For infection of lentiviral constructs into the LX-2 line, the cells were plated in 10-cm dishes at 30–50% confluence. The lentiviral stocks of either DDX5 WT, DDX5 SNP, or the control vector were diluted 10-fold with culture medium and incubated with the cells. After 48 h of co-culture, the infected cells were selected in medium containing 500 μg/ml ZeocinTM (Invitrogen). The colonies were expanded and either stored in liquid nitrogen or used directly for experiments.
Expression of Fibrogenic Genes and Their Promoter Activities in DDX5 WT- and SNP-expressing Cells
Stable HSCs lines expressing DDX5 WT or SNP were plated in 24-well plates, 6-well plates, or 10-cm dishes at a density of 2 × 105 cells/ml of 0.5, 2.5, and 10 ml per well or dish, respectively. They were grown overnight and treated with fresh medium containing 2 ng/ml TGF-β1 for 12 or 24 h. The cells and the culture supernatants were collected for mRNA and protein analysis.
For the assessment of promoter activities, the HSC lines plated in 24-well plates were transfected with the α2(I) collagen, TIMP-1, and TGF-β1 promoter luciferase reporter plasmids, along with a Renilla luciferase expression construct (in a ratio of 1:0.005) for 12 h using Fugene HD transfection reagent (Roche Appled Science). The transfected cells were treated with TGF-β1 or vehicle and collected at 12 h thereafter. The promoter activities were assessed as described below.
In separate experiments, the promoter reporter plasmids were co-transfected with cDNAs that express JunD, c-Fos, Sp1, or Smad3 into either LacZ-, DDX5 WT-, or SNP-expressing cells (plasmids of promoter/transcriptional factor/Renilla in a ratio of 1:1:0.005) for 24 h and then treated with TGF-β1 or vehicle for 12 h prior to collection for the assay of promoter activities. Alternatively, the promoter plasmids containing mutations of the AP-1, Sp1, or Smad3 binding sites were transfected, and then cells were assayed for the relative promoter activities, as described below.
In order to test the relative effects of DDX5 WT or SNP sequences on the promoter activity, LX-2 cells were plated into 48-well plates. In each well, a total of 0.8 μg containing varying ratios of DDX5 WT and SNP cDNAs (0.8:0, 0.6:0.2, 0.4:0.4, 0.2:0.6, and 0:0.8) or LacZ control vector were co-transfected with 0.2 μg of promoter luciferase reporter plasmids of α2(I) collagen, TIMP-1, or TGF-β1. The cells were assayed for the promoter activities at 24 h post-transfection.
Reverse Transcription and Real-time Quantitative PCR
RNA was extracted from the cells and reverse transcribed into cDNA using RNeasy® kit (Qiagen, Valencia, CA) and Omniscript reverse transcription kit (Qiagen), respectively, and analyzed by real-time quantitative PCR using SYBR Green real-time quantitative PCR Master Mix (Roche Applied Science) on the lightCycler®480 System (Roche Applied Science). The primers used for α2(I) collagen, TIMP-1, TGF-β1, and glyceraldehyde-3-phosphate dehydrogenase are listed in Table 1.
Immunocytochemistry
LX-2 cells were seeded onto glass coverslips and cultured for 24 h. They were fixed with 4% paraformaldehyde/phosphate-buffered saline and permeabilized by 0.2% Triton/phosphate-buffered saline for 5 min. The immunostaining was performed according to the manufacturer's instructions (DAKO Strept ABC complex horseradish peroxidase kit, Dako (Carpinteria, CA)), using a primary mouse anti-human DDX5 monoclonal antibody (Upstate, Temecula, CA) (1:200 dilution). The coverslips were then incubated with biotinylated link antibody, followed by incubation with peroxidase-labeled streptavidin for 30 min and subsequently stained with diaminobenzidine-H2O2 solution. The cells were dehydrated in ethanol with ascending concentrations and cleared with xylene before being mounted. The staining was captured under a Nikon microscope (Nikon Instruments Inc., Melville, NY).
The localization of FLAG-tagged exogenous human DDX5 WT or SNP protein in LX-2 cells was detected with direct immunofluorescence on cells that were plated in chamber slides (Nalgene Nunc International, Naperville, IL) with anti-FLAG M2-fluorescein isothiocyanate mAb (Sigma) at 10 μg/ml (1:200 in phosphate-buffered saline) at 4 °C overnight, followed by examination under a fluorescence microscope (Nikon Eclipse, Nikon Instruments Inc.).
Western Blot Analysis
Western blots of cell extracts were prepared by pelleting the cells with lysis buffer (Roche Applied Science) complemented with protease inhibitor mixture (Roche Applied Science) and protein phosphatase inhibitor mixtures (Upstate). Protein concentration was determined with the Bradford method using the Bio-Rad DC protein assay kit. Equal amounts of protein were separated by SDS-PAGE; transferred to Hybond C membranes; immunoblotted with specific primary antibodies for α2(I) collagen (Rockland Immunochemicals, Inc., Gilbertsville, PA), TGF-β1, α-SMA (Promega), DDX5 (mAb from Upstate or polyclonal antibody from Bethyl Laboratory Inc. (Montgomery, TX)), FLAG M2, and β-actin (Sigma); and visualized using secondary horseradish peroxidase-conjugated antibodies (GE Healthcare) and the Western chemiluminescent detection system (Millipore Corp., Temecula, CA).
Enzyme-linked Immunosorbent Assay
The culture supernatants were collected from cells in 6-well plates for TIMP-1 analysis at 24 h following incubation with or without TGF-β1 (2 ng/ml) treatment. TIMP-1 was quantified using enzyme-linked immunosorbent assay kits (R&D Systems) according to the manufacturer's instructions.
Luciferase Reporter Assay
The treated cells in 24- or 48-well plates were washed three times with cold phosphate-buffered saline, and cell lysates were prepared using a dual luciferase reporter assay system (Promega). The luciferase activity in 20 μl of each cell lysate was measured using the LumiCount microplate luminometer (Dynex Technologies, Worthing, West Sussex, UK). Changes in firefly luciferase activity were calculated and plotted after normalizing for Renilla luciferase activity in the same sample.
Nuclear Extract Preparation and Co-immunoprecipitation
Nuclear protein extracts were prepared from DDX5 WT- and SNP-expressing HSCs and cells expressing LacZ using a nuclear protein extraction and co-immunoprecipitation kit, following the manufacturer's instructions (Active Motif, Carlsbad, CA). They were quantified for the protein concentrations by Bio-Rad protein assay kit. A portion of the nuclear protein was aliquoted and stored as “input protein,” and the others were used for the assay of the co-precipitation of DDX5 WT and SNP proteins with nuclear transcriptional factors. Each 100 μg of nuclear protein was combined with 5 μg of anti-FLAG M2 mAb (Chemicon International, Temecula, CA) or non-relevant (to the host of primary antibodies) normal serum as negative control in a final volume of 500 μl of complete co-immunoprecipitation/wash buffer provided by the kit and incubated overnight on a rolling shaker at 4 °C. 50 μl of the protein G beads (Active Motif) were added to each tube and incubated for 1 h at 4 °C on a rotator. The beads were collected and carefully washed with co-immunoprecipitation buffer four times, followed by resuspension in 50 μl of 2× reducing loading buffer (130 mm Tris, pH 6.8, 4% SDS, 0.02% bromphenol blue, 20% glycerol, 100 mm dithiothreitol). The samples were boiled for 5 min and microcentrifuged for 30 s at full speed. The supernatants were transferred to a fresh tube. The immunoprecipitated proteins in the supernatants were separated by SDS-PAGE and Western blot using standard methods as described with appropriate primary and secondary antibodies. Primary antibodies used were for human DDX5 (Bethyl Laboratory Inc.), phospho-JunD (Cell Signaling), c-Fos (Cell Signaling), Runx2 (Active Motif), Sp1 (Active Motif), Smad3 (Cell Signaling), and glyceraldehyde-3-phosphate dehydrogenase (Santa Cruz Biotechnology, Inc.).
In order to characterize the formation of DDX5 WT- and/or SNP-containing homodimers in the nuclei of HSCs, LX-2 cells were transfected with the same amount of FLAG-tagged DDX5 WT or SNP cDNAs, along with a construct expressing the GAL4-DBD linked DDX5. Nuclear protein was extracted at 24 h post-transfection. The proteins were immunoprecipitated with anti-FLAG M2 mAb (Sigma), and then Western blot was performed with an antibody to the GAL4-DBD (Santa Cruz Biotechnology, Inc.). The interaction between exogenous DDX5 and HDAC1 was also assayed in this experiment using an HDAC1 monoclonal antibody (Millipore Corp.).
Chromatin Immunoprecipitation (ChIP) Assay
DDX5 WT- and SNP-expressing HSCs and cells expressing LacZ were plated in three 15-cm plates each and grown to 80% confluence. The cells were processed by formaldehyde fixation, chromatin isolation, and enzymatic shearing using a ChIP-ITTM Express kit (Active Motif), following the manufacturer's instructions. An aliquot of the freshly prepared chromatin was reserved as “input DNA,” and the remainder was captured by protein G magnetic beads (Active Motif) with anti-FLAG M2 mAb (Chemicon International) or anti-histone H3 (as a positive control, Active Motif) and nospecific IgG (as a negative control) at 4 °C for 12 h on a rolling shaker. The beads were then carefully washed, and the combined chromatin was eluted. The chromatin in the elution was reimmunoprecipitated by protein G magnetic beads with antibodies for phospho-JunD (Cell Signaling) or Sp1 (Active Motif) at 4 °C for 12 h on a rolling shaker. After washing and eluting, the chromatin DNA was reverse cross-linked and treated with proteinase K at 37 °C for 1 h. The resultant supernatant containing ChIP DNA was immediately used in PCR or stored at −20 °C.
DNA from each ChIP reaction and the input DNA were subjected to PCR amplification using SYBR Green real-time quantitative PCR Master Mix (Roche Applied Science) on the lightCycler®480 system (Roche Applied Science). The primers used were designed to cover the promoter regions that contain cis-acting elements of the Sp1/Smad3/AP-1 binding site on the α2(I)-collagen promoter, AP-1/Runx2/Sp1 sites on the TIMP-1 promoter, and AP-1/Sp1 binding sites on the TGF-β1 promoter, as listed in Table 1.
In a separate experiment, LX-2 cells plated in 15-cm plates were transfected with the same amount of FLAG-tagged DDX5 WT or SNP cDNAs linked to a GAL4-DBD-expressing plasmid. The chromatin was extracted 24 h later and dually immunoprecipitated with anti-FLAG M2 and GAL4-DBD antibodies as described above. PCR amplification was performed using primers for fibrogenic gene promoters.
GAL4 Transactivation Assays
LX-2 cells were plated in 24-well plates and cultured overnight. Plasmids encoding fusion proteins of Sp1 or JunD linked to a GAL4-DBD and the TATA-luciferase reporter plasmid were co-transfected with DDX5 WT- or SNP-expressing vectors or a LacZ-expressing control plasmid and a GAL4-responsive TATA-luciferase reporter, as well as the Renilla luciferase plasmid (in a ratio of 1:1:1:0.005) into the cells using Fugene HD transfection reagent (Roche Applied Science). Cell lysates were collected 24 h later, and the luciferase assays were carried out as described above.
