

Abstract
The farnesoid X receptor (FXR) is a member of the nuclear receptor superfamily that regulates bile acid homeostasis. It is expressed in the liver and the gastrointestinal tract, but also in several non-enterohepatic tissues including testis. Recently, FXR was identified as a negative modulator of the androgen-estrogen-converting aromatase enzyme in human breast cancer cells. In the present study we detected the expression of FXR in Leydig normal and tumor cell lines and in rat testes tissue. We found, in rat Leydig tumor cells, R2C, that FXR activation by the primary bile acid chenodeoxycholic acid (CDCA) or a synthetic agonist GW4064, through a SHP-independent mechanism, down-regulates aromatase expression in terms of mRNA, protein levels, and its enzymatic activity. Transient transfection experiments, using vector containing rat aromatase promoter PII, evidenced that CDCA reduces basal aromatase promoter activity. Mutagenesis studies, electrophoretic mobility shift, and chromatin immunoprecipitation analysis reveal that FXR is able to compete with steroidogenic factor 1 in binding to a common sequence present in the aromatase promoter region interfering negatively with its activity. Finally, the FXR-mediated anti-proliferative effects exerted by CDCA on tumor Leydig cells are at least in part due to an inhibition of estrogen-dependent cell growth. In conclusion our findings identify for the first time the activators of FXR as negative modulators of the aromatase enzyme in Leydig tumor cell lines.
Keywords: Bile Acid, Cytochrome P450, Nuclear Receptors, Transcription Factors, Tumor, Aromatase, Chenodeoxycholic Acid, Farnesoid X Receptor, Leydig Tumor Cells, Steroidogenic Factor-1
Introduction
The farnesoid X receptor (FXR)3 (NR1H4) is a member of the nuclear receptor superfamily of ligand-dependent transcription factors, normally produced in the liver and the gastrointestinal tract, where it acts as a bile acid sensor (1–3). FXR regulates the expression of a wide variety of target genes involved in bile acid, lipid, and glucose metabolism by binding either as monomer or as a heterodimer with the retinoid X receptor (RXR) to FXR response element (FXREs) (4–7). FXR induces the up-regulation of nuclear receptor SHP (small heterodimer partner), which interacts with other nuclear receptors preventing their activation (8–10).
Recently, new functions of FXR beyond its roles in metabolism were discovered in several nonenterohepatic tissues, including its control in regulating cell growth and carcinogenesis (11–14). For instance, it has been demonstrated that FXR activation inhibits breast cancer cell proliferation and negatively regulates aromatase activity reducing local estrogen production, which sustains tumor growth and progression (13).
Estrogen dependence is also a feature of testicular tumor, which is the most frequent solid malignant tumor diagnosed in young men (20–40 years old) accounting for up to 20% of all malignancies diagnosed at this age. Ninety-five percent of all human testicular neoplasms arise from germinal cells, whereas Leydig cell tumors are the most common tumors of the gonadal stroma (15). The molecular basis of testicular cell malignant transformation is poorly defined. It has been reported that estrogen serum levels are elevated in patients with testicular germ cell cancer as a consequence of increased local estrogen production reflecting an higher aromatase activity present in Sertoli and Leydig cells (16). Several studies on both rodents and humans indicate that prenatal, early postnatal, and adult exposure to an excess of estrogens might have a central role in the mechanism leading to male reproductive tract malformations such as testicular and prostatic tumors (17). The biological significance of estrogen-induced testicular tumorogenesis has been suggested by transgenic mice overexpressing aromatase and exhibiting enhancement of 17β-estradiol (E2) circulating levels (18). About half of these male mice were infertile and/or had enlarged testis and showed Leydig cell hyperplasia and Leydig cell tumors (18). Recently, we demonstrated aromatase and ERs expression in testis from patients affected by Leydigioma in which high estradiol levels in the presence of ERα could significantly contribute to tumor cell growth and progression (19). Besides, we also reported that one of the molecular mechanisms determining Leydig cell tumorogenesis is an excessive estrogen production that stimulates a short autocrine loop determining cell proliferation (20).
Aromatase activity is regulated primarily at the level of gene expression by tissue-specific promoters and is present in testicular somatic cells and along the maturative phases of male germ cells (21, 22). A promoter proximal to the translation start site, called promoter II (PII) regulates aromatase expression in fetal and adult testis, R2C and H540 rat Leydig tumor cells, and in purified preparations of rat Leydig, Sertoli, and germ cells (23, 24). Specific sequences seem to be mainly involved in aromatase expression: cAMP-responsive element (CRE)-like sequences binding CREB/ATF protein families (25, 26) and a sequence containing half-site binding nuclear receptors (AGGTCA) in position −90 binding steroidogenic factor 1 (SF-1) (27), which is essential for sex differentiation and development of gonads (28).
On the basis of all these observations, in this study we investigated in rat tumor Leydig cells R2C whether FXR activation by specific ligand chenodeoxycholic acid (CDCA) or a synthetic agonist GW4064 may modulate aromatase expression and antagonize estrogen signaling, inhibiting testicular tumor growth and progression. We demonstrated that the molecular mechanism by which FXR ligands inhibit aromatase gene expression in R2C cells is mediated by a direct binding of FXR to the SF-1 response element present in the aromatase promoter region.
EXPERIMENTAL PROCEDURES
Reagents
Nutrient mixture Ham's F-10, Dulbecco's modified Eagle's medium/Ham's F-12 nutrient mixture, Dulbecco's modified Eagle's medium, l-glutamine, penicillin, streptomycin, fetal bovine serum (FBS), horse serum, phosphate-buffered saline, aprotinin, leupeptin, phenylmethylsulfonyl fluoride, bovine serum albumin, and sodium orthovanadate were purchased by Sigma. TRIzol and Lipofectamine 2000 were from Invitrogen and FuGENE 6 by Roche Applied Science. Taq DNA polymerase, RETROscript kit, 100-bp DNA ladder, Dual Luciferase kit, TnT master mixture, and thymidine kinase Renilla luciferase plasmid were provided by Promega (Madison, WI). SYBR Green Universal PCR Master Mix was from Bio-Rad. Antibodies against FXR, β-actin, GAPDH, cyclin D1, cyclin E, and lamin B were from Santa Cruz Biotechnology (Santa Cruz, CA), antibody against Aromatase from Serotec (Raleigh, NC), antibody against SF-1 was kindly provided from Dr. K. Morohashi (National Institute Basic Biology, Myodaiji-cho, Okazaki, Japan), and anti-LRH-1 antibody was kindly provided by Dr. Luc Belanger (Laval University, Quebec, Canada). The ECL system and Sephadex G-50 spin columns were from Amersham Biosciences. [1β-3H]Androst-4-ene-3,17-dione, [γ-32P]ATP, and [3H]thymidine were from PerkinElmer Life Sciences. Salmon sperm DNA/protein A-agarose was from UBI (Chicago, IL).
Plasmids
The plasmids containing different segments of the rat aromatase PII sequence ligated to a luciferase reporter gene (−1037/+94 (p-1037), −688/+94 (p-688), −475/+94 (p-475), −183/+94 (p-183), and −688/+94 mut (p-688m) (SF-1 site mutant)) were previously described (27). The FXR-responsive reporter gene (FXRE-IR1) and FXR-DN (dominant negative) expression plasmids were provided by Dr. T. A. Kocarek (Institute of Environmental Health Sciences, Wayne State University) (29). The FXR expression plasmid was provided by Dr. D. J. Mangelsdorf (Southwestern Medical Center, TX). SF-1 expression plasmid and the CYP17 gene reporter were obtained from Dr. W. E. Rainey (Medical College of Georgia). XETL plasmid is a construct containing an estrogen-responsive element from the Xenopus vitellogenin promoter, driving expression of the luciferase gene.
Cell Cultures and Animals
Rat Leydig tumor cells (R2C) were cultured in Ham's F-10 supplemented with 15% horse serum, 2.5% FBS, and antibiotics. Mouse Leydig cells (TM3) were cultured in Dulbecco's modified Eagle's medium/Ham's F-12 supplemented with 5% horse serum, 2.5% FBS, and antibiotics. Human cervix tumor cells (HeLa) and hepatoma cells (HepG2) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 1% l-glutamine and antibiotics. The cells were starved in serum-free medium 24 h before treatments. Male Fisher 344 rats (a generous gift of Sigma-Tau), 6 (FRN) and 24 (FRT) months of age, were used for studies. Twenty-four-month-old animals presented spontaneously developed Leydig cell tumors, which were absent in younger animals. Testes of all animals were surgically removed by qualified, specialized animal care staff in accordance with the Guide for Care and Use of Laboratory Animals (NIH) and used for experiments.
Aromatase Activity Assay
The aromatase activity in subconfluent R2C cells culture medium was measured by the tritiated water release assay using 0.5 μm [1β-3H]androst-4-ene-3,17-dione as substrate (30). The incubations were performed at 37 °C for 2 h under an air/CO2 (5%) atmosphere. The results obtained were expressed as picomole/h and normalized to mg of protein (pmol/h/mg of protein).
Total RNA Extraction and Reverse Transcription-PCR Assay
Total RNA was extracted from R2C and TM3 cells using TRIzol reagent and evaluation of gene expression was performed by the reverse transcription-PCR method using a RETROscript kit. The cDNAs obtained were amplified by PCR using the following primers: forward 5′-CAGCTATACTGAAGGAATCCACACTGT-3′ and reverse 5′-AATCGTTTCAAAAGTGTAACCAGGA-3′ (P450 aromatase); forward 5′-TTTCTACCCGCAACAACCGGAA-3′ and reverse 5′-GTGACAAAGAAGCCGCGAATGG-3′ (FXR); forward 5′-CAGCCACCAGACCCACCACAA-3′ and reverse 5′-GAGGCACCGGACCCCATTCTA-3′ (rat-SHP); forward 5′-CGTCCGACTATTCTGTATGC-3′ and reverse 5′-CTTCCTCTAGCAGGATCTTC-3′ (mouse-SHP); or forward 5′-GAAATCGCCAATGCCAACTC-3′ and reverse 5′-ACCTTCAGGTACAGGCTGTG-3′ (L19). The PCR was performed for 25 cycles for P450 aromatase (94 °C for 1 min, 58 °C for 1 min, and 72 °C for 2 min), 35 cycles for FXR (94 °C for 1 min, 65 °C for 1 min, and 72 °C for 2 min), 28 cycles for SHP (94 °C for 1 min, 65 °C for 1 min, and 72 °C for 2 min), and 25 cycles for L19 (94 °C for 1 min, 60 °C for 1 min, and 72 °C for 2 min) in the presence of 1 μl of first strand cDNA, 1 μm each of the primers, 0.5 mm dNTP, Taq DNA polymerase (2 units/tube), and 2.2 mm magnesium chloride in a final volume of 25 μl. DNA quantity in each lane was analyzed by scanning densitometry.
Immunoblot Analysis
R2C, TM3, HepG2 cells, or total tissue of FRNT and FRTT were lysed in 500 μl of 50 mm Tris-HCl, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 2 mm sodium fluoride, 2 mm EDTA, 0.1% SDS, containing a mixture of protease inhibitors (aprotinin, phenylmethylsulfonyl fluoride, and sodium orthovanadate) for protein extraction. Nuclear extracts were prepared as previously described (31). Equal amounts of proteins were resolved on a 11% SDS-polyacrylamide gel, transferred to a nitrocellulose membrane, and probed with FXR, aromatase, cyclin D1, and cyclin E antibodies. To ensure equal loading all membranes were stripped and incubated with anti-lamin B antibody for nuclear extracts or anti-GADPH and anti-β-actin antibodies for total extracts. The antigen-antibody complex was detected by incubation of the membranes with peroxidase-coupled goat anti-mouse, goat anti-rabbit, or donkey anti-goat IgG and revealed using the ECL system. The bands of interest were quantified by the Scion Image laser densitometry scanning program.
Immunofluorescence
R2C cells seeded on glass coverslips were treated with 50 and 100 μm CDCA for 24 h, washed with PBS, and then fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. Next, cells were permeabilized with 0.2% Triton X-100 in PBS for 5 min, blocked with 5% bovine serum albumin for 30 min, and incubated overnight with anti-aromatase antibody (1:100) in PBS overnight at 4 °C. The day after the cells were washed three times with PBS and incubated with the secondary antibody anti-mouse IgG-fluorescein isothiocyanate (1:200) for 1 h at room temperature. To check the specificity of immunolabeling the primary antibody was replaced by normal mouse serum (negative control). Immunofluorescence analysis was carried out on a OLYMPUS BX51 microscope using a ×40 objective.
Transient Transfection Assay
R2C cells were transiently transfected using FuGENE 6 reagent with the FXR reporter gene (FXRE-IR1) in the presence or absence of FXR-DN or XETL plasmid. A set of experiments was performed transfecting rat aromatase PII constructs p-1037, p-688, p-475, p-183, and p-688m. HeLa cells were transiently cotransfected with the CYP17 gene promoter and FXR or SF-1 expression plasmids. After transfection, R2C and HeLa cells were treated with 50 μm CDCA or 3 μm GW4064 for 24 h. Empty vectors were used to ensure that DNA concentrations were constant in each transfection. Thymidine kinase Renilla luciferase plasmid was used to normalize the efficiency of the transfection. Firefly and Renilla luciferase activities were measured by the Dual Luciferase kit. The firefly luciferase data for each sample were normalized based on transfection efficiency measured by Renilla luciferase activity.
RNA Interference (RNAi)
R2C cells were transfected with the RNA duplex of the stealth RNAi targeted rat SHP mRNA sequence, 5′-ACUGAACUGCUUGAAGACAUGCUUU-3′, with RNA duplex of stealth RNAi targeted for the rat FXR mRNA sequence, 5-UCUGCAAGAUCUACCAGCCCGAGAA-3 (Invitrogen), with RNA duplex of the validated RNAi- targeted rat aromatase mRNA sequence, 5-GCUCAUCUUCCAUACCAGGtt-3 (Ambion), or with a stealth RNAi control to a final concentration of 50 nm using Lipofectamine 2000 as recommended by the manufacturer. After 5 h the transfection medium was changed with serum-free medium and then the cells were exposed to treatments.
Electrophoretic Mobility Shift Assay
Nuclear extracts from R2C cells were prepared as previously described (31). The probe was generated by annealing single-stranded oligonucleotides, labeled with [γ-32P]ATP using T4 polynucleotide kinase, and purified using Sephadex G-50 spin columns. The DNA sequences used as probe or as cold competitors were (nucleotide motifs of interest are underlined and mutations are shown as lowercase letters): SF-1, CAGGACCTGAGTCTCCCAAGGTCATCCTTGTTTGACTTGTA; and mutated SF-1, TCTCCCAAtaTCATCCTTGT. In vitro transcribed and translated SF-1 and FXR proteins were synthesized using the T7 polymerase in the rabbit reticulocyte lysate system. The protein-binding reactions were carried out in 20 μl of buffer (20 mmol/liter of HEPES (pH 8), 1 mmol/liter of EDTA, 50 mmol/liter of KCl, 10 mmol/liter of dithiothreitol, 10% glycerol, 1 mg/ml of bovine serum albumin, 50 μg/ml of poly(dI-dC)) with 50,000 cpm of labeled probe, 20 μg of R2C nuclear protein, or an appropriate amount of SF-1 or FXR proteins and 5 μg of poly(dI-dC). The mixtures were incubated at room temperature for 20 min in the presence or absence of unlabeled competitor oligonucleotides. For experiments involving anti-SF-1 and anti-FXR antibodies, the reaction mixture was incubated with these antibodies at 4 °C for 12 h before addition of labeled probe. The entire reaction mixture was electrophoresed through a 6% polyacrylamide gel in 0.25 × Tris borate-EDTA for 3 h at 150 V.
Chromatin Immunoprecipitation and Re-ChIP Assays
R2C cells were treated with 50 μm CDCA for 1 h and then cross-linked with 1% formaldehyde and sonicated. Supernatants were immunocleared with salmon sperm DNA/protein A-agarose for 1 h at 4 °C. The precleared chromatin was immunoprecipitated with specific anti-FXR or anti-polymerase II antibodies, and re-immunoprecipitated with anti-SF-1 or anti-LRH-1 antibodies. A normal mouse serum IgG was used as negative control. Pellets were washed as reported, eluted with elution buffer (1% SDS, 0.1 m NaHCO3), and digested with proteinase K. DNA was obtained by phenol/chloroform/isoamyl alcohol extractions and precipitated with ethanol; 3 μl of each sample were used for PCR amplification with the primers flanking the SF-1 sequence present in the P450arom PII promoter region: 5′- ATGCACGTCACTCTACCCACTCAA-3′ and 5′-TAGCACGCAAAGCAGTAGTTTGGC-3′ and 5-CAGAGGAGAACAGGAAGAGTG-3 and 5-TGATAACGACTCCAGCGTCTT-3 upstream of the SF-1 site. The amplification products were analyzed in a 2% agarose gel and visualized by ethidium bromide staining. Moreover, a 5-μl volume of each sample and input were used for real time PCR.
PCR were performed in the iCycler iQ Detection System (Bio-Rad), using 0.1 μm of each primer, in a total volume of 50 μl of reaction mixture following the manufacturer's recommendations. SYBR Green Universal PCR Master Mix with the dissociation protocol was used for gene amplification. Negative controls contained water instead of DNA. Final results were calculated using the DDCt method as previously reported (20), using input Ct values instead of the 18 S. The basal sample was used as calibrator.
[3H]Thymidine Incorporation
R2C cells were treated with 50 and 100 μm CDCA for 24 and 48 h, respectively. For the last 6 h, [3H]thymidine (1 μCi/ml) was added to the culture medium. After rinsing with PBS, the cells were washed once with 10% and three times with 5% trichloroacetic acid, lysed by adding 0.1 n NaOH, and then incubated for 30 min at 37 °C. Thymidine incorporation was determined by scintillation counting. In other experiments R2C cells were transiently transfected with FXR-DN expression plasmid or transfected with siRNA for FXR or aromatase before starting with the same treatments mentioned above.
Anchorage-independent Soft Agar Growth Assays
R2C cells were plated in 4 ml of Ham's F-10 with 0.5% agarose and 5% charcoal-stripped FBS, in 0.7% agarose base in six-well plates. Two days after plating, medium containing hormonal treatments (androst-4-ene-3,17-dione and CDCA) was added to the top layer, and the appropriate medium was replaced every 2 days. After 14 days, 150 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide was added to each well and allowed to incubate at 37 °C for 4 h. Plates were then placed at 4 °C overnight and colonies >50 μm diameter from triplicate assays were counted. Data are the mean colony number of three plates and representative of two independent experiments.
Statistical Analysis
Each data point represents the mean ± S.D. of three different experiments. Statistical analysis was performed using analysis of variance followed by Newman-Keuls testing to determine differences in means. p < 0.05 was considered as statistically significant.
RESULTS
FXR Expression in Normal and Tumor Testicular Cells
We first aimed to evaluate, by Western blotting analysis, the expression of FXR receptor in Leydig normal (TM3) and tumor (R2C) cell lines and in testes tissue from younger (FRNT) and older (FRTT) Fisher rats. The latter group have a high incidence of spontaneous Leydig cell neoplasma (32, 33), a phenomenon not observed in younger animals. Immunoblot analysis revealed the presence of a FXR-immunoreactive protein band at ∼60 kDa in all samples examined, particularly, the FXR receptor seems to be more expressed in R2C cells with respect to TM3 and in FRTT with respect to its control FRNT (Fig. 1A). Human hepatocyte cells (HepG2) were used as a positive control for FXR expression. In R2C cells, incubation for 24 h with 50 and 100 μm CDCA, a natural ligand of FXR, increased the level of the receptor at both mRNA and protein levels (Fig. 1, B and C). Because CDCA may also exert FXR-independent effects (34), the influence of GW4064, a synthetic FXR agonist, was also investigated. We observed that GW4064 (3 μm) increased FXR mRNA and protein levels to a similar order of magnitude as CDCA (Fig. 1, B and C). Moreover, to assess the ability of CDCA and GW4064 to transactivate endogenous FXR, we transiently transfected R2C cells with the FXR-responsive reporter gene (FXRE-IR1). As reported in Fig. 1D, CDCA and GW4064 induced a significant enhancement in transcriptional activation of the reporter plasmid to a higher extent under GW treatment. In the presence of dominant negative FXR the GW-induced transactivation was completely abrogated.
FIGURE 1.

FXR expression and activation in R2C cells. A, Western blot analysis of FXR was done on 50 μg of total proteins extracted from normal (TM3), tumor Leydig cells (R2C), and human hepatocytes cells (HepG2), or from tissues of normal (FRNT) and tumor (FRTT) Fisher rat testes. β-Actin was used as a loading control. B, total RNA was extracted from R2C cells treated with vehicle (−) or 50 and 100 μm CDCA or 3 μm GW4064 for 24 h and reverse transcribed. cDNA was subjected to PCR using primers specific for FXR or L19 (ribosomal protein). NC, negative control, RNA sample without the addition of reverse transcriptase. The histograms represent the mean ± S.D. (error bars) of three separate experiments in which band intensities were evaluated in terms of optical density arbitrary units and expressed as percentages of the control, which was assumed to be 100%. *, p < 0.05; **, p < 0.01 compared with vehicle. C, nuclear proteins were extracted from R2C cells treated with vehicle (−), 50 and 100 μm CDCA, or 3 μm GW4064 for 24 h and then Western blotting analysis was performed using anti-FXR antibody. Lamin B was used as loading control. The histograms represent the mean ± S.D. of three separate experiments in which band intensities were evaluated in terms of optical density arbitrary units and expressed as percentages of the control, which was assumed to be 100%. *, p < 0.05 compared with vehicle. D, R2C cells were transiently transfected with the FXR reporter gene (FXRE-IR1) and treated as reported above or co-transfected with FXR-DN and treated with vehicle (−) or 3 μm GW4064. The values represent the mean ± S.D. of three different experiments performed in triplicate. *, p < 0.01 compared with vehicle.
Inhibitory Effects of FXR Agonists on Aromatase Expression in R2C Cells
Starting from previous findings showing that FXR activation represses aromatase expression in breast cancer cells (13) we investigated the ability of FXR agonists to modulate aromatase enzyme in R2C cells that have been shown to have high aromatase expression and activity (27). Treatment with 50 and 100 μm CDCA for 24 h showed a down-regulation of aromatase mRNA and protein content in a dose related manner (Figs. 2, A and B). Similar results were observed upon treatment with GW4064 (3 μm) for 24 h (Fig. 2, A and B). The down-regulatory effects of CDCA on the expression of aromatase was further confirmed by immunofluorescence analysis. The strong P450 aromatase immunoreactivity was detected in the cytoplasm as well as in the perinuclear region of untreated R2C cells and was drastically decreased upon CDCA at the doses of 50 and 100 μm for 24 h (Fig. 2C). Next, we evaluated the effects of CDCA on aromatase enzymatic activity by tritiated water release assay. As reported in Fig. 2D, exposure to 50 and 100 μm CDCA for 24 h reduced enzymatic activity in a dose-dependent manner in R2C cells.
FIGURE 2.

Effects of CDCA on aromatase expression and activity in R2C cells. A, total RNA was extracted from R2C cells treated with vehicle (−), 50 and 100 μm CDCA or 3 μm GW4064 for 24 h and reverse transcribed. cDNA was subjected to PCR using primers specific for P450 aromatase or L19. NC, negative control, RNA sample without the addition of reverse transcriptase. The histograms represent the mean ± S.D. (error bars) of three separate experiments in which band intensities were evaluated in terms of optical density arbitrary units and expressed as percentages of the control, which was assumed to be 100%. *, p < 0.05; **, p < 0.01 compared with vehicle. B, total proteins extracted from R2C cells treated with vehicle (−), 50 and 100 μm CDCA, or 3 μm GW4064 for 24 h were used for immunoblot analysis of aromatase. GAPDH was used as a loading control. The histograms represent the mean ± S.D. (error bars) of three separate experiments in which band intensities were evaluated in terms of optical density arbitrary units and expressed as percentages of the control, which was assumed to be 100%. *, p < 0.01 compared with vehicle. C, R2C cells were treated with vehicle (−) or 50 and 100 μm CDCA for 24 h and aromatase expression was determined by immunofluorescence analysis. 4′,6-Diamidino-2-phenylindole (DAPI) staining was used to visualize the cell nucleus. Each experiment is representative of at least 4. D, R2C were cultured in the presence of vehicle (−) or 50 and 100 μm CDCA for 24 h. Aromatase activity was performed as described under “Experimental Procedures.” The results obtained were expressed as picomole of [3H]H2O/h of release and were normalized for milligrams of protein (pmol/mg of proteins/h). The values represent the mean ± S.D. (error bars) of three different experiments each performed with triplicate samples. *, p < 0.01 compared with vehicle.
A direct involvement of FXR in modulating aromatase expression was provided by the evaluation of aromatase mRNA, protein content, and its enzymatic activity after knocking down FXR in R2C cells with a specific siRNA. In preliminary experiments we evaluated, after 24, 48, and 72 h of siRNA transfection, that FXR protein expression was effectively silenced as revealed by Western blotting (Fig. 3A). As shown in Fig. 3, B–D, silencing the FXR gene reversed the down-regulatory effects induced by CDCA on aromatase expression and its enzymatic activity, whereas no change was observed after transfection of cells with scramble siRNA upon identical experimental conditions.
FIGURE 3.

Effects of FXR silencing on aromatase expression in R2C cells. A, FXR protein in R2C cells that were not transfected (−) or transfected with siRNA targeted rat FXR mRNA sequence as reported under “Experimental Procedures” for 24, 48, and 72 h. GAPDH was used as loading control. The histograms represent the mean ± S.D. (error bars) of three separate experiments in which band intensities were evaluated in terms of optical density arbitrary units and expressed as percentages of the control, which was assumed to be 100%. *, p < 0.01 compared with not transfected cells. B–D, R2C cells were transfected with control siRNA or FXR siRNA for 24 h, and then treated with vehicle (−), 50 μm CDCA, or 3 μm GW4064 for 24 h. B, total RNA was extracted and reverse transcription-PCR analysis was performed to evaluate the expression of aromatase. L19 was used as loading control. NC, negative control, RNA sample without the addition of reverse transcriptase. C, total proteins were extracted and Western blotting analysis was performed. GAPDH was used as loading control. The histograms represent the mean ± S.D. of three separate experiments in which band intensities were evaluated in terms of optical density arbitrary units and expressed as percentages of the control, which was assumed to be 100%. *, p < 0.01 compared with vehicle. D, aromatase activity was performed as described under “Experimental Procedures.” The results obtained were expressed as picomole of [3H]H2O/h of release and were normalized for milligrams of protein (pmol/mg of proteins/h). The values represent the mean ± S.D. of three different experiments each performed with triplicate samples. *, p < 0.01 compared with vehicle.
SHP Is Not Involved in the Down-regulatory Effects Induced by FXR Ligand on Aromatase
Induction of SHP expression is considered one of the canonical features of FXR transactivation. SHP has been shown to be expressed in the interstitial compartment of the adult testis, including steroidogenic Leydig cells (35).
We evidenced that SHP mRNA expression was significantly higher in R2C cells compared with very low levels detected in the TM3 cell line, but administration of CDCA or GW4064 did not induce an increase of SHP mRNA in both cell lines (data not shown). However, to explore the role of SHP in CDCA-mediated repression of the aromatase gene, we knocked SHP by siRNA. SHP mRNA expression was effectively silenced as revealed by reverse transcription-PCR after 24, 48, and 72 h of siRNA transfection (Fig. 4A). As shown in Fig. 4, B and C, silencing of the SHP gene failed to reverse the inhibition of aromatase expression induced by the specific FXR ligand in R2C cells ruling out any SHP involvement in the inhibitory effects of CDCA on aromatase expression.
FIGURE 4.
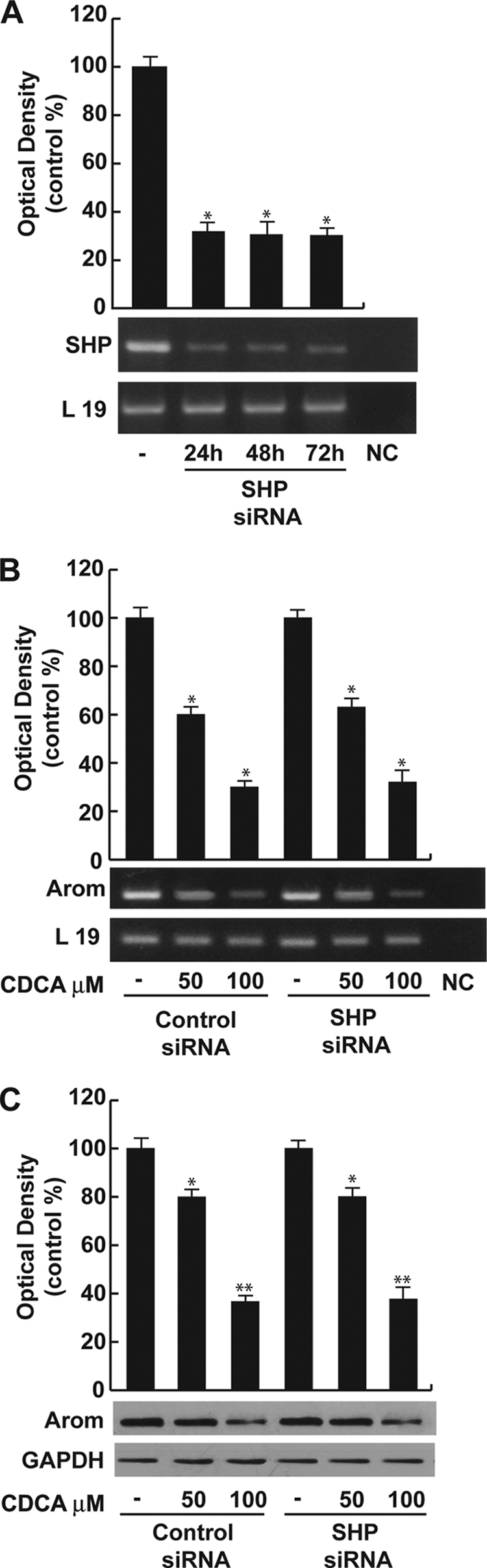
SHP is not involved in CDCA-mediated down-regulation of aromatase. A, SHP mRNA expression in R2C cells that were not transfected (−) or transfected with the siRNA-targeted rat SHP mRNA sequence as described under “Experimental Procedures” for 24, 48, and 72 h. L19 was used as loading control. NC, negative control, RNA sample without the addition of reverse transcriptase. The histograms represent the mean ± S.D. (error bars) of three separate experiments in which band intensities were evaluated in terms of optical density arbitrary units and expressed as percentages of the control, which was assumed to be 100%. *, p < 0.01 compared with not transfected cells. B, R2C cells were transfected with control siRNA or SHP siRNA for 24 h, and then treated with vehicle (−) or 50 and 100 μm CDCA for 24 h. Total RNA was extracted and reverse transcription-PCR analysis was performed to evaluate the expression of aromatase. L19 was used as loading control. The histograms represent the mean ± S.D. of three separate experiments in which band intensities were evaluated in terms of optical density arbitrary units and expressed as percentages of the control, which was assumed to be 100%. *, p < 0.01 compared with vehicle. C, in the same experimental condition as B, total proteins were extracted and Western blotting analysis was performed. GAPDH was used as loading control. The histograms represent the mean ± S.D. of three separate experiments in which band intensities were evaluated in terms of optical density arbitrary units and expressed as percentages of the control, which was assumed to be 100%. *, p < 0.05; **, p < 0.01 compared with vehicle.
CDCA Down-regulates Aromatase Promoter Activity through SF-1 Site
The aforementioned observations led us to ascertain if the down-regulatory effects of CDCA on aromatase expression were due to its direct inhibitory influence in regulating aromatase gene transcriptional activity. Thus, we transiently transfected in R2C cells plasmids containing different segments of rat PII aromatase (Fig. 5A). A significant reduction of promoter activity was observed in cells transfected with p-1037, p-688, p-475, and p-183 exposed to 50 μm CDCA for 24 h. It is worth noting that the construct p-688m bearing the SF-1 mutated site displays significantly lower basal activity compared with the p-688 plasmid, whereas no inhibitory effects were noticeable upon CDCA treatment (Fig. 5B). This latter result highlights the importance of the SF-1 binding site in regulation of aromatase expression in the R2C cells and suggests that the inhibitory effect of CDCA requires AGGTCA sequence motif.
FIGURE 5.

Functional interaction between FXR and the SF-1 site. A, schematic map of the P450arom proximal promoter PII constructs used in this study. All of the promoter constructs contain the same 3′ boundary (+94). The 5′ boundaries of the promoter fragments varied from −1037 to −183. Three putative CRE motifs (5′-CRE at −335; 3′-CRE at −231; XCRE at −169) are indicated as squares. The AGGTCA site (SF-1 RE at-90) is indicated as a rectangle. A mutated SF-1 binding site (SF-1 mut) is present in p-688m (black rectangle). B, aromatase transcriptional activity of R2C cells transfected with promoter constructs are shown. After transfection, cells were treated in the presence of vehicle (−) or 50 μm CDCA for 24 h. These results represent the mean ± S.D. (error bars) of three different experiments performed in triplicate. *, p < 0.01 with respect to the vehicle; **, p < 0.01 with respect to the control of p688. C, HeLa cells were transiently cotransfected with the CYP17 promoter and SF-1 plasmid or empty vector (EV) in the presence of increasing amounts of FXR expression plasmid. These results represent the mean ± S.D. of three different experiments performed in triplicate. In each experiment, the activities of the transfected plasmids were assayed in triplicate transfections. *, p < 0.01 with respect to the EV; **, p < 0.01 with respect to the SF-1 alone.
SF-1 is closely related to liver receptor homologue-1 (LRH-1) and both proteins recognize the same canonical DNA motif (36). However, because LRH-1 is not expressed in R2C cells (supplemental Fig. S1) we focused our attention on the SF-1 transcriptional factor.
To further demonstrate the functional interaction of FXR with SF-1 binding site, we transiently cotransfected HeLa cells, which do not express significant levels of SF-1 (37) with the CYP17 promoter construct containing multiple SF-1 response elements (38) with or without SF-1 plasmid in the presence of increasing amounts of the FXR expression plasmid. The SF-1 expression vector strongly increased the CYP17 promoter activity, which was progressively reduced by increasing amounts of FXR (Fig. 5C). We observed a similar result also in HeLa cells overexpressing FXR and treated with CDCA (data not shown). These data support the competitive role of FXR in the SF-1 binding site.
FXR Protein Binds to SF-1 RE in Vitro and in Vivo
On the basis of the evidence that the inhibitory effect of CDCA on aromatase requires the crucial presence of SF-1 RE, electrophoretic mobility shift assay experiments were performed using the SF-1 motif present in the aromatase promoter as probe. We observed the formation of a complex in nuclear extract from R2C cells (Fig. 6A, lane 1), which was abrogated by 100-fold molar excess of unlabeled probe (Fig. 6A, lane 2) demonstrating the specificity of the DNA binding complex. This inhibition was no longer observed when mutated oligodeoxyribonucleotide was used as competitor (Fig. 6A, lane 3). 50 μm CDCA for 6 h induced an increase in the DNA binding complex compared with control samples (Fig. 6A, lane 4). The inclusion of anti-SF-1 and anti-FXR antibodies in the reactions attenuated the specific bands, suggesting the presence of SF-1 and FXR proteins in the complex (Fig. 6A, lanes 5 and 6). Using SF-1 and FXR proteins transcribed and translated in vitro, we obtained complexes migrating at the same level as that of R2C nuclear extracts (Fig. 6A, lanes 7 and 8). Competition binding studies revealed that both transcribed and translated SF-1 and FXR DNA binding complexes were abrogated by 100-fold molar excess of unlabeled probe (Fig. 6B, lanes 2 and 7). Finally the specificity of these bands was proved by drastic attenuation of the complex in the presence of the anti-SF-1 antibody, whereas the inclusion of anti-FXR antibody completely immunodepleted the binding (Fig. 6B, lanes 3 and 8). IgG did not affect either SF-1 or FXR complex formation (Fig. 6B, lanes 4 and 9).
FIGURE 6.

FXR binds to SF-1 site within the aromatase promoter region. A, nuclear extract from R2C cells were incubated with a double-stranded SF-1-specific sequence probe labeled with [γ-32P]ATP and subjected to electrophoresis in a 6% polyacrylamide gel (lane 1). Competition experiments were performed adding as competitor a 100-fold molar excess of unlabeled probe (lane 2) or a 100-fold molar excess of unlabeled oligonucleotide containing a mutated SF-1 RE (lane 3). Lane 4, nuclear extracts from CDCA (50 μm)-treated R2C cells. Lanes 5 and 6, CDCA-treated nuclear extracts were incubated with anti-SF-1 or anti-FXR antibodies, respectively. We used as positive controls transcribed and translated in vitro SF-1 (lane 7) and FXR (lane 8) proteins. Lane 9 contains probe alone. B, SF-1 protein (lane 1) and FXR protein (lane 6) was incubated with a double-stranded SF-1 sequence probe labeled with [γ-32P]ATP and subjected to electrophoresis in a 6% polyacrylamide gel. Competition experiments were performed adding as competitor a 100-fold molar excess of unlabeled probe (lanes 2 and 7). SF-1 and FXR proteins were incubated with anti-SF-1 antibody (lane 3), anti-FXR antibody (lane 8), or IgG (lanes 4 and 9). Lanes 5 and 10 contain probe alone. C, R2C cells were treated in the presence of vehicle (−) or 50 and 100 μm CDCA for 1 h, then cross-linked with formaldehyde, and lysed. The precleared chromatin was immunoprecipitated with anti-FXR, and anti-RNA Pol II antibodies and normal mouse serum (NC) as negative control. Chromatin immunoprecipitated with the anti-FXR antibody was re-immunoprecipitated with anti-SF-1 antibody. The PII promoter sequence including the SF-1 site and that located upstream of the SF-1 site were detected by PCR with specific primers, as described under “Experimental Procedures,” and D, a 5-μl volume of each sample and input were used for real time PCR. To determine input DNA, the PII promoter fragment was amplified from 30 μl of initial preparations of soluble chromatin before immunoprecipitations. Similar results were obtained in multiple independent experiments. *, p < 0.01 compared with vehicle.
The interaction of FXR with the aromatase gene promoter was further investigated by the ChIP assay. Using specific antibody against FXR and RNA-POL II, formaldehyde cross-linked protein-chromatin complexes were immunoprecipitated from R2C cells cultured with or without 50 and 100 μm CDCA. The resulting genomic DNA precipitated by using anti-FXR was then reprecipitated with the anti-SF-1 antibody. The results analyzed by PCR indicated that FXR was weakly constitutively bound to the aromatase promoter in untreated cells and this recruitment was increased upon CDCA treatment, which was correlated with a reduced association of RNA polymerase II (Fig. 6C). Interestingly, by a re-ChIP assay, we observed upon CDCA stimulation a significant reduction in SF-1 recruitment to the aromatase promoter (Fig. 6C). Moreover, as expected, no involvement of LRH-1 was observed in the re-ChIP experiment (data not shown). Next, the anti-FXR antibody did not immunoprecipitate a region upstream of the SF-1 site located within the aromatase promoter gene (Fig. 6C). The ChIP assay was quantified by real time PCR as shown in Fig. 6D.
CDCA Inhibits R2C Cell Proliferation through FXR Activation
Finally, we evaluated the effect of CDCA on growth of R2C cells by measuring changes in the rate of DNA synthesis ([3H]thymidine incorporation). As shown in Fig. 7A, treatment with CDCA for 24 and 48 h reduced R2C cell proliferation in a dose- and time-dependent manner. The specific involvement of FXR in the antiproliferative response of R2C cells to CDCA was demonstrated by evidence that such inhibitory effects were completely reversed in the presence of the FXR dominant negative plasmid as well as after knocking down FXR with a specific siRNA (Fig. 7, B and C).
FIGURE 7.
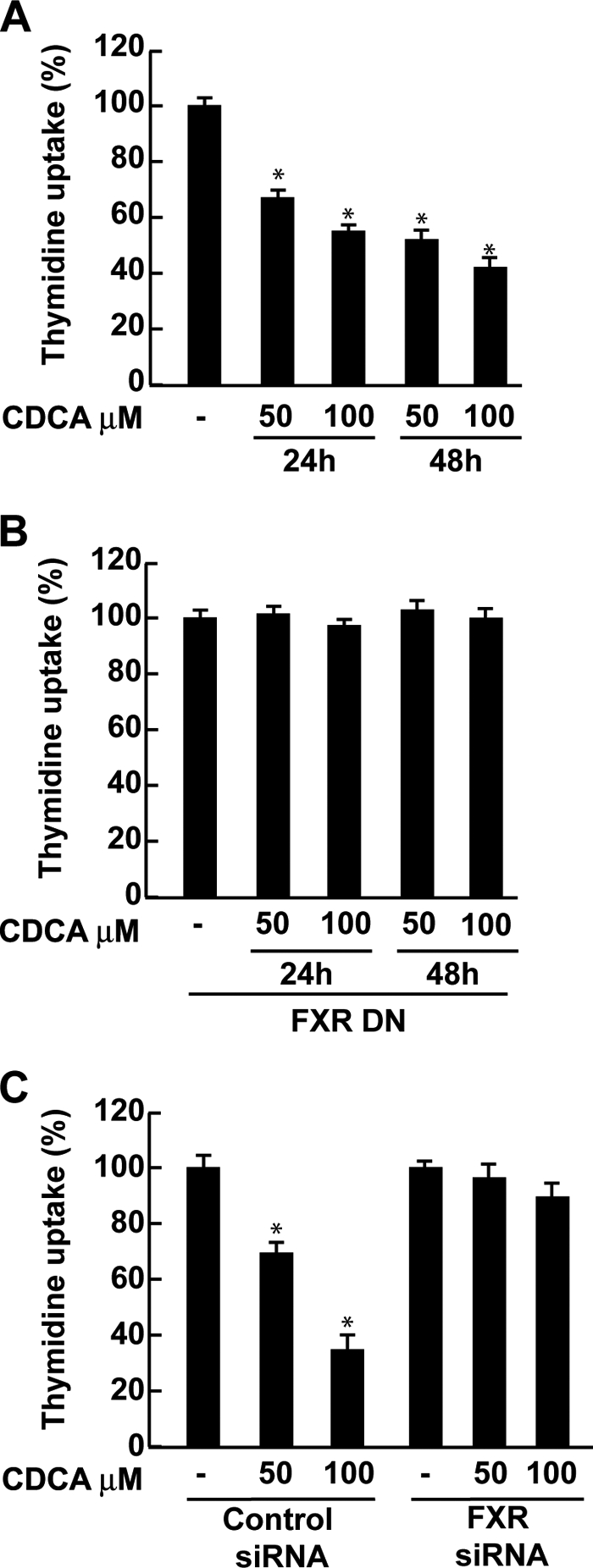
CDCA effects on R2C cell proliferation. A, R2C cells were treated with vehicle (−) or 50 and 100 μm CDCA for 24 and 48 h, or B, transiently transfected with FXR dominant negative (FXR-DN) for 24 h and then treated as reported above, or C, transfected with control siRNA or FXR siRNA for 24 h and treated for 24 h with 50 and 100 μm CDCA. Thymidine incorporation assay was performed. The results represent the mean ± S.D. (error bars) of three different experiments each performed with triplicate samples, and expressed as percentage of growth versus control, which was assumed to be 100%. *, p < 0.01 compared with vehicle.
It is well known that aromatase overexpression in tumor Leydig cells leads to a consequent excess of in situ estradiol production that sustains tumor cell growth and proliferation (18). Because we demonstrated the ability of CDCA to down-regulate aromatase expression and activity in R2C cells, we wondered if CDCA was able to antagonize the effect of an aromatizable androgen androst-4-ene-3,17-dione (AD) on estradiol/ERα signaling in R2C cells. To this aim we performed a transient transfection experiment using the XETL plasmid, which carries firefly luciferase sequences under control of an estrogen response element upstream of the thymidine kinase promoter. As shown in Fig. 8A we observed that the exposure to CDCA (50 μm) per se did not elicit any changes in luciferase activity but completely reversed XETL activation induced by AD. CDCA antagonizes the effect of AD on estradiol/ERα signaling by a FXR-dependent pathway because this effect was not observed when the FXR gene was knocked down (Fig. 8A). Moreover, we examined if CDCA was able to inhibit the effect of AD on R2C cell proliferation using two experimental approaches, thymidine incorporation and anchorage independent soft agar growth assay. As expected, treatment with 100 nm AD, through its conversion into estradiol, increased thymidine incorporation as well as the number of colonies present in soft agar (Fig. 8, B and C) concomitantly with increased levels of cell cycle regulators cyclin D1 and cyclin E (Fig. 8D). All these events were completely reversed by CDCA exposure (Fig. 8, B–D). Finally, we evaluated the effects of AD and/or CDCA on the R2C cell proliferation assay by thymidine incorporation after knocking down aromatase with a specific siRNA (Fig. 8E). As shown in Fig. 8F, as expected AD does not exert proliferative effects on R2C cells, whereas the addition of CDCA can only slightly decrease cell growth. These data demonstrated that the FXR ligand, through a down-regulation of aromatase activity, plays an important role in inhibiting R2C cell proliferation.
FIGURE 8.

CDCA reverses the effects of AD on R2C cell proliferation. A, R2C cells were transfected with control siRNA or FXR siRNA for 24 h and then transiently transfected with the XETL promoter plasmid. Cells were treated with 50 μm CDCA in the with or without 100 nm AD for 24 h. These results represent the mean ± S.D. of three different experiments. In each experiment, the activities of the transfected plasmids were assayed in triplicate transfections. *, p < 0.01 with respect to the vehicle. **, p < 0.01 CDCA + AD treated versus AD alone. B, R2C cells were treated with 100 nm AD in the presence or not of 50 μm CDCA for 24 h. Thymidine incorporation assay was performed. The results represent the mean ± S.D. of three different experiments each performed with triplicate samples. *, p < 0.01 AD treated compared with vehicle. **, p < 0.01 CDCA + AD treated versus AD alone. C, R2C cells were seeded (10,000/well) in 0.5% agarose and treated as described above. Cells were allowed to grow for 14 days and then the number of colonies >50 μm were quantified and the results graphed. The results represent the mean ± S.D. of three different experiments each performed with triplicate samples. *, p < 0.01 AD treated compared with vehicle. **, p < 0.01 CDCA + AD treated versus AD alone. D, total proteins extracted from R2C cells treated with vehicle (−), 100 nm AD, 50 μm CDCA, and AD + CDCA for 24 h were used for immunoblot analysis of cyclin D1 and cyclin E. β-Actin was used as a loading control. The histograms represent the mean ± S.D. of three separate experiments in which band intensities were evaluated in terms of optical density arbitrary units and expressed as percentages of the control, which was assumed to be 100%. *, p < 0.01 AD treated compared with vehicle. **, p < 0.01 CDCA + AD treated versus AD alone. E, aromatase protein in R2C cells that were not transfected (−) or transfected with siRNA targeted rat aromatase mRNA sequence as described under “Experimental Procedures” for 24, 48, and 72 h. GAPDH was used as loading control. F, R2C cells were transfected with control siRNA or Arom siRNA for 48 h and then treated with 100 nm AD in the presence or not of 50 μm CDCA for 24 h. Thymidine incorporation assay was performed. The results represent the mean ± S.D. of three different experiments each performed with triplicate samples. *, p < 0.01 AD treated compared with vehicle. **, p < 0.01 CDCA + AD treated versus AD alone.
DISCUSSION
FXR is highly expressed in the enterohepatic system where it drives bile acid absorption and secretion, lipid, glucose metabolism, and immunological response to intestinal bacterial overgrowth (2, 4, 39–41). In hepatocytes, activation of FXR causes both feedback inhibition of cholesterol 7α-hydroxylase (CYP7A1), the rate-limiting enzyme in bile acid biosynthesis from cholesterol, and activation of intestinal bile acid-binding protein (42). In addition, several observations suggest that FXR may also be involved in control of steroid metabolism (13, 43). Indeed, FXR activation results in modulation of genes encoding androgen precursor-synthesizing enzymes, namely dehydroepiandrosterone sulfotransferase (SULT2A1), 5α-reductase, and 3β-hydroxysteroid dehydrogenase in the liver (44, 45). Recently, FXR was shown to inhibit androgen glucuronidation in prostatic cancer cell lines (46) and suppress the activity of aromatase in human breast cancer cells (13). The enzyme aromatase coded by the CYP19 gene, converts androgens into estrogens and is involved in progression and growth of various estrogen hormonal-induced neoplasms. For instance, overexpression of aromatase plays a significant role in excessive estrogen production sustaining tumorogenesis in Leydig cells (18).
Here, we have documented that FXR is expressed in tissues of normal and tumor Fisher rat testis and in Leydig normal and tumor cell lines. In R2C cells, FXR activators CDCA and GW4064 down-regulate aromatase expression at both mRNA and protein levels, together with the inhibition of its enzymatic activity. Moreover, we demonstrated a direct involvement of FXR in regulating aromatase expression using a specific FXR siRNA.
One of the well characterized mechanisms by which FXR down-regulates gene expression is through induction of SHP (10), an atypical nuclear receptor lacking both a DNA-binding domain and the NH2-terminal ligand-independent activation domain (8). This receptor interacts with other nuclear receptors, including peroxisome proliferator-activated receptor, RXR, ER, and LRH-1, preventing their activation of gene transcription (8–10). In preadipocytes of cancerous breast tissue, LRH-1 can regulate via an alternate promoter (II) the expression of aromatase induced by prostaglandin E2 (47, 48). Moreover, SHP can inhibit LRH-1 induction of aromatase (49). LRH-1 is most homologous to SF1, which is essential for sex differentiation and development of gonads (28), because they share a highly conserved DNA-binding domain (DBD) (>90% identity) and a moderately conserved ligand-binding domain (56% identity). SHP is detected in the interstitial cells of the adult testis and its expression has been shown to be induced by FXR (35).
Our current study revealed that FXR activation does not induce SHP expression in Leydig tumor cells in which inhibition of the aromatase protein by CDCA occurs even when this nuclear receptor was knocked down. These results suggest that SHP is not required for the effect of the FXR ligand to down-regulate aromatase expression, at least in R2C cells. On the basis of these observations, we focused our attention on the direct effect of FXR on the transcriptional activity of aromatase gene.
Distinctive tissue-specific promoters are employed to direct the expression of aromatase mRNA driving from a single aromatase gene. The promoter located immediately upstream of the transcriptional initiation site (PII) regulates aromatase expression in rat Leydig, Sertoli, and germ cells and in R2C Leydig tumor cells (23, 24). A number of functional motifs have been identified in the PII aromatase promoter: three motifs resembling CRE and an SF-1 binding site (27, 28).
We demonstrated by functional studies, using constructs containing different 5′-deleted regions of rat PII aromatase promoter, that CDCA treatment induces a decreased transcriptional activity. The observed inhibitory effect of CDCA was abrogated when a promoter fusion containing a mutated SF-1 element was employed. These results clearly suggest that the integrity of the SF-1 sequence is a prerequisite for the down-regulatory effects of the FXR ligand on aromatase promoter activity. These findings raise the possibility that FXR and SF-1 are competing for binding to a common site within this regulatory region. This assumption is further supported by the observation that FXR expression vector is able to abrogate the induction of SF-1 on the human CYP17 promoter, which contains multiple SF-1 response elements. As a transcription factor, FXR binds to a specific consensus sequence (inverted repeat of 2 AGGTCA half-sites) either as a monomer or as a heterodimer with a common partner for NRs, as RXR to regulate the expression of various genes (6, 7).
Location of an AGGTCA sequence at the −90 position supports a possible binding of FXR to this promoter region, which we verified by electrophoretic mobility shift assay experiments. Nuclear extracts from R2C cells treated with CDCA revealed an increase in the DNA binding complex that was immunodepleted by both anti-SF-1 and anti-FXR antibodies suggesting how the two proteins are able to bind the AGGTCA sequence located in the PII aromatase promoter. The specificity of the binding was proved by the attenuation, in the presence of anti-SF-1 and anti-FXR antibodies, of the DNA complex observed using SF-1 and FXR transcribed and translated in a cell-free system. In addition, the in vivo interaction between FXR and the aromatase promoter was further supported by ChIP assay, where upon CDCA treatment we observed a reduced recruitment of RNA-POLII to this promoter addressing a negative transcriptional regulation mediated by FXR. All together these data suggest that FXR is able to compete with SF-1 in binding to a common sequence within the PII promoter of aromatase interfering negatively with its activity. Finally, in our study we demonstrated that FXR activator CDCA induces growth inhibition in R2C cells, which was reversed in the presence of FXR dominant negative as well as after knocking down FXR with a specific siRNA addressing a FXR dependence of this event. However, it is worth mentioning, on the basis of our recent findings, that aromatase overexpression, in Leydig tumor cells, determines an excessive local estradiol production that is able to stimulate the expression of genes involved in cell cycle regulation sustaining cell proliferation (20).
Here, we evidenced the ability of CDCA to reverse the stimulatory effects of an aromatizable androgen AD at three different levels: 1) E2/ERα signaling; 2) an anchorage dependent and independent R2C cell growth proliferation; and 3) expression of cell cycle regulators cyclin D1 and cyclin E. Finally, knocking down aromatase enzyme reduces estradiol production by R2C cells upon AD exposure and exhibits as biological counterpart a decreased cell proliferation. In the same experimental condition the addition of CDCA can only slightly decrease cell growth demonstrating that the FXR activator through an inhibition of aromatase expression exerts an important role in reducing R2C cell proliferation. In conclusion, our results elucidate, for the first time, a new molecular mechanism through which FXR antagonizes estrogen signaling and inhibits Leydig tumor growth and progression.
Acknowledgments
We thank Dr. T. A. Kocarek, Dr. D. J. Mangelsdorf, and Dr. W. E. Rainey for generously providing the FXR-responsive reporter gene, and FXR-DN, FXR, SF-1, and CYP17 gene reporter plasmids, respectively. We thank Dr. K. Morohashi for providing the anti-SF-1 antibody and Dr. Luc Belanger for the antibody anti-LRH-1.
This work was supported by PRIN-MIUR (Programmi di Ricerca Scientifica di Rilevante Interesse Nazionale-Ministero dell'Istruzione dell'Università della Ricerca) and Associazione Italiana per la Ricerca sul Cancro (AIRC).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- FXR
- farnesoid X receptor
- RXR
- retinoid X receptor
- FXRE
- FXR response element
- SHP
- small heterodimer partner
- E2
- 17β-estradiol
- CDCA
- chenodeoxycholic acid
- FBS
- fetal bovine serum
- RNAi
- RNA interference
- FXRE-IR1
- FXR-responsive reporter gene
- AD
- androst-4-ene-3,17-dione
- ER
- estrogen receptor
- LRH-1
- liver receptor homolog-1
- CRE
- cAMP response element
- CREB
- cAMP response element-binding protein
- SF-1
- steroidogenic factor 1
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- siRNA
- small interfering RNA
- ChIP
- chromatin immunoprecipitation
- PII
- promoter II
- DN
- dominant negative.
REFERENCES
- 1.Forman B. M., Goode E., Chen J., Oro A. E., Bradley D. J., Perlmann T., Noonan D. J., Burka L. T., McMorris T., Lamph W. W., Evans R. M., Weinberger C. (1995) Cell 81, 687–693 [DOI] [PubMed] [Google Scholar]
- 2.Makishima M., Okamoto A. Y., Repa J. J., Tu H., Learned R. M., Luk A., Hull M. V., Lustig K. D., Mangelsdorf D. J., Shan B. (1999) Science 284, 1362–1365 [DOI] [PubMed] [Google Scholar]
- 3.Parks D. J., Blanchard S. G., Bledsoe R. K., Chandra G., Consler T. G., Kliewer S. A., Stimmel J. B., Willson T. M., Zavacki A. M., Moore D. D., Lehmann J. M. (1999) Science 284, 1365–1368 [DOI] [PubMed] [Google Scholar]
- 4.Kalaany N. Y., Mangelsdorf D. J. (2006) Annu. Rev. Physiol. 68, 159–191 [DOI] [PubMed] [Google Scholar]
- 5.Ananthanarayanan M., Balasubramanian N., Makishima M., Mangelsdorf D. J., Suchy F. J. (2001) J. Biol. Chem. 276, 28857–28865 [DOI] [PubMed] [Google Scholar]
- 6.Laffitte B. A., Kast H. R., Nguyen C. M., Zavacki A. M., Moore D. D., Edwards P. A. (2000) J. Biol. Chem. 275, 10638–10647 [DOI] [PubMed] [Google Scholar]
- 7.Claudel T., Sturm E., Duez H., Torra I. P., Sirvent A., Kosykh V., Fruchart J. C., Dallongeville J., Hum D. W., Kuipers F., Staels B. (2002) J. Clin. Invest. 109, 961–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seol W., Choi H. S., Moore D. D. (1996) Science 272, 1336–1339 [DOI] [PubMed] [Google Scholar]
- 9.Seol W., Hanstein B., Brown M., Moore D. D. (1998) Mol. Endocrinol. 12, 1551–1557 [DOI] [PubMed] [Google Scholar]
- 10.Goodwin B., Jones S. A., Price R. R., Watson M. A., McKee D. D., Moore L. B., Galardi C., Wilson J. G., Lewis M. C., Roth M. E., Maloney P. R., Willson T. M., Kliewer S. A. (2000) Mol. Cell. Biol. 6, 517–526 [DOI] [PubMed] [Google Scholar]
- 11.Wang Y. D., Chen W. D., Moore D. D., Huang W. D. (2008) Cell Res. 18, 1087–1095 [DOI] [PubMed] [Google Scholar]
- 12.Journe F., Laurent G., Chaboteaux C., Nonclercq D., Durbecq V., Larsimont D., Body J. J. (2008) Breast Cancer Res. Treat. 107, 49–61 [DOI] [PubMed] [Google Scholar]
- 13.Swales K. E., Korbonits M., Carpenter R., Walsh D. T., Warner T. D., Bishop-Bailey D. (2006) Cancer Res. 66, 10120–10126 [DOI] [PubMed] [Google Scholar]
- 14.Modica S., Murzilli S., Salvatore L., Schmidt D. R., Moschetta A. (2008) Cancer Res. 68, 9589–9594 [DOI] [PubMed] [Google Scholar]
- 15.Hawkins C., Miaskowski C. (1996) Oncol. Nurs. Forum 23, 1203–1211 [PubMed] [Google Scholar]
- 16.Carroll P. R., Whitmore W. F., Jr., Herr H. W., Morse M. J., Sogani P. C., Bajorunas D., Fair W. R., Chaganti R. S. (1987) J. Urol. 137, 420–423 [DOI] [PubMed] [Google Scholar]
- 17.Bosland M. C. (1996) Prog. Clin. Biol. Res. 394, 309–352 [PubMed] [Google Scholar]
- 18.Fowler K. A., Gill K., Kirma N., Dillehay D. L., Tekmal R. R. (2000) Am. J. Pathol. 156, 347–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carpino A., Rago V., Pezzi V., Carani C., Andò S. (2007) Eur. J. Endocrinol. 157, 239–244 [DOI] [PubMed] [Google Scholar]
- 20.Sirianni R., Chimento A., Malivindi R., Mazzitelli I., Andò S., Pezzi V. (2007) Cancer Res. 67, 8368–8377 [DOI] [PubMed] [Google Scholar]
- 21.Aquila S., Sisci D., Gentile M., Carpino A., Middea E., Catalano S., Rago V., Andò S. (2003) Hum. Reprod. 18, 1650–1659 [DOI] [PubMed] [Google Scholar]
- 22.Inkster S., Yue W., Brodie A. (1995) J. Clin. Endocrinol. Metab. 80, 1941–1947 [DOI] [PubMed] [Google Scholar]
- 23.Young M., Lephart E. D., McPhaul M. J. (1997) J. Steroid Biochem. Mol. Biol. 63, 37–44 [DOI] [PubMed] [Google Scholar]
- 24.Lanzino M., Catalano S., Genissel C., Ando S., Carreau S., Hamra K., McPhaul M. J. (2001) Biol. Reprod. 64, 1439–1443 [DOI] [PubMed] [Google Scholar]
- 25.Fitzpatrick S. L., Richards J. S. (1994) Mol. Endocrinol. 8, 1309–1319 [DOI] [PubMed] [Google Scholar]
- 26.Carlone D. L., Richards J. S. (1997) Mol. Endocrinol. 11, 292–304 [DOI] [PubMed] [Google Scholar]
- 27.Young M., McPhaul M. J. (1998) Endocrinology 139, 5082–5093 [DOI] [PubMed] [Google Scholar]
- 28.Parker K. L., Schimmer B. P. (1997) Endocr. Rev. 18, 361–377 [DOI] [PubMed] [Google Scholar]
- 29.Kocarek T. A., Shenoy S. D., Mercer-Haines N. A., Runge-Morris M. (2002) J. Pharmacol. Toxicol. Methods 47, 177–187 [DOI] [PubMed] [Google Scholar]
- 30.Lephart E. D., Simpson E. R. (1991) Methods Enzymol. 206, 477–483 [DOI] [PubMed] [Google Scholar]
- 31.Andrews N. C., Faller D. V. (1991) Nucleic Acids Res. 19, 2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coleman G. L., Barthold W., Osbaldiston G. W., Foster S. J., Jonas A. M. (1977) J. Gerontol. 32, 258–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacobs B. B., Huseby R. A. (1967) J. Natl. Cancer Inst. 39, 303–309 [PubMed] [Google Scholar]
- 34.Nguyen A., Bouscarel B. (2008) Cell. Signal. 20, 2180–2197 [DOI] [PubMed] [Google Scholar]
- 35.Volle D. H., Duggavathi R., Magnier B. C., Houten S. M., Cummins C. L., Lobaccaro J. M., Verhoeven G., Schoonjans K., Auwerx J. (2007) Genes Dev. 21, 303–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pezzi V., Sirianni R., Chimento A., Maggiolini M., Bourguiba S., Delalande C., Carreau S., Andò S., Simpson E. R., Clyne C. D. (2004) Endocrinology 145, 2186–2196 [DOI] [PubMed] [Google Scholar]
- 37.Sugawara T., Holt J. A., Kiriakidou M., Strauss J. F., 3rd (1996) Biochemistry 35, 9052–9059 [DOI] [PubMed] [Google Scholar]
- 38.Hanley N. A., Rainey W. E., Wilson D. I., Ball S. G., Parker K. L. (2001) Mol. Endocrinol. 15, 57–68 [DOI] [PubMed] [Google Scholar]
- 39.Modica S., Moschetta A. (2006) FEBS Lett. 580, 5492–5499 [DOI] [PubMed] [Google Scholar]
- 40.Jung D., Inagaki T., Gerard R. D., Dawson P. A., Kliewer S. A., Mangelsdorf D. J., Moschetta A. (2007) J. Lipid Res. 48, 2693–2700 [DOI] [PubMed] [Google Scholar]
- 41.Inagaki T., Moschetta A., Lee Y. K., Peng L., Zhao G., Downes M., Yu R. T., Shelton J. M., Richardson J. A., Repa J. J., Mangelsdorf D. J., Kliewer S. A. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 3920–3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiang J. Y. (2002) Endocr. Rev. 23, 443–463 [DOI] [PubMed] [Google Scholar]
- 43.Lee F. Y., Lee H., Hubbert M. L., Edwards P. A., Zhang Y. (2006) Trends Biochem. Sci. 31, 572–580 [DOI] [PubMed] [Google Scholar]
- 44.Pircher P. C., Kitto J. L., Petrowski M. L., Tangirala R. K., Bischoff E. D., Schulman I. G., Westin S. K. (2003) J. Biol. Chem. 278, 27703–27711 [DOI] [PubMed] [Google Scholar]
- 45.Miyata M., Matsuda Y., Tsuchiya H., Kitada H., Akase T., Shimada M., Nagata K., Gonzalez F. J., Yamazoe Y. (2006) Drug Metab. Pharmacokinet. 21, 315–323 [DOI] [PubMed] [Google Scholar]
- 46.Kaeding J., Bouchaert E., Bélanger J., Caron P., Chouinard S., Verreault M., Larouche O., Pelletier G., Staels B., Bélanger A., Barbier O. (2008) Biochem. J. 410, 245–253 [DOI] [PubMed] [Google Scholar]
- 47.Clyne C. D., Speed C. J., Zhou J., Simpson E. R. (2002) J. Biol. Chem. 277, 20591–20597 [DOI] [PubMed] [Google Scholar]
- 48.Zhou J., Suzuki T., Kovacic A., Saito R., Miki Y., Ishida T., Moriya T., Simpson E. R., Sasano H., Clyne C. D. (2005) Cancer Res. 65, 657–663 [PubMed] [Google Scholar]
- 49.Kovacic A., Speed C. J., Simpson E. R., Clyne C. D. (2004) Mol. Endocrinol. 18, 252–259 [DOI] [PubMed] [Google Scholar]