

Abstract
The Gram-positive pathogen Streptococcus pyogenes injects a β-NAD+ glycohydrolase (SPN) into the cytosol of an infected host cell using the cytolysin-mediated translocation pathway. In this compartment, SPN accelerates the death of the host cell by an unknown mechanism that may involve its β-NAD+-dependent enzyme activities. SPN has been reported to possess the unique characteristic of not only catalyzing hydrolysis of β-NAD+, but also carrying out ADP-ribosyl cyclase and ADP-ribosyltransferase activities, making SPN the only β-NAD+ glycohydrolase that can catalyze all of these reactions. With the long term goal of understanding how these activities may contribute to pathogenesis, we have further characterized the enzymatic activity of SPN using highly purified recombinant protein. Kinetic studies of the multiple activities of SPN revealed that SPN possessed only β-NAD+ hydrolytic activity and lacked detectable ADP-ribosyl cyclase and ADP-ribosyltransferase activities. Similarly, SPN was unable to catalyze cyclic ADPR hydrolysis, and could not catalyze methanolysis or transglycosidation. Kinetic analysis of product inhibition by recombinant SPN demonstrated an ordered uni-bi mechanism, with ADP-ribose being released as a second product. SPN was unaffected by product inhibition using nicotinamide, suggesting that this moiety contributes little to the binding energy of the substrate. Upon transformation, SPN was toxic to Saccharomyces cerevisiae, whereas a glycohydrolase-inactive SPN allowed for viability. Taken together, these data suggest that SPN functions exclusively as a strict β-NAD+ glycohydrolase during pathogenesis.
Keywords: Enzymes, Enzymes/Catalysis, Enzymes/Kinetics, Organisms/Bacteria, Toxins, Toxins/Drugs/Xenobiotics/Bacterial, Streptococcus
Introduction
The enzymes that cleave the nicotinamide-ribosyl bond of β-NAD+ to produce numerous small molecules are able to modulate various aspects of cellular function, including signal transduction, vascular activity, gene expression, calcium homeostasis, and cell death (reviewed in Refs. 1, 2). All of these enzymes hydrolyze β-NAD+ to produce nicotinamide and adenosine diphosphoribose (ADPR)3 (see Fig. 1 and Table 2). Interestingly, most enzymes within this class are multifunctional and can be further classified on the basis of the additional reactions that they can catalyze following the release of nicotinamide (see Fig. 1 and Table 2). The ADP-ribosyl cyclases (EC 3.2.2.6) convert enzyme-bound ADPR to cyclic ADPR (cADPR) (reviewed in Refs. 3, 4), and the mono-ADP-ribosyltransferases (EC 2.4.4.30, EC 2.4.4.31, and EC 2.4.4.36) covalently link ADPR onto one of several different amino acid acceptors on target proteins (reviewed in Ref. 5). Only the strict β-NAD+ glycohydrolases (EC 3.2.2.5) are not capable of further catalysis of the products of the initial reaction, and, as a general rule, the cyclases and transferases do not catalyze each other's reactions (Fig. 1).
FIGURE 1.

Different classes of β-NAD+ glycohydrolases. Enzymes that hydrolytically cleave the nicotinamide-ribosyl bond of β-NAD+ (shown in red) to form nicotinamide (blue) and ADPR (green and black) are known as β-NAD+ glycohydrolases and are further classified by the additional bonds (if any) whose formation they can catalyze (also shown in red). These include: A, covalent addition of ADPR to a specific substrate (mono-ADP-ribosyltransferases); B, cyclization of ADPR (ADP-ribosyl cyclases); or C, no additional reaction (strict β-NAD+ glycohydrolases). Thin lines indicate other reactions catalyzed by the ADP-ribosyl cyclases. The thin dashed line for the cADPR hydrolase reaction indicates that the cADPR is not a sequential intermediate during the hydrolytic conversion of β-NAD+ to ADPR. *, β to α inversion of the ribosyl linkage after the transfer of ADPR (21, 34).
TABLE 2.
Enzymes that cleave the nicotinamide-ribosyl bond of β-NAD+
Enzymes were based on the classification scheme of Oppenheimer and Handlon (34).
| Propertya | Mono-ADP-ribosyltransferases | NAD+ glycohydrolases |
|
|---|---|---|---|
| Strict NAD+ glycohydrolases | ADP-ribosyl cyclases | ||
| EC number(s) | EC 2.4.2.30 | EC 3.2.2.5 | EC 3.2.2.6 |
| EC 2.4.2.31 | |||
| EC 2.4.2.36 | |||
| kcat β-NAD+ hydrolysis (min−1) | <10 | 103–105 | 103–104 |
| Insensitive to nicotinamide | No | Yes | No |
| β-NAD+ methanolysis | No | No | Yes |
| Transglycosidation | No | No | Yes |
| Generation of cADPR | No | No | Yes |
| cADPR hydrolysis | No | No | Yes |
| NADP+ hydrolysis | No | Yes | Yes |
| ADP-ribosylation of substrates | Yes | No | No |
An exception to the above rule, the Streptococcus pyogenes β-NAD+ glycohydrolase (SPN, also known as Nga) is the only enzyme reported to possess all three activities (6–9). These activities can contribute to S. pyogenes virulence as, after SPN is exported, the enzyme is injected across the host cell membrane into the cytosol by a process called cytolysin-mediated translocation (10, 11). Once in the cytosol, SPN likely contributes to the pathogenesis of the numerous different diseases that S. pyogenes can cause, including pharyngitis, impetigo, necrotizing faciitis rheumatic fever, or acute glomerulonephritis (12). The contribution of SPN to pathogenesis has been demonstrated in several model systems. For example, mutants of S. pyogenes engineered to lack SPN were avirulent in animal models of streptococcal infection (13), and cytosolic SPN was highly cytotoxic to yeast and to cultured epithelial cells (10, 11, 14). However, the mechanism for pathogenesis by SPN remains unknown. Given the number and diversity of the different diseases that S. pyogenes can cause, an understanding of the contribution of SPN to pathogenesis of any streptococcal disease requires a detailed characterization of its kinetic and catalytic properties.