Determination of HDAC Activity
DDX5 WT and SNP cDNAs and a LacZ control vector were transiently transfected into LX-2 cells with Fugene HD reagent. The cells were harvested at 24 h. The nuclear proteins were extracted, and the FLAG-tagged DDX5 proteins were immunoprecipitated with EZviewTM Red anti-FLAG®M2 affinity gel (Sigma). The beads were washed and eluted with assay buffer to assess HDAC activity. The effects of FLAG-tagged DDX5 WT and SNP proteins on HDAC activity were determined by an HDAC (HDAC1 and HDAC2) colorimetric assay kit (BIOMOL International LP) according to the manufacturer's instructions. The color development reflecting HDAC activity was measured at 405 nm and expressed as OD value versus input protein concentration.
Statistics
Results are expressed as mean ± S.E. p values (Student's two-tailed, unpaired t test) from at least three independent determinations, calculated using Microsoft Excel software. Data were considered to be statistically significant with p < 0.05.
RESULTS
HSCs Express DDX5, Which Represses Fibrogenic Gene Expression
Prior to examining the impact of DDX5 on HSC behavior, we first validated its endogenous expression in LX-2 cells, an immortalized human stellate cell line that has previously been well characterized (28). Reverse transcription-PCR (with direct sequencing to confirm product identity), Western blot, and immunocytochemistry (supplemental Fig. 1) confirmed expression of endogenous DDX5. Of note, the DDX5 sequence is wild type in this cell line (not shown).
We examined the biologic activity of DDX5 by transiently knocking down its expression either in LX-2 cells or in primary human stellate cells. Cells were transiently transfected with a Custom DDX5 Stealth siRNA. This was followed by real-time PCR and Western blot for several key fibrogenic genes, including α-SMA, collagen type I, and TGF-β1. As shown in Fig. 1, there was a time-dependent increase in key fibrogenic transcripts in both the human LX-2 cell line (Fig. 1A), and primary human HSCs (Fig. 1B). Similar effects were seen on their protein levels (Fig. 1C).
FIGURE 1.

TGF-β1, α-SMA, and α2(I) collagen mRNA expression is increased after DDX5 knockdown by siRNA in LX-2 and primary human HSCs. A, LX-2 cells were serum-starved for 24 h and then transfected with duplexes of Stealth siRNA® against DDX5 or a non-targeting siRNA control in parallel cultures for 12 h, all at a final concentration of 100 mm. The cells were then cultured in 1% fetal bovine serum-supplemented Dulbecco's modified Eagle's medium and harvested at 12, 24, and 48 h, followed by real-time PCR analysis of TGF-β1, α-SMA, and α2(I) collagen mRNA levels. The results are shown as means ± S.E. of at least three independent experiments (*, p < 0.05; **, p < 0.01 when compared with the control siRNA treated group). B, primary human HSCs of passage 4 were also transfected with the same DDX5 siRNAs, and a non-targeting siRNA was included as a control (100 mm). Cells were treated identically to LX-2 analysis in A and cultured in 10% fetal bovine serum-supplemented Dulbecco's modified Eagle's medium. The mRNA expression was quantified by real-time PCR. The results are shown as means ± S.E. of at least three independent experiments (*, p < 0.05; **, p < 0.01). C, LX-2 cells were transfected with either non-targeting control siRNA (CsiRNA) or siRNA against DDX5 (siRNA) for 12 h after serum starvation for 24 h. The cells were harvested at 12, 24, and 48 h and analyzed by Western blotting with DDX5, α-SMA, collagen type I (Col I), and TGF-β1 antibodies. Their expressions were increased markedly when the DDX5 was knocked down (siRNA) in comparison with non-targeting siRNA (CsiRNA)-treated control cells.
Divergent Activities of DDX5 WT and SNP cDNAs in Hepatic Stellate Cells
We next sought to distinguish the impact of the DDX5 SNP variant on fibrogenic gene expression in LX-2 cells. To accomplish this, we first knocked down endogenous DDX5 in LX-2. Because this siRNA specifically targeted the 5′-untranslated region of endogenous DDX5, we could then reconstitute either WT or risk variant DDX5 by transfecting their respective plasmids. As shown in Fig. 2, comparable expression of either the reconstituted DDX5 WT or variant could be achieved using this approach. Reconstitution of DDX5 using the variant sequence led to higher α-SMA as well as collagen type I protein expression at 24 h compared with that of DDX5 WT.
FIGURE 2.
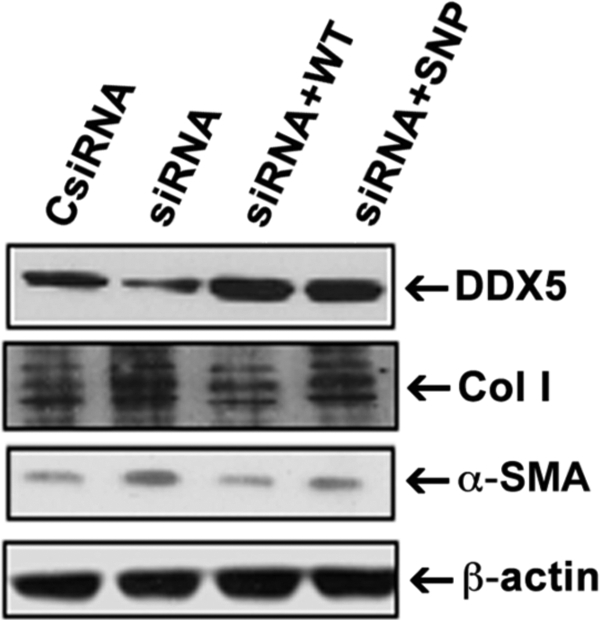
LX-2 cells reconstituted with DDX5 SNP variant have higher collagen type I and α-SMA expression. The siRNA to the 5′-untranslated region of DDX5 was transfected into LX-2 cells to knock down endogenous DDX5 mRNA expression. The cells were then transfected with DDX5 WT- or SNP-expressing cDNAs to reconstitute expression. Cells were lysed 24 h thereafter and analyzed by Western blot for DDX5, α-SMA, and collagen type I. β-Actin was used as a protein loading control. The siRNA-transfected cells display marked reduction in endogenous DDX5, whereas reconstitution by transfection restored expression of either DDX5 wild type-containing (siRNA+WT) or SNP-containing (siRNA+SNP) proteins to equal levels. Expression of α-SMA and collagen type I in LX-2 cells reconstituted with DDX5 SNP protein is higher than that of cells reconstituted with DDX5 WT. The results are representative of three independent experiments.
We next explored the impact of the DDX5 variant on the expression of fibrogenic proteins by stably expressing either wild type or variant DDX5 via lentivirus infection in LX-2 cells. As shown in Fig. 3, using immunofluorescence, high efficiency expression of both the DDX5 WT and variant proteins was observed. Using these lines, we compared fibrogenic gene expression between cells expressing DDX5 WT or risk variant, with or without exogenous TGF-β1. As shown in Fig. 4, although total DDX5 protein was the same in both lines, DDX5 variant-expressing cells had higher expression of basal and TGF-β1-stimulated cellular production of collagen type I and TGF-β1 proteins by Western blots (Fig. 4A) and secreted TIMP-1 by enzyme-linked immunosorbent assay (Fig. 4B).
FIGURE 3.

Expression of exogenous DDX5 in the LX-2 line. Using immunoflurorecence, DAPI nuclear stain, and anti-FLAG M2-fluorescein isothiocyanate monoclonal antibody, exogenous DDX5 protein expression was detectable in LX-2 cells stably transfected with DDX5 WT (a–c) or SNP (d–f) cDNA but not in cells transfected with a LacZ-expressing control vector (g–i). The localization of DDX5 is both to perinuclei and nuclei.
FIGURE 4.

Effect of DDX5 SNP on collagen type I, TIMP-1, and TGF-β1 protein expression in LX-2 cells. A, Western blot for collagen type I (Col I) and TGF-β1 in the cell lysates. B, enzyme-linked immunosorbent assay for TIMP-1 protein level in culture supernatant of LX-2 cells that were stably transfected with DDX5 WT and SNP as well as a control LacZ cDNA with or without TGF-β stimulation. DDX5 SNP-expressing cells had higher basal and TGF-β1-stimulated cellular production of these key fibrogenic genes. Each column represents the mean ± S.E. of n = 6 per group in three independent experiments. *, p < 0.05 when compared with the non-TGF-β1-treated control cells. #, p < 0.01 when compared with the DDX5 WT-expressing cells.
DDX5 SNP-expressing HSCs Have Enhanced Promoter Activities of Key Fibrogenic Genes
We next examined whether the increased expression of fibrogenic mRNAs and proteins in DDX5 SNP-expressing cells was correlated with enhanced transcriptional activity. As shown in Fig. 5, DDX5 variant-expressing cells had enhanced activities of α2(I) collagen, TIMP-1, and TGF-β1 promoter luciferase reporters compared with DDX5 WT-expressing cells or cells expressing LacZ.
FIGURE 5.

Effects of DDX5 SNP on the promoter activities of α2(I) collagen, TIMP-1, and TGF-β1 genes in LX-2 cells. Shown are the promoter activities (expressed as relative light units (RLU)) of α2(I) collagen (A), TIMP-1 (B), TGF-β1 (C) in the cell lysates. LX-2 cells that were stably transfected with DDX5 WT and SNP as well as control LacZ cDNAs with or without TGF-β1 stimulation. Each column represents the mean ± S.E. of n = 6 per group in three independent experiments. Differences between the DDX5 WT- and SNP-expressing cells were significant. *, p < 0.05 when compared with the non-TGF-β1 treated control cells. #, p < 0.01 when compared with the DDX5 WT-expressing cells. ¥, p < 0.05 when compared with the LacZ cDNA-transfected control cells. Differences between LacZ- and SNP-expressing cells were also significant in the absence or presence of TGF-β1 (p < 0.01; no symbols included on the figure to indicate these differences).
Smad3 and AP-1 Signaling Pathways Are Differentially Affected by the DDX5 SNP
A comprehensive analysis was undertaken to implicate specific transcription factors in the differential response to the DDX5 variant. To do so, either the α2(I) collagen, TIMP-1, or TGF-β1 promoter reporter was co-transfected with cDNAs that expressed JunD/c-Fos (two components of the AP-1 complex), Sp1, Smad3, or a control vector PCMV5 into either LacZ-, DDX5 WT-, or SNP-expressing cells (supplemental Fig. 2). Among these, JunD, c-Fos, Sp1, and Smad3 co-transfection specifically increased α2(I) collagen promoter, and JunD and c-Fos increased TIMP-1 and TGF-β1 promoter activities to a greater extent in cells expressing the DDX5 variant than in the DDX5 WT-expressing cells in the presence of TGF-β1. To confirm the role of these transcription factors, similar experiments were performed, but instead we transfected α2(I) collagen or TIMP-1 promoter reporters in which cognate binding sides were mutated for either AP-1, Sp1, or Smad3. As shown in supplemental Fig. 3, mutations in each of these sites abrogated promoter activity.
Based on the altered transcriptional activity of α2(I) collagen, TIMP-1, and TGF-β1, we further tested two key signaling intermediates, Smad3 and AP-1, which regulate each of these promoters, to determine if these convergent pathways might be differentially affected by the DDX5 SNP. To do so, the stable cell lines were transfected with either a Smad3-responsive or AP-1-responsive reporter in the presence or absence of TGF-β1. As shown in Fig. 6, activity of both reporters was markedly increased in response to TGF-β1. Cells expressing the DDX5 variant have significantly greater reporter activities than DDX5 WT-expressing cells.
FIGURE 6.

Effects of DDX5 SNP on Smad3 and AP-1 responsive reporter activities in LX-2 cells. Shown are the activities of Smad3-responsive reporter (A) and AP-1-responsive reporter (B) in the LacZ-, DDX5 WT-, or SNP-expressing cells with or without TGF-β1 stimulation. DDX5 SNP-expressing cells had higher basal and TGF-β1-stimulated induction of Smad3- and AP-1-responsive reporter genes than WT DDX5-expressing cells (expressed as relative light units (RLU)). Each column represents the mean ± S.E. of n = 6 per group in three independent experiments. *, p < 0.01 when compared with the non-TGF-β1-treated control cells. #, p < 0.01 when compared with the DDX5 WT-expressing cells. ¥, p < 0.01 when compared with the LacZ cDNA-transfected control cells.
We further examined whether the altered transcriptional activity of Smad3- and AP-1-responsive reporters by the DDX5 SNP might be due to differential interaction between the DDX5 WT protein and the respective transcription factors compared with the SNP protein. As shown in supplemental Fig. 4, both DDX5 WT and SNP proteins interacted to the same extent with Sp1, Smad3, JunD, and Runx2 based on co-immunoprecipitation. Similarly, both DDX5 WT and SNP were present with JunD and Sp1 to the same extent on the α2(I) collagen, TIMP-1, and TGF-β1 promoters based on a ChIP assay (supplemental Fig. 5).
DDX5 SNP Confers Greater Transactivation of GAL4-Sp1 and GAL4-JunD Reporters
Next, we used a GAL4 one-hybrid system to compare co-regulatory activity of DDX5 WT and variant cDNAs. As shown in Fig. 7, LX-2 cells cotransfected with DDX5 variant cDNA and vectors expressing fusion proteins of Sp1 or JunD linked to a GAL4 DNA-binding domain showed higher transactivation of a GAL4 TATA-responsive reporter than cells that were transfected with DDX5 WT cDNA.
FIGURE 7.

DDX5 SNP confers greater transactivation of a GAL4- responsive reporter. LX-2 cells cotransfected with DDX5 SNP cDNA and vectors expressing fusion proteins of Sp1 (A) or JunD (B) linked to a GAL4-DBD showed higher transactivation of a GAL4-responsive TATA luciferase reporter than that of cells transfected with DDX5 WT cDNA. Each column represents the mean ± S.E. (expressed as relative light units (RLU)) of n = 6 per group in three independent experiments. #, p < 0.01 when compared with the DDX5 WT cDNA-transfected cells. ¥, p < 0.01 when compared with the LacZ cDNA-transfected control cells.
DDX5 WT and SNP both Recruit HDAC1 Activity
We examined whether the increased fibrogenic gene expression associated with DDX5 SNP was due to reduced ability to recruit the HDAC1 co-repressor molecule. However, co-immunoprecititation and ChIP analysis demonstrated that both DDX5 WT and SNP interact equally well with HDAC1 in the cell nuclei (Fig. 8) and in the context of fibrogenic gene promoter regions within HSCs (Fig. 9). Moreover, each was associated with an equal amount of HDAC activity (supplemental Fig. 6).
FIGURE 8.

DDX5 WT and SNP both interact with HDAC1 and form nuclear homodimers. LX-2 cells were co-transfected with the same amount of GAL4 and FLAG fusion DDX5 WT- or SNP cDNA-expressing plasmids. Nuclear protein was immunoprecipitated with anti-FLAG M2 mAb and blotted with antibodies to either GAL4-DBD or HDAC1. Similar interactions between HDAC1 and either DDX5 WT or DDX5 SNP are visible. In contrast, less homodimer complex was detected in cells transfected with only the DDX5 SNP cDNAs (SNP-SNP) or DDX5 WT and SNP cDNAs (WT-SNP) compared with those expressing WT cDNAs alone (WT-WT). Non-relevant normal serum was used as a negative control (Control). The figure is representative of three independent experiments that all yielded similar results. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
FIGURE 9.

Reduced interaction of DDX5 SNP-containing homodimers with the promoter regions of α2(I) collagen, TIMP-1, and TGF-β1. LX-2 cells were co-transfected with the same amount of GAL4 and FLAG-tagged DDX5 WT or SNP cDNA-expressing plasmids. Binding of DDX5 WT or SNP homodimers or of these in complex with HDAC1 on the promoters of α2(I)-collagen, TIMP-1, and TGF-β1 was analyzed by ChIP using anti-FLAG M2 antibody, followed by reimmunoprecipitation with GAL4-DBD or HDAC1 antibodies, and then PCR-amplified. Nonspecific IgG was used as a negative control (Control). Binding of DDX5 SNP protein homodimers to the three promoters was reduced compared with that of the DDX5 WT. Interactions of DDX5 WT or SNP with HDAC1 on the promoter regions were similar. The figure is representative of three independent experiments that all yielded similar results.
Reduced Nuclear Homodimer Formation by DDX5 SNP Protein
As shown in Figs. 8 and 9, following co-transfection of GAL4-DBD and FLAG-tagged-DDX5 WT or SNP cDNAs, there was decreased nuclear homodimer formation and binding on the promoters of fibrogenic genes in DDX5 SNP-transfected cells compared with cells expressing DDX5 WT cDNA, as assessed by co-immunoprecipitation and ChIP. Progressive substitution of DDX5 WT with DDX5 SNP increasingly abrogated the repressive effect of DDX5 WT on fibrogenic promoter activities (Fig. 10), indicating that the repressive effect of DDX5 WT is attenuated by the presence of DDX5 SNP protein in a dimeric complex.
FIGURE 10.

Replacement of DDX5 WT with SNP protein abrogates the repressive effect of DDX5 WT on fibrogenic gene activity. LX-2 cells were plated into 48-well plates. In each well, a total of 0.8 μg of DDX5 WT and SNP cDNAs with different ratios (0.8:0, 0.6:0.2, 0.4:0.4, 0.2:0.6, and 0:0.8) or LacZ control vector were co-transfected with 0.2 μg of promoter luciferase reporter plasmids of α2(I) collagen, TIMP-1, or TGF-β1. The cells were assayed for promoter-luciferase activities (expressed as relative light units (RLU)) at 24 h post-transfection. Introduction of the cDNA combinations containing progressively more DDX5 SNP in place of DDX5 WT gradually abrogated the repressive effect of DDX5 WT on the promoter activities. Each bar represents the mean ± S.E. of n = 8 per group in three independent experiments. *, p < 0.05 when compared with that of the 0.8 μg of WT cDNA-transfected cells. ¥, p < 0.01 when compared with the LacZ cDNA-transfected control cells.
DISCUSSION
Genome-wide scanning has been proven to be an effective approach to discover gene polymorphisms that illuminate underlying disease pathways, are predictive for disease prognosis and treatment response, and permit the generation of clinical risk-predictive models. In large, well characterized clinical cohorts with chronic hepatitis C, DDX5 S480A SNP was identified as a novel genetic component that was significantly associated with advanced fibrosis (7). DDX5 (previously called p68) is a highly conserved and multifunctional (ribosome biogenesis, splicesome, transcription, nonsense-mediated decay, etc.) protein. This DEAD box protein is expressed in multiple tissues, including the liver (38). Moreover, DDX5 interacts with NS5B, the HCV RNA-dependent RNA polymerase, and depletion of endogenous DDX5 correlates with a reduction in the transcription of HCV RNA (23). However, because the DDX5 risk SNP has been associated with fibrosis progression in a non-viral liver disease, its interaction with HCV is not sufficient to explain the observed association of DDX5 with hepatic fibrosis risk. Indeed, our data indicate that DDX5 directly affects the expression of several fibrogenic genes in activated HSCs via interaction with several key transcriptional factors in the context of fibrogenic gene promoters. The findings indicate a critical role of DDX5 in regulating fibrogenic gene transcription in HSCs and validate the power of unbiased genetic studies to identify novel biologic pathways affecting the risk of hepatic fibrosis progression.
Activated HSCs are the major source of extracellular matrix (ECM) during fibrogenesis (1, 25, 39). HSCs also release profibrogenic cytokines with autocrine and paracrine effects, including TGF-β1, and overexpress tissue inhibitors of metalloproteinase (expecially TIMP-1 and TIMP-2), which promote ECM accumulation by inhibiting matrix degradation. In the present study, DDX5 was found to be expressed by primary isolated HSCs and LX-2 cells, an immortalized activated HSC line, and immunolocalized primarily in the nucleus and perinuclear region, consistent with a role of DDX5 in regulating gene transcription in HSCs. DDX5 knockdown by siRNAs in LX-2 cells increased basal and TGF-β1-stimulated up-regulation of α2(I) collagen, TIMP-1, and TGF-β1 expression, whereas overexpression of DDX5 in LX-2 cells yielded opposite effects, suggesting a repressive function of DDX5 on these key fibrogenic genes.
Our data uncover a repressive effect of DDX5 on α2(I) collagen, TIMP-1, and TGF-β1 expression due to an inhibition of promoter activities through the interaction of several fibrogenesis-related transcription factors and HDAC1. In contrast, the DDX5 S480A SNP variant had attenuated repressor activity. Among the candidate transcription factors, we focused on AP-1 because it is a downstream effector of MAPK signaling that contributes to TGF-β1- and platelet-derived growth factor-induced HSC fibrogenesis and proliferation, respectively. Jun factors are critical components of transcriptionally active AP-1 dimers. JunD is the predominant Jun protein expressed in activated HSCs and an AP-1 factor that significantly stimulates TIMP-1. Runx2 regulates the transcription of genes that control ECM remodeling, such as MMP-1 (matrix metalloproteinase 1) and TIMP-1 (35). Sp1 is a ubiquitous transcriptional factor that contributes to basal expression of ECM genes, including several collagens, TIMP-1, and decorin (40), and mediates the transcriptional regulatory function of Smad3 toward the collagen type I promoter (41) by binding to three responsive elements located between bp −303 and −271 (42). Smads are downstream effector molecules of TGF-β1 signaling that regulate cell proliferation, differentiation and the production of ECM. Several transcription factors could combine to form complexes that synergistically mediate the transcription of fibrogenic related genes. Examples of these critical combinations include the cooperation of Smad3/4 complex with AP-1 in mediating TGF-β1-induced transcription (43); JunD and Runx factors assemble at the TIMP-1 promoter to stimulate gene transcription (35), and Sp1 synergistically combines with Smad3/Smad4 to stimulate TGF-β1-mediated induction of α2(I)-collagen transcription (36). HDAC1 belongs to the class 1 HDAC family, which regulates transcription by altering the chromatin structure and affecting histone acetylation/deacetylation. HDAC1 reportedly interacts with DDX5 and DDX17, repressing transcription in a promoter-specific manner. The HDAC inhibitor trichostatin displays strong antifibrogenic effects in cultured skin fibroblasts and in several fibrogetic diseases (44–46), indicating a generally permissive effect of HDAC on fibrogenesis. In our study, however, DDX5 decreases HDAC activity, yet it still represses transcription of key fibrogenic genes.
Our findings add to the series of combinatorial interactions of DDX5 by demonstrating that it binds to JunD, Sp1, Smad3, Runx2, and HDAC1 in nuclear extracts of LX-2 cells. However, there was no difference in binding between DDX5 WT or SNP variant with either transcription factors or HDAC1 on fibrogenic promoters, suggesting that their divergent promoter activities are not due to altered recruitment of DDX5 transcriptional factors or DDX5-HDAC1 complex on the promoters but rather result from another mechanism. Specifically, DDX5 homodimers containing the SNP protein display impaired recruitment to target promoters, leading to enhanced fibrogenic gene expression.
In conclusion, DDX5 is a transcriptional repressor of key fibrogenic genes in hepatic stellate cells, including α2(I)-collagen, TIMP-1, and TGF-β1. This DEAD box protein interacts with transcriptional complexes containing components of JunD, Runx2, Sp1, and Smad3 on gene promoter regions. The enhanced fibrogenic activity of the DDX5 SNP is linked to the loss of repressor function toward these target genes by antagonizing DDX5 WT in a dominant negative fashion. These findings unearth a novel repressor that regulates HSC biology and provide a mechanistic link that explains how a specific DDX5 variant confers enhanced risk of fibrosis progression. These studies also validate the power of genetic association studies to identify gene variants associated with disease risk and to uncover novel pathways of disease pathogenesis.
Acknowledgments
We gratefully thank Drs. Seong Jin Kim, Yutaka Inagaki, Francesco Ramirez, Frank Anania, and Betty Schwartz for providing plasmids and Dr. Hongjin Huang for early discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant DK65521. This work was also supported by a research contract from Celera.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–6.
- HCV
- hepatitis C virus
- α-SMA
- α-smooth muscle actin
- ChIP
- chromatin immunoprecipitation
- ECM
- extracellular matrix
- GAL4-DBD
- GAL4 DNA binding domain
- HSC
- hepatic stellate cell
- HDAC
- histone deacetylase
- mAb
- monoclonal antibody
- siRNA
- small interfering RNA
- SNP
- single nucleotide polymorphism
- TGF-β1
- transforming growth factor β1
- TIMP
- tissue inhibitor of metalloproteinase
- MAPK
- mitogen-activated protein kinase
- WT
- wild type.
REFERENCES
- 1.Friedman S. L. (2008) Gastroenterology 134, 1655–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wise M., Bialek S., Finelli L., Bell B. P., Sorvillo F. (2008) Hepatology 47, 1128–1135 [DOI] [PubMed] [Google Scholar]
- 3.Bataller R., North K. E., Brenner D. A. (2003) Hepatology 37, 493–503 [DOI] [PubMed] [Google Scholar]
- 4.Huang H., Shiffman M. L., Friedman S., Venkatesh R., Bzowej N., Abar O. T., Rowland C. M., Catanese J. J., Leong D. U., Sninsky J. J., Layden T. J., Wright T. L., White T., Cheung R. C. (2007) Hepatology 46, 297–306 [DOI] [PubMed] [Google Scholar]
- 5.Hellier S., Frodsham A. J., Hennig B. J., Klenerman P., Knapp S., Ramaley P., Satsangi J., Wright M., Zhang L., Thomas H. C., Thursz M., Hill A. V. (2003) Hepatology 38, 1468–1476 [DOI] [PubMed] [Google Scholar]
- 6.Martinelli A., Knapp S., Anstee Q., Worku M., Tommasi A., Zucoloto S., Goldin R., Thursz M. (2008) J. Gastroenterol. Hepatol. 23, 1403–1409 [DOI] [PubMed] [Google Scholar]
- 7.Huang H., Shiffman M. L., Cheung R. C., Layden T. J., Friedman S., Abar O. T., Yee L., Chokkalingam A. P., Schrodi S. J., Chan J., Catanese J. J., Leong D. U., Ross D., Hu X., Monto A., McAllister L. B., Broder S., White T., Sninsky J. J., Wright T. L. (2006) Gastroenterology 130, 1679–1687 [DOI] [PubMed] [Google Scholar]
- 8.Nelson J. E., Bhattacharya R., Lindor K. D., Chalasani N., Raaka S., Heathcote E. J., Miskovsky E., Shaffer E., Rulyak S. J., Kowdley K. V. (2007) Hepatology 46, 723–729 [DOI] [PubMed] [Google Scholar]
- 9.Schott E., Witt H., Neumann K., Taube S., Oh D. Y., Schreier E., Vierich S., Puhl G., Bergk A., Halangk J., Weich V., Wiedenmann B., Berg T. (2007) J. Hepatol. 47, 203–211 [DOI] [PubMed] [Google Scholar]
- 10.Wilfred de Alwis N. M., Day C. P. (2007) Semin. Liver Dis. 27, 44–54 [DOI] [PubMed] [Google Scholar]
- 11.Wasmuth H. E., Werth A., Mueller T., Berg T., Dietrich C. G., Geier A., Schirin-Sokhan R., Gartung C., Lorenzen J., Matern S., Lammert F. (2004) J. Mol. Med. 82, 64–69 [DOI] [PubMed] [Google Scholar]
- 12.Huang H., Cheung R. (2008) Textbook of Hepatology: From Basic Science to Clinical Practice (Rodes J., Benhamou J. P., Blei A. T., Reichen J., Rizzetto M. eds) 3rd Ed., pp. 356–363, Blackwell Publishers, Oxford, UK [Google Scholar]
- 13.Camats M., Guil S., Kokolo M., Bach-Elias M. (2008) PLoS ONE 3, e2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caretti G., Schiltz R. L., Dilworth F. J., Di Padova M., Zhao P., Ogryzko V., Fuller-Pace F. V., Hoffman E. P., Tapscott S. J., Sartorelli V. (2006) Dev. Cell 11, 547–560 [DOI] [PubMed] [Google Scholar]
- 15.Fuller-Pace F. V., Ali S. (2008) Biochem. Soc. Trans. 36, 609–612 [DOI] [PubMed] [Google Scholar]
- 16.Bates G. J., Nicol S. M., Wilson B. J., Jacobs A. M., Bourdon J. C., Wardrop J., Gregory D. J., Lane D. P., Perkins N. D., Fuller-Pace F. V. (2005) EMBO J. 24, 543–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warner D. R., Bhattacherjee V., Yin X., Singh S., Mukhopadhyay P., Pisano M. M., Greene R. M. (2004) Biochem. Biophys. Res. Commun. 324, 70–76 [DOI] [PubMed] [Google Scholar]
- 18.Jensen E. D., Niu L., Caretti G., Nicol S. M., Teplyuk N., Stein G. S., Sartorelli V., van Wijnen A. J., Fuller-Pace F. V., Westendorf J. J. (2008) J. Cell. Biochem. 103, 1438–1451 [DOI] [PubMed] [Google Scholar]
- 19.Rossow K. L., Janknecht R. (2003) Oncogene 22, 151–156 [DOI] [PubMed] [Google Scholar]
- 20.Wilson B. J., Bates G. J., Nicol S. M., Gregory D. J., Perkins N. D., Fuller-Pace F. V. (2004) BMC Mol. Biol. 5, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H., Gao X., Huang Y., Yang J., Liu Z. R. (2009) Cell Res. 19, 1388–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogilvie V. C., Wilson B. J., Nicol S. M., Morrice N. A., Saunders L. R., Barber G. N., Fuller-Pace F. V. (2003) Nucleic Acids Res. 31, 1470–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goh P. Y., Tan Y. J., Lim S. P., Tan Y. H., Lim S. G., Fuller-Pace F., Hong W. (2004) J. Virol. 78, 5288–5298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bambha K., Abar O., Chang M., Schrodi S., Ross D., Catanese J., Lagier R., Rowland C., Merriman R., Aouizerat B., Sninsky J., Bass N. (2007) Hepatology 48, 815A [Google Scholar]
- 25.Friedman S. L. (2008) Physiol. Rev. 88, 125–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedman S. L., Rockey D. C., McGuire R. F., Maher J. J., Boyles J. K., Yamasaki G. (1992) Hepatology 15, 234–243 [DOI] [PubMed] [Google Scholar]
- 27.Friedman S. L., Roll F. J. (1987) Anal. Biochem. 161, 207–218 [DOI] [PubMed] [Google Scholar]
- 28.Xu L., Hui A. Y., Albanis E., Arthur M. J., O'Byrne S. M., Blaner W. S., Mukherjee P., Friedman S. L., Eng F. J. (2005) Gut 54, 142–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inagaki Y., Truter S., Greenwel P., Rojkind M., Unoura M., Kobayashi K., Ramirez F. (1995) Hepatology 22, 573–579 [PubMed] [Google Scholar]
- 30.Bahr M. J., Vincent K. J., Arthur M. J., Fowler A. V., Smart D. E., Wright M. C., Clark I. M., Benyon R. C., Iredale J. P., Mann D. A. (1999) Hepatology 29, 839–848 [DOI] [PubMed] [Google Scholar]
- 31.Lin S., Saxena N. K., Ding X., Stein L. L., Anania F. A. (2006) Mol. Endocrinol. 20, 3376–3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim S. J., Glick A., Sporn M. B., Roberts A. B. (1989) J. Biol. Chem. 264, 402–408 [PubMed] [Google Scholar]
- 33.Kim S. J., Jeang K. T., Glick A. B., Sporn M. B., Roberts A. B. (1989) J. Biol. Chem. 264, 7041–7045 [PubMed] [Google Scholar]
- 34.Smart D. E., Vincent K. J., Arthur M. J., Eickelberg O., Castellazzi M., Mann J., Mann D. A. (2001) J. Biol. Chem. 276, 24414–24421 [DOI] [PubMed] [Google Scholar]
- 35.Bertrand-Philippe M., Ruddell R. G., Arthur M. J., Thomas J., Mungalsingh N., Mann D. A. (2004) J. Biol. Chem. 279, 24530–24539 [DOI] [PubMed] [Google Scholar]
- 36.Zhang W., Ou J., Inagaki Y., Greenwel P., Ramirez F. (2000) J. Biol. Chem. 275, 39237–39245 [DOI] [PubMed] [Google Scholar]
- 37.Kim Y., Ratziu V., Choi S. G., Lalazar A., Theiss G., Dang Q., Kim S. J., Friedman S. L. (1998) J. Biol. Chem. 273, 33750–33758 [DOI] [PubMed] [Google Scholar]
- 38.Rössler O. G., Hloch P., Schütz N., Weitzenegger T., Stahl H. (2000) Nucleic Acids Res. 28, 932–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo J., Friedman S. L. (2007) Semin. Liver Dis. 27, 413–426 [DOI] [PubMed] [Google Scholar]
- 40.Verrecchia F., Rossert J., Mauviel A. (2001) J. Invest. Dermatol. 116, 755–763 [DOI] [PubMed] [Google Scholar]
- 41.Inagaki Y., Truter S., Ramirez F. (1994) J. Biol. Chem. 269, 14828–14834 [PubMed] [Google Scholar]
- 42.Tamaki T., Ohnishi K., Hartl C., LeRoy E. C., Trojanowska M. (1995) J. Biol. Chem. 270, 4299–4304 [DOI] [PubMed] [Google Scholar]
- 43.Chung K. Y., Agarwal A., Uitto J., Mauviel A. (1996) J. Biol. Chem. 271, 3272–3278 [DOI] [PubMed] [Google Scholar]
- 44.Rombouts K., Niki T., Greenwel P., Vandermonde A., Wielant A., Hellemans K., De Bleser P., Yoshida M., Schuppan D., Rojkind M., Geerts A. (2002) Exp. Cell Res. 278, 184–197 [DOI] [PubMed] [Google Scholar]
- 45.Wang J. C., Chen C., Dumlao T., Naik S., Chang T., Xiao Y. Y., Sominsky I., Burton J. (2008) Leuk. Lymphoma 49, 2321–2327 [DOI] [PubMed] [Google Scholar]
- 46.Pang M., Kothapally J., Mao H., Tolbert E., Ponnusamy M., Chin Y. E., Zhuang S. (2009) Am. J. Physiol. Renal Physiol. 297, F996–F1005 [DOI] [PMC free article] [PubMed] [Google Scholar]