Until recently, characterization of the kinetic properties of SPN has been hindered due to difficulties in expression of recombinant SPN owing to its toxicity. Toxicity is so severe that expression plasmids containing the gene for SPN cannot be introduced into Escherichia coli even under conditions that repress its expression (15, 16). However, identification of an endogenous streptococcal protein that inhibits SPN′s β-NAD+ glycohydrolase activity, immunity factor for SPN (IFS), lead to a coexpression system that allows for production of SPN in E. coli (15, 16). Further characterization of the structure of SPN has revealed that SPN is a modular protein consisting of at least two distinct functional domains. The amino-terminal domain (residues 41–190) aids in SPN translocation into the host cell via cytolysin-mediated translocation (17). The carboxyl-terminal domain (residues 191–451) contains the β-NAD+ glycohydrolase activity, which when expressed alone has activity that is indistinguishable from the full-length protein (17). Identification of the enzymatic domain of SPN has allowed a more refined comparison of its primary structure to those of other multifunctional β-NAD+ glycohydrolases, including ADP-ribosyl cyclases and ADP-ribosyltransferases.
Previous studies of the enzymatic properties of SPN have demonstrated that the enzyme is multifunctional (6–9). However, these studies utilized protein purified from S. pyogenes culture supernatants, which contains numerous bioactive proteins (18) that have high specific activities (12). Thus, definitive conclusions concerning the enzymatic properties of SPN will require careful kinetic analysis of the recombinant enzyme using well established assays that can distinguish between the different classes of enzymes (strict glycohydrolases, cyclases, and transferases) (3, 19, 20). Interestingly, these enzyme classes share similarity in reaction chemistry, in that all mechanisms require an essential glutamic acid to stabilize an oxycarbenium ion intermediate, which subsequently is attacked by a nucleophile (5, 21, 22). For the glycohydrolase reaction, water is the nucleophile; for the transferase reaction, an amino acid residue of the target protein supplies the nucleophile, and for the cyclase reaction, the N1 position of the ADPR adenine ring contributes the nucleophile (see Fig. 1). However, whether an enzyme is an ADP-ribosyl cyclase or transferase has important considerations for the global structure. The cyclases share a highly similar overall architecture and a highly homologous primary sequence that is considerably different from the transferases (22). In contrast, the ADP-ribosyltransferases share little primary sequence homology among themselves, but do share a common overall fold and several short conserved sequence motifs that are involved in binding β-NAD+ (5, 23, 24). Interestingly, SPN shares some primary sequence similarity with both the ADP-ribosyl cyclases (17) and transferases (see below).
In the present study, the enzymatic properties SPN were re-evaluated using recombinant enzyme to further distinguish the β-NAD+ glycohydrolase of SPN from ADP-ribosyl cyclase and transferase activities (Table 2). Recombinant SPN was found to have high β-NAD+ glycohydrolase activity but no detectable ADP-ribosyl cyclase or transferase activity, suggesting that the mechanism for SPN pathogenesis is β-NAD+ depletion within the host cell. Consistent with this, the cytotoxic effect of SPN following expression in the yeast Saccharomyces cerevisiae was found to be associated with β-NAD+ glycohydrolase activity, as an enzymatically inactive variant was not cytotoxic. Analyses of these reactions are important for understanding how SPN is related to other β-NAD+ cleaving enzymes and for understanding how it affects host cells during infection. In addition, analysis of the recombinant protein prepared in the absence of any other streptococcal toxins allowed an unambiguous examination of the type of reactions that the enzyme may catalyze under physiological conditions. The compilation of this information is of critical importance for understanding how SPN contributes to the pathogenesis of S. pyogenes diseases.
EXPERIMENTAL PROCEDURES
Plasmids, Strains, Culture Conditions, and Chemicals
The plasmids used in this study are described in supplemental Table S1. All plasmid-cloning experiments utilized E. coli Top10 (Invitrogen). Recombinant proteins were expressed in Top10 (Invitrogen), the yeast S. cerevisiae strain INVSc1 (Invitrogen) or the methylotropic yeast Pichia pastoris strain X-33 (Invitrogen). SPN-deficient S. pyogenes strain SPN1 (17) derived from wild-type strain JRS4 was used for expression of various mutated versions of SPN. Luria-Bertani broth (LB), and BactoTM Todd-Hewitt broth supplemented with 0.02% BBLTM autolyzed yeast extract (ThyB) were used for routine cultures of Top10 and SPN1, respectively, whereas yeast extract, peptone, and dextrose broth (YPD) was used for routine culture of INVSc1 and X-33. Specific culture conditions for induction of protein expression and transformation are described below. Where appropriate, antibiotics were used to supplement media at the following concentrations: chloramphenicol (Fluka), 7.5 mg/ml for E. coli and 3 mg/ml for S. pyogenes; ampicillin (MidWest Sci, 100 mg/ml) and kanamycin (MidWest Sci, 50 mg/ml) for E. coli and Zeocin (Invitrogen) and 100 mg/ml for P. pastoris. Unless otherwise indicated, all chemicals, solvents, and commercially available enzymes were obtained from Sigma.
Manipulation of DNA
For Transformation of Top10 we used the method of Kushner (25), for S. pyogenes we used the method of Caparon and Scott (26), and for S. cerevisiae and P. pastoris we used the methods recommended by the vendor (Invitrogen). Transformed S. cerevisiae were selected on supplemented SC medium lacking uracil (SC-ura), as described by the vendor (Invitrogen). Plasmid DNA was isolated by standard techniques, and all enzymes, including restriction endonucleases, T4 DNA ligase (New England Biolabs), and DNA polymerases (Pfx, Invitrogen) were used according to the manufacturers' recommendations. All site-specific mutations (except SPN E344G in pJOY48, generated by an inside-out PCR strategy using 5′-phosphorylated primers) described in the text were generated using the QuikChange XL kit (Stratagene) with the oligonucleotide primers listed in supplemental Table S2. Fidelity of all DNA sequences generated by PCR was verified by DNA sequence analyses performed by a commercial vendor (SeqWright, Galveston, TX).
Expression and Purification of Recombinant SPNs
His6-tagged recombinant SPN and SPN E391Q were expressed in E. coli, from pMAM3.18 (16) and pJOY104, respectively. A plasmid for expression of a naturally occurring β-NAD+ glycohydrolase activity-deficient allele of SPN from strain HSC5 (16) was constructed by PCR amplification of genomic DNA (primers listed in supplemental Table S2), followed by exchange of DNA fragments between the BbsI and BstEII sites of the SPN coding region in pMAM3.18. For expression, strains were cultured in low salt LB (0.5% NaCl) supplemented with ampicillin at 25 °C. When the culture reached an A600 of 0.3, expression was induced by the addition of l-arabinose (0.02% final concentration) and allowed to incubate for 16 h at 25 °C. SPN was targeted into the periplasmic space, and extruded by osmotic shock, as described previously (16). This material was concentrated to one-fifth of its original volume by ultrafiltration (Amicon Ultra 15, molecular mass 30,000 kDa), and exhaustively dialyzed against 50 mm sodium phosphate, 300 mm NaCl, pH 7.0 at 4 °C. The protein was bound to cobalt-agarose (TALONTM resin, Clontech) and washed extensively following the manufacturer's protocol. Bound protein was step-eluted with buffer containing 250 mm imidazole. The eluted SPNs were exhaustively dialyzed against 50 mm potassium phosphate, pH 7.4, at 4 °C. The protein was concentrated to a final volume of 1.5 ml by ultrafiltration (Amicon Ultra 4, molecular weight of 10,000) and stored at 4 °C. Concentration of the purified proteins was determined using a BCA assay (Pierce), with a molecular mass of 48.6 kDa, and a single band was visualized on SDS-PAGE stained with Coomassie Brilliant Blue (supplemental Fig. S1).
Expression and Purification of Recombinant CD38
Expression and secretion of a stable and monomeric soluble recombinant CD38 was adapted from a previously described strategy (27). A region encoding the soluble enzymatic domain of human CD38 (residues Arg-45 to Ile-300) was amplified by PCR from pCMV6-XL4-CD38 (Origene Technologies, Cat. no. TC119047; accession number NM_001775.2) using the primers listed in supplemental Table S2. The resulting CD38 PCR fragment with nucleotides coding for 8 additional amino-terminal residues (His6-Gly-Thr) was inserted between the NcoI and XbaI sites of pET24d(+) (Novagen) generating plasmid pJOY 116. To prevent post-translational N-glycosylation of CD38 in P. pastoris, for consistency with previous studies (27), four site-specific mutations (N100D, N164A, N209D, and N219D) were serially introduced using pJOY116 as the initial template. The resulting plasmid (pJOY121, supplemental Table S1) was then used as a template to amplify the region encoding His6-Gly-Thr-CD38 (Arg-45 to Ile-300 with N100D, N164A, N209D, and N219D) (recombinant CD38). This amplimer was digested and inserted between the XhoI and XbaI sites of the P. pastoris expression-secretion vector pPICZα B (Invitrogen, pJOY123). To facilitate chromosomal integration, the plasmid was linearized by digestion with SacI and used to transform P. pastoris. One resulting isolate was chosen for subsequent expression of secreted recombinant CD38, which was conducted in batches of 1.6 liters according to the recommendations of the vendor (Invitrogen). For purification, the supernatant from a 48-h post-induction culture was harvested by centrifugation, filter sterilized (0.22-μm filter) to remove cell debris, and concentrated to a final volume of 30 ml (Centricon Plus-70, molecular mass 10,000). Recombinant CD38 was further purified by metal affinity chromatography as described above. Eluted protein was extensively dialyzed against 50 mm potassium phosphate, pH 7.4, concentrated to a final volume of 1.5 ml (Amicon Ultra 4, molecular mass 10,000), and stored at 4 °C. Concentration of the purified protein was determined by using a BCA assay (Pierce), with a molecular mass of 31 kDa for monomeric CD38, and purity was assessed to be homogeneous by SDS-PAGE (supplemental Fig. S1).
β-NAD+ Glycohydrolase Activity of SPN from S. pyogenes
The β-NAD+ glycohydrolase activities of the various SPN constructs were determined in culture supernatants using a fluorometric assay (11) modified as described (17). The concentrations of SPN in the culture supernatants were normalized by immunoblotting to detect a hemagglutinin epitope tag as previously described (17). Specific activities reported are relative to the rate obtained for the wild-type strain. Data presented are derived from three independent experiments, each performed in triplicate.
β-NAD+ and β-NADP+ Glycohydrolase Activities of Recombinant Enzymes
The glycohydrolase activities were monitored by analytical HPLC. Briefly, initial rates of hydrolysis of β-NAD+ were measured by incubation of various concentrations of β-NAD+ with SPN (0.02 pmol), SPN E391Q (20.6 pmol), or CD38 monomer (0.16 pmol) in a 530-μl final volume of assay buffer (50 mm potassium phosphate, pH 7.4, plus 0.5 mg/ml BSA) at 37 °C. Aliquots of the reactions were quenched by addition to an equal volume of 20% ice-cold perchloric acid, to precipitate the enzyme and the BSA. The protein precipitates were removed by centrifugation, and the supernatants were diluted in deionized water. All samples were analyzed by reversed-phase HPLC (SunFire, C18 column, Waters, 5 μm, 4.6 × 250 mm), which was developed isocratically with 1% (v/v) acetonitrile in buffer A (10 mm diammonium phosphate buffer, pH 6.4) with a flow rate of 1.5 ml/min over 22 min. The concentrations of β-NAD+ left at various times during the reaction were determined by comparison to a standard β-NAD+ concentration curve run under the same conditions. The initial rate of β-NAD+ hydrolysis for each substrate concentration was determined from linear regression analyses of plots of substrate depletion (less than 10% substrate consumption) versus time. Plots of initial rates of substrate depletion as a function of substrate concentration were analyzed using the Michaelis-Menten equation to determine the Km,app and the kcat for the reaction. β-NAD+ glycohydrolase product inhibition experiments were performed in the presence of increasing concentrations of free nicotinamide or ADP-ribose. Analysis of the inhibition of the enzyme by ADP-ribose was analyzed using the product inhibition equation for an ordered uni-bi system (19, 28) as follows.
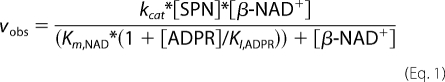 |
Reciprocal plots of the data were also constructed using the reciprocal of the above equation.
β-NADP+ glycohydrolase activity was also monitored by analytical HPLC with the following modifications. SPN (0.4 pmol) was incubated in assay buffer at 37 °C with various concentrations of β-NADP+. Aliquots of the reactions from different time points were quenched by addition to 9 volumes of ice-cold 1 m phosphoric acid to precipitate the enzyme and BSA. After removal of the protein precipitates by centrifugation, samples were analyzed by HPLC to determine the Km,app and the kcat for the reaction as described above. Non-linear least squares analyses of the data were performed using either Prism 5 software (GraphPad) or Scientist software (MicroMath). The errors of the fitted parameters are reported as ±2 S.D.
cADPR Hydrolase Activity
cADPR hydrolase activity of SPNs were evaluated using a similar HPLC method described above. The enzymes (SPN, SPN E391Q, or recombinant CD38, 0.5 ng/μl) were incubated with 1.0 mm cADPR in assay buffer at 37 °C for 6 h (SPN and SPNE391Q) or 1 h (CD38). Reactions were quenched by ultrafiltration (Microcon Ultra YM3, molecular weight 3000) to obtain cADPR substrate and ADPR products in the flow through. Filtrates were diluted 10-fold in deionized water and analyzed by HPLC as described above.
ADP-ribosyl Cyclase Activity
Cyclase activity of SPNs was evaluated by adaption of a previously described cycling assay (29) and also by measuring the conversion of nicotinamide guanine di-nucleotide into cyclic GDP-ribose (30). In the cycling assay, enzymes (SPN, SPN E391Q, recombinant CD38, or Aplysia cyclase, 0.2 ng/μl) were mixed with 10 mm β-NAD+ in buffer (100 mm sodium phosphate, pH 8.0, plus 0.1 mg/ml BSA) and incubated at 37 °C for 20 min. Products generated from the cyclase activity were obtained by ultrafiltration (Microcon YM3, molecular mass 3000). Filtrates containing products were incubated with 2× hydrolytic enzyme mix (0.016 unit/ml Neurospora crassa β-NAD+ glycohydrolase, 0.88 unit/ml Crotalus adamanteus venom nucleotide pyrophosphatase, 25 units/ml calf intestinal alkaline phosphatase, 0.2 mg/ml BSA, and 5 mm MgCl2) at 37 °C for 18 h to remove all nucleotides except cADPR, which was then detected by a coupled fluorescence assay as previously described (29). Increases in resorufin fluorescence (λex 544 nm/λem 590 nm) were monitored every 5 min for 4 h on a PerkinElmer LS55 with a plate reader accessory.
β-NAD+ Methanolysis Activity
SPNs were evaluated for their ability to methanolyze β-NAD+ using a previously described method (31). Briefly, SPN, SPN E391Q, and control CD38 (0.1 ng/μl) were mixed with β-NAD+ (500 μm) in assay buffer containing 10% (v/v) methanol. Reactions were incubated at 37 °C for 20 min and quenched by removal of the proteins with ultrafiltration (MicroCon Ultracel YM3, molecular weight 3000). Filtrates were diluted and analyzed by reversed-phase HPLC with minor modifications to the method as described above. The column (μBondapak C18, Waters, 10 μm, 3.9 × 300 mm) was developed with 0.9% acetonitrile in buffer A, and the presence or absence of α-methyl- and β-methyl-ADP-ribose were determined by comparison to the respective standards prepared as described (31). Non-enzymatic methanolysis was conducted at 85 °C for 45 min.
Transglycosidation Activity
The transglycosidation activity of SPNs was evaluated in the following manner. Either SPN, SPN E391Q, or recombinant CD38 (0.1 ng/μl) was mixed with β-NADP+ (1 mm) and nicotinic acid (20 mm) in buffer (68.2 mm potassium phosphate, pH 6.0, 0.5 mg/ml BSA) for 30 min at 37 °C to allow transglycosidation to occur. Reactions were quenched by ultrafiltration, and filtrates were analyzed by reversed-phase HPLC (Atlantis T3 C18, Waters, 5 μm, 4.6 × 250 mm). The column was developed with buffer A for 5 min, followed by a 25-min linear gradient to 2.5% acetonitrile in buffer A, concluding with a 5-min wash step of buffer A. Transglycosidase activity was evaluated by the appearance of nicotinic acid adenine dinucleotide phosphate within the chromatogram.
Analysis of ADP-ribosylation Activity
The ability of the enzymes to ADP-ribosylate target proteins was evaluated using poly-l-arginine as described (6). A second ADP-ribosyltransferase assay was performed using HeLa whole cell extracts (Active Motif, Carlsbad, CA). Briefly, enzymes (10 ng/μl) were incubated with HeLa whole cell extract (1.25 mg/ml) and [32P]adenylate-NAD+ (1 μm, from PerkinElmer) in assay buffer containing BSA (0.1 mg/ml) for 1 h at 37 °C. Control reactions were performed with exotoxin A from Pseudomonas aeruginosa (List Biological Laboratories, Campbell, CA). Reactions were quenched by addition of SDS-PAGE sample buffer and boiling for 10 min. Samples were analyzed by SDS-PAGE. Gels were fixed, dried, and subjected to autoradiography to visualize the ADP-ribosylated proteins.
Cytotoxicity for Yeast
Genes encoding SPN and SPN lacking β-NAD+ glycohydrolase activity (from strains JRS4 and HSC5, respectively) were inserted into pYES2 (Invitrogen) in the absence of IFS and under the control of a galactose-inducible promoter (supplemental Table S1) and used to transform S. cerevisiae. Growth characteristics of transformants were analyzed as follows: an overnight culture in SC-ura containing 2% glucose was used to inoculate 50 ml of SC-ura supplemented with 2% galactose to an A600 of 0.4. This culture was incubated at 30 °C with shaking, and 5-ml aliquots were removed at the times indicated in the text. The A600 was determined for each sample, the cells were then harvested by centrifugation and resuspended in 500 μl of breaking buffer (50 mm sodium phosphate, pH 7.4, 1 mm EDTA, 5% glycerol, 1 mm phenylmethylsulfonyl fluoride) along with an equal volume of glass beads (cat. no. G8772, Sigma). The cells were disrupted using a reciprocating shaking device (FastPrep-24, MP Biomedicals) at a speed setting of 6 for 3 times at 30 s. Debris was removed by centrifugation, and the supernatants were analyzed by Western blotting as described above.
RESULTS
Identification of the Putative Catalytic Glutamic Acid Residue
Identification of the catalytic residues involved in hydrolysis of β-NAD+ were preformed based on sequence homologies to other ADP-ribosyl cyclases and transferases. Although these two classes of enzymes do not share significant sequence homology, their active sites both contain an essential catalytic glutamic acid residue. ADP-ribosyl cyclases have very high similarities in their primary sequences and three-dimensional structures. We previously noted some homology between SPN and several ADP-ribosyl cyclases (17). From this, we predicted Glu-344 of SPN as the essential catalytic residue (Fig. 2A). However, this assignment was confounded by a number of significant differences. The ADP-ribosyl cyclase monomers have 10 conserved cysteine residues forming five intra-molecular disulfide bonds (3, 4), whereas SPN contains only a single cysteine residue, SPN is monomeric in solution (16), but ADP-ribosyl cyclases are dimeric. Also missing in SPN are two tryptophan residues conserved in the ADP-ribosyl cyclases that are involved in substrate binding (17). In contrast to the ADP-ribosyl cyclases, the ADP-ribosyltransferases do not share extensive homology among themselves but do share a conserved ARTT motif (ADP-ribosylating turn-turn motif) (24). This motif is involved in binding substrate and typically contains a bi-glutamic acid containing a catalytic glutamic acid residue (24). Examination of the SPN primary sequence revealed a putative ARTT motif (Fig. 2B), implicating additional potential catalytic glutamic acid residues.
FIGURE 2.

SPN has homology with both ADP-ribosyl cyclases and ADP-ribosyltransferases. The carboxyl-terminal enzymatic domain of SPN (residues 234–451) shares some homology with the enzymatic domains of eukaryotic ADP-ribosyl cyclase (A), and prokaryotic ADP-ribosyltransferases (B). Alignment of the catalytic glutamic acid residue of the ADP-ribosyl cyclases is the highlighted box and open arrow in A. Additional glutamic acid residues of SPN subjected to mutagenesis are indicated by the open arrows (A). The putative ARTT domain of SPN is highlighted by the bar over the sequence, and the putative catalytic glutamic acid residue and second glutamic acid/glutamine residue are shown by the closed arrows (A). A comparison of the putative ARTT motif of SPN to those characterized ADP-ribosyltransferases (24) is shown (B). The catalytic and associated glutamic acid/glutamine residues are highlighted. The two “turns” of the ARTT motif (5) are indicated (T1 and T2). Alignments (A) were generated using ClustalW (45).
To determine which, if any, of these glutamic acid residues may represent the catalytic residue of SPN, Glu-344, predicted from comparison to cyclases, and Glu-389 and Glu-391, predicted to be in the putative ARTT motif (Fig. 2), were mutated to glycine and glutamine residues. Two more potential glutamic acid residues (Glu-326 and Glu-348) flanking the putative cyclase active glutamic acid (Glu-344) were also mutated, to ensure coverage (Fig. 2A). Initially, the mutants were analyzed for their ability to hydrolyze β-NAD+ using the fluorescence assay. Mutation of the cyclase-like residues (Glu-326, Glu-344, and Glu-348) had only a modest effect on glycohydrolase activity as compared with wild type (Fig. 3). In contrast, changing either residue of putative ARTT motif (Glu-389 or Glu-391) to glycine reduced activity to near background levels (Fig. 3). Conversion of the first SPN residue (Glu-389) to glutamine did not reduce activity, whereas a similar mutation at the second position (Glu-391) reduced activity to <1.0% as compared with the wild-type enzyme (Fig. 3). These results are consistent with data observed for other ARTT motifs. In general, the first conserved residue contributes to β-NAD+ binding and is sometimes a glutamine residue (Fig. 2B), while the second conserved residue functions as the catalytic residue and is invariantly a glutamic acid (24). These data suggest that SPN contains a functional ARTT motif and implicates Glu-391 as a catalytic residue.
FIGURE 3.

SPN has a functional ARTT motif. Mutant versions of SPN were constructed to change specific glutamic acid residues (E) of the putative “cyclase” or “ARTT” motifs highlighted in Fig. 3 to glycine (G) or glutamine (Q) residues, as indicated. Mutant proteins were expressed in S. pyogenes SPN1, and culture supernatants were analyzed for β-NAD+ glycohydrolase activity relative to wild-type JRS4 SPN (WT), as shown. Data presented represents the mean and standard error of the mean derived from three independent experiments.
Kinetic Parameters for SPN Hydrolysis of β-NAD+
To further distinguish the catalytic activities of SPN, the enzyme was subjected to kinetic analysis. Initially, the substrate specificity of SPN for α-NAD+ or β-NAD+ was tested. Cyclases, transferases, and strict glycohydrolases all share one common characteristic in that they all readily hydrolyze β-NAD+, but none of them cleave α-NAD+ (20, 32, 33). Purified recombinant SPN readily hydrolyzed β-NAD+, but no hydrolysis of α-NAD+ could be detected (data not shown). The strict glycohydrolases and the ADP-ribosyl cyclases can be distinguished from the ADP-ribosyltransferases by comparison of their turnover numbers (kcat) for β-NAD+ hydrolysis, which for the latter are typically low (34). The kinetic parameters for the β-NAD+ glycohydrolase reaction were determined using a sensitive HPLC-based assay (Table 1). The observed turnover number for SPN (Table 1) was similar to that determined for a related β-NAD+ glycohydrolase purified from a group C streptococcal species (12,000 s−1) (35). Comparison of this value to the reported kcat for the ADP-ribosyltransferase diphtheria toxin fragment A (0.0083 s−1) (33) shows the turnover number for SPN is ∼1 × 107-fold higher, suggesting the activity may resemble that of the strict β-NAD+ glycohydrolases and not that of ADP-ribosyltransferases. The Km,app (Table 1) determined using the HPLC assay was ∼2-fold lower than we previously reported for SPN purified from culture supernatant (379 μm) (16). The decrease in the Michaelis constant seen here can be attributed to using purified enzyme produced in E. coli and a more sensitive method of product detection.
TABLE 1.
Comparison of the kinetic properties of SPN and CD38
| Enzymea | Variantb | Propertyc |
|
|---|---|---|---|
| Km | kcat | ||
| μm | s−1 | ||
| SPN | WT | 188 ± 23 | 8,390 ± 360 |
| E391Q | 294 ± 47 | 2.2 ± 0.1 | |
| CD38 | 18 ± 3 | 148 ± 8 | |
a Recominant proteins purified as described under “Experimental Procedures” and shown in Fig. 4.
b Wild-type (WT) and E391Q SPN are derived from S. pyogenes JRS4. Strain HSC5 has a naturally-occuring SPN allele with reduced activity (16).
c Determined using the HPLC-based NAD+ glycohydrolase assay. Data presented represent the means ± S.E. of the mean derived from at least three independent experiments.
The kinetic parameters for β-NAD+ hydrolysis with the SPN active site mutant (E391Q), from a naturally occurring β-NAD+ glycohydrolase-negative variant of SPN (from strain HSC5) (16, 36), and recombinant CD38 (as a control) were also compared. The active site mutant (E391Q) retained minimal activity with an increase in the Michaelis constant of ∼1.5-fold and an ∼3,800-fold decrease in turnover (Table 1), suggesting that altering the catalytic residue allows the enzyme to retain binding of the substrate but limits further hydrolysis of the substrate. Hydrolysis of β-NAD+ by the natural variant HSC5 SPN was undetectable under these conditions even using higher enzyme concentration (up to 2.1 μm). Control reactions with CD38 showed similar Km,app and kcat (Table 1) to those determined previously (16 μm and 96 s−1, respectively) (37). Overall, these results further implicate a role for Glu-391 in hydrolysis of β-NAD.
SPN β-NAD+ Glycohydrolase Activity Has Little Sensitivity to Nicotinamide
The microbial strict β-NAD+ glycohydrolase enzymes are unique for their apparent low sensitivity to product inhibition by free nicotinamide, except at very high concentrations (>100 mm) (20). To determine the effect of nicotinamide on the β-NAD+ glycohydrolase activity of SPN, product inhibition of SPN was performed in the presence of increasing concentrations of nicotinamide (Fig. 4A). Inhibition of SPN β-NAD+ hydrolysis by nicotinamide was undetectable under these conditions, up to 20 mm nicotinamide (∼100-fold Km,app for β-NAD+). Global analysis of the data using several inhibition models yielded poor fits and unrealistic parameters (results not shown), implying little effect of nicotinamide on the enzyme.
FIGURE 4.

SPN is insensitive to free nicotinamide and competitively inhibited by ADP-ribose. SPN hydrolysis of β-NAD+ was subjected to product inhibition to determine further classify the enzyme. In the presence of increasing concentrations of nicotinamide (A), SPN activity was unaffected. Increasing concentrations of ADP-ribose (B) inhibited the reaction in a competitive manner, indicative of an ordered uni-bi mechanism. As there was little or no inhibition seen with increasing concentrations of nicotinamide, the data in the absence of the nicotinamide was fitted to the Michaelis-Menten equation with parameters described in the text. Analysis of the ADP-ribose inhibition data were fit as described under “Experimental Procedures” with the parameters in the text. Results were also fitted to the reciprocal of the equation under “Experimental Procedures” to obtain a Lineweaver-Burk plot (inset).
The mechanism of SPN was further characterized using product inhibition by ADP-ribose (ADPR). SPN was inhibited in a competitive manner by ADPR with an inhibition constant (KI,ADPR) of 3.6 ± 0.7 mm, and the apparent Km,NAD and kcat values were similar to the values reported in the absence of any inhibitor (177 ± 26 μm and 8282 ± 424 s−1, respectively) (Fig. 4B). Results were also fitted to the reciprocal of Equation 1 under “Experimental Procedures” to obtain a Lineweaver-Burk plot (Fig. 4B, inset). This analysis yielded parameters that were similar to the parameters obtained using the global analysis method, confirming ADPR is a competitive inhibitor of the reaction. Overall, these results are consistent with ADPR leaving the enzyme active site last (ordered uni-bi mechanism) as previously seen for the calf spleen enzyme (19, 28). Analysis of these data using other inhibition models yielded fits that were of inferior quality (data not shown). These results suggest that SPN belongs to the strict β-NAD+ glycohydrolase class of enzymes, because the ADP-ribosyl cyclases and ADP-ribosyltransferases are inhibited by both nicotinamide and ADPR (32, 33, 38).
SPN Is Deficient in Both cADPR Hydrolase and ADP-ribosyl Cyclase Activities
To further characterize the ADP-ribosyl cyclase activity of SPN, the cyclase activity was compared with that of CD38. Previously, SPN purified from culture supernatant has been reported to produce cADPR at levels similar to CD38 (7, 34). Chromatographic analysis of the reaction products of purified recombinant SPN did not reveal any evidence for the formation of cADPR (data not shown). However, ADP-ribosyl cyclases can also hydrolyze cADPR (32, 39), thus lowering the yield of cADPR. ADPR was not detected following incubation of cADPR with either SPN or the E391Q mutant even after 6 h of incubation (Fig. 5A). In contrast, hydrolysis of cADPR by CD38 was readily apparent as determined by the production of ADPR following 1 h of incubation (Fig. 5A, second panel).
FIGURE 5.

SPN lacks cADPR hydrolysis and cyclase activity. A, analysis of cADPR hydrolase activity. Shown are HPLC chromatograms of the products generated by incubation of the various proteins indicated in the Fig. with 1 mm cADPR for 1 h (recombinant CD38) or for 6 h (all other proteins shown). Locations of the peaks representing cADPR and ADPR are shown at the top. Note that commercially obtained cADPR contained some ADPR, as shown in the chromatogram lacking enzyme. B, cycling assay to measure cADPR synthesis. Shown are plots of resorufin fluorescence over time generated from the indicated standard concentrations of cADPR used as reference standards (cADPR Stds) or from cADPR synthesized by enzymatic reactions conducted with the proteins listed in the Fig. Data shown are from a single experiment representative of at least three independent experiments with identical results.
As noted above, using HPLC analysis, purified recombinant SPN did not produce any detectable cADPR. However, this method may not show small changes in the concentration of cADPR. Therefore a second highly sensitive cycling assay for detection of cADPR at nanomolar levels was used to test for ADP-ribosyl cyclase activity. This assay makes use of the ADP-ribosyl cyclase activity from the marine mollusk Aplysia californica to catalyze the reverse stoichiometric synthesis of β-NAD+ from cADPR and nicotinamide. The resulting β-NAD+ is coupled to a cycling assay that generates fluorescent resorufin from resazurin (29). For comparison, CD38 and the Aplasia ADP-ribosyl cyclase, whose reaction product is almost exclusively cADPR (3), were also analyzed (Fig. 5B). As expected, both CD38 and the Aplysia enzyme produced readily detectable levels of cADPR (Fig. 5B). In contrast, recombinant SPN was unable to produce any detectable increase in resorufin fluorescence and appeared identical to the E391Q mutant and the control reaction lacking any enzyme (Fig. 5B). Similarly, many cyclases are able to produce highly fluorescent cyclic GDP-ribose (cGDPR) from nicotinamide guanine dinucleotide (30). Recombinant SPN was unable to produce any demonstrable cGDPR from nicotinamide guanine di-nucleotide (data not shown). These results corroborate that recombinant SPN is a member of the strict β-NAD+ glycohydrolases.
SPN Lacks β-NAD+ Methanolysis Activity
The ADP-ribosyl cyclases have the unique ability to use methanol as an acceptor for the enzyme-ADP-ribosyl intermediate to generate β-methyl ADP-ribose from β-NAD+ (methanolysis) (3). On the other hand, the strict β-NAD+ glycohydrolases and the ADP-ribosyltransferases are not capable of catalyzing this reaction (21). Recombinant SPN lacked detectable methanolysis activity, forming only ADPR and nicotinamide (Fig. 6), whereas for CD38, the formation of the methanolytic product (β-methyl-ADPR) in addition to the hydrolytic product (ADPR) was evident (Fig. 6). The active site mutant E391Q had no detectable activity similar to the reaction in the absence of any enzyme (Fig. 6). These results further verify that SPN′s activity falls into the strict β-NAD+ glycohydrolase class.
FIGURE 6.

SPN lacks methanolysis activity. Presented are HPLC chromatograms showing the products obtained from non-enzymatic methanolysis of β-NAD+ or from reactions with the various proteins indicated. The identities of the various peaks are noted at the top. Data shown are from a single experiment representative of at least three independent experiments with identical results.
SPN Is Unable to Catalyze Transglycosidation
Another characteristic of the ADP-ribosyl cyclases is their ability to catalyze a pyridine-base exchange reaction (called transglycosidation), which results in the formation of nicotinic acid adenine dinucleotide phosphate from β-NADP+ and nicotinic acid (3). SPN did not catalyze transglycosidation (Fig. 7, third panel), whereas in contrast CD38 readily produced nicotinic acid adenine dinucleotide phosphate (Fig. 7, second panel) resulting in an increase in the respective peak area. For comparison, the E391Q mutant and a reaction lacking enzyme yielded similar chromatograms to that seen with wild-type SPN (Fig. 7), further confirming the lack of transglycosidation activity of SPN. Interestingly, SPN was found to rapidly hydrolyze β-NADP+ much more efficiently than CD38, even in the presence of 20 mm nicotinic acid (Fig. 7). This was an unexpected result, because the β-NAD+ glycohydrolases from the related group C streptococcal species do not hydrolyze β-NADP+ (35). However, analysis of Michaelis-Menten parameters revealed that NADP+ is a poor substrate for SPN with an apparent Km of 1.7 ± 0.4 mm and observed kcat of 384 ± 56 s−1. These results are ∼9-fold higher and ∼22-fold lower, respectively, compared with β-NAD+ (see Table 1), indicating β-NAD+ is a better substrate than β-NADP+.
FIGURE 7.

SPN is deficient in transglycosidase activity toward NADP+ but is capable of hydrolysis. Shown are HPLC chromatograms of the reaction products obtained from incubation of 1 mm NAD(P)+ and 20 mm nicotinic acid with the various proteins indicated. The identities of the various peaks are noted at the top. Of note, the commercially available nicotinic acid contained low levels of nicotinic acid adenine dinucleotide phosphate (NAADP+). Data shown are from a single experiment representative of at least three independent experiments with identical results.
SPN Does Not ADP-ribosylate Substrates
SPN was further tested for ADP-ribosyltransferase activity by two independent methods. ADP-ribosyltransferase activity has been reported for SPN in filter-sterilized culture supernatants acting on a synthetic poly-l-arginine substrate (6). However, using recombinant SPN from E. coli, this activity was undetectable (data not shown). Similarly, the ability of recombinant SPN to ADP-ribosylate substrates in the cytosol was analyzed using a whole cell extract prepared from HeLa cells. For comparison, the well characterized ADP-ribosyltransferase exotoxin A from P. aeruginosa (40) was also subjected to this analysis. The pattern of labeled bands produced by recombinant SPN (JRS4, Fig. 8A) was similar to that of the naturally occurring glycohydrolase negative allele (HSC5, Fig. 8A) and that produced in the absence of any added toxin (None, Fig. 8A). Control reactions with exotoxin A demonstrated labeling of a protein that migrates with a size consistent with eukaryotic elongation factor-2, a known substrate (Fig. 8A). Thus, under these conditions, SPN did not demonstrate any detectable ability to catalyze ADP ribosylation. These results provide additional evidence for classification of SPN into the strict β-NAD+ glycohydrolase class.
FIGURE 8.

SPN does not ADP-ribosylate proteins, but β-NAD+ glycohydrolase activity is required for cytotoxicity. A, presented is an autoradiogram of an SDS-PAGE analysis of the reactions conducted with the various proteins indicated or with no added protein (none), 32P-β-NAD+, and either the presence (+) or absence (−) of a whole cell extract prepared from HeLa cells (Extract). JRS4 and HSC5 are recombinant SPN generated from naturally occurring β-NAD+ glycohydrolase-positive and -negative alleles, respectively. The migration of the protein molecular mass (kDa) standards is shown on the left while the migration of the ADP-ribosylated elongation factor-2 is indicated by the filled arrowhead on the right. The two open arrowheads show proteins ADP-ribosylated by an endogenous ADP-ribosyltransferase activity present in the HeLa whole cell extract. Data shown are from a single experiment representative of at least three independent experiments with identical results. B, shown is a Western blot analysis of whole cell extracts prepared from S. cerevisiae yeast expressing SPN that lacks β-NAD+ glycohydrolase activity (SPN[NADase−]) or the plasmid vector alone (vector). SPN is expressed from the pYES2 vector under a galactose-inducible promoter. Expression was assessed at the times indicated (in hours) following growth in the presence of 2% glucose (Glc) or 2% galactose (Gal). C, growth of the indicated strains in the presence of 2% galactose.
β-NAD+ Glycohydrolase Activity Is Associated with Yeast Cytotoxicity
Previous results have demonstrated expression of SPN in the yeast S. cerevisiae resulted in rapid depletion of cellular β-NAD+ and loss of viability (14). However, because β-NAD+ depletion is a common feature of cell death pathways triggered by a number of different stimuli (41), the cytotoxicity may or may not be a direct result of the action of SPN β-NAD+ glycohydrolase activity or a consequence of some other cytotoxic activity of SPN. Consistent with its highly toxic nature, when a plasmid encoding β-NAD+ glycohydrolase-active SPN was used to transform S. cerevisiae, viable transformants capable of expressing SPN were not obtained. In contrast, a plasmid containing the HSC5 β-NAD+ glycohydrolase-inactive SPN (SPN[NADase−]) transformed yeast at a frequency equivalent to the vector alone. Furthermore, upon induction of SPN expression, SPN was expressed at high levels (Fig. 8B) and, when compared with yeast transformed by vector alone, had an identical microscopic morphology (data not shown). The yeast also demonstrated an identical growth rate and yield (Fig. 8C). The results are consistent with the previously reported data demonstrating β-NAD+ depletion of S. cerevisiae by β-NAD+ glycohydrolase activity (14) and indicate that the presence of SPN glycohydrolase activity alone is enough to confer cytotoxicity on these cells.
DISCUSSION
The results of this study distinguished recombinant SPN, in the absence of extraneous proteins from S. pyogenes, from the multifunctional enzymes that cleave the nicotinamide-ribosyl bond of β-NAD+. For each of the parameters analyzed, the data are consistent with classifying SPN as a member of the family of strict β-NAD+ glycohydrolases that function exclusively to hydrolyze β-NAD+. The activity of recombinant SPN maintained the key properties of a strict β-NAD+ glycohydrolase, most notably a failure to detect formation of cADPR or ADP ribosylation of substrates, which are signatures of the other two enzyme classes (Table 2). Understanding of the specific metabolites produced by SPN are important in understanding the modulation of host cell function, contributing to pathogenesis caused by S. pyogenes. Classification of SPN into the strict class of β-NAD+ glycohydrolases suggests that, in the absence of any cyclase or ribosylation activities, once SPN is translocated into the host cell, the stores of β-NAD+ may be depleted resulting in host cell death.
Our assignment of SPN to the strict β-NAD+ glycohydrolase class would benefit from a more detailed comparison with other representatives of this class among microbial species. However, within bacterial species, the strict enzymes are among the least studied, and the genes that encode any other member of this class have yet to be identified. Our interrogation of genomic information available for those microbial species that have been reported to possess a strict β-NAD+ glycohydrolase failed to identify any gene that could encode a protein with any similarity to SPN. If other members of this class within share SPN′s extreme toxicity for expression in a heterologous host, this property would likely complicate sequencing strategies based on templates cloned in E. coli. However, apparently, the strict glycohydrolases from other bacterial species are associated with a specific inhibitor, similar to SPN (42–44). Yet to date, no gene with significant homology to that encoding SPN′s inhibitor, IFS, has been identified (16). Nevertheless, an inhibitor from Bacillus subtilis has many features in common with IFS, including that they are similar in size as determined by gel-filtration chromatography (31.5 kDa, IFS; 26 kDa, Bacillus inhibitor), that they bind their cognate glycohydrolase in a 1:1 stoichiometry, and that a consequence of binding is inhibition of β-NAD+ hydrolysis (16, 44). The fact that inhibitors are typically not associated with either ADP-ribosyl cyclases or ADP-ribosyltransferases lends additional support to the assignment of SPN to the strict β-NAD+ glycohydrolase family.
SPN is the first bacterial strict β-NAD+ glycohydrolase for which complete sequence information is available. Results from the mutagenesis of the glutamic acid residues demonstrate that SPN contains a functional ARTT motif, suggesting that the bacterial strict β-NAD+ glycohydrolases may share common ancestry with the bacterial ADP-ribosyltransferases (24). Although members of this latter class do not necessarily share extensive homology among themselves, they do share several other short motifs in common and share a similar overall topology (5). However, the primary sequence of SPN does not reveal any obvious candidates for other conserved motifs found in the active sites of various ADP-ribosyltransferases, including the STS motif of the diphtheria toxin subclass or the Y-X10-Y motif of the cholera toxin subclass (5). Thus, further analysis of the structural basis for the distinctive characteristics of SPN, including its high catalytic efficiency, will likely provide important insights into the evolution of the ADP-ribosyltransferases.
SPN has been demonstrated to be translocated into the cytosol of several different cell types (Madden et al. (11)). Translocation of this enzyme into cells increased the β-NAD+ glycohydrolase activity in the cell lysate (Madden et al. (11)), suggesting that this activity is important in the pathogenesis of S. pyogenes. Our current results demonstrate that expression of active recombinant SPN did not allow sustainable growth within yeast cells, whereas expression of inactive SPN showed growth similar to that in the absence of any exogenous proteins. These results further suggest that regulation of the concentration of β-NAD+ within the cell is important in the regulation of disease caused by these bacteria. Depletion of β-NAD+ within the cell disrupts many cellular processes and is a primary pathological feature of oncosis, as cellular energy stores become exhausted by the cell's attempt to replenish β-NAD+ resulting in cell death (41). We hypothesize that the ability of SPN to rapidly turn over β-NAD+ within the cell begins the process of host cell death and ultimately disease progression. Although this process is critical for host cell death, the mechanism of pathogenesis of S. pyogenes remains complicated as there are multiple virulence factors involved in maturation of the bacteria within the host. The interplay between these virulence factors contributes to the global pathogenic cycle of S. pyogenes. Understanding this pathogenic cycle, given the wide variety of diseases that S. pyogenes causes, will be important in further advancement of treatment.
Acknowledgments
We sincerely thank Dr. Francis Schuber (Université, de Strasbourg, France) for his enthusiasm, encouragement, and countless suggestions, Dr. Hon C. Lee (University of Hong Kong, China) for his gift of purified Aplysia cyclase and suggestions for the cADPR cycling assay, Dr. Uh-Hyun Kim (Chonbuk National University, Korea) for his gift of N. crassa β-NAD+ glycohydrolase, Dr. Joseph T. Barbieri (Medical College of Wisconsin) for his suggestions for the ADP-ribosyltransferase assay, and Dr. Tamara Doering (Washington University) and her laboratory members for sharing their HPLC expertise and their HPLC system.
This work was supported, in whole or in part, by National Institutes of Health Grant AI064721 (to M. G. C.) from the United States Public Health Service. This work was also supported by an American Heart Association National Scientist Development Award (0530110N) and subsequently a Grant-in-aid (09GRNT2220282 to P. J. A.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1, Tables 1 and 2, and additional Refs. 1–3.
- ADPR
- adenosine diphosphoribose
- cADPR
- cyclic adenosine diphosphoribose
- SPN
- S. pyogenes β-NAD+ glycohydrolase
- IFS
- immunity factor for SPN
- HPLC
- high-performance liquid chromatography
- BSA
- bovine serum albumin.
REFERENCES
- 1.Lin H. (2007) Org. Biomol. Chem. 5, 2541–2554 [DOI] [PubMed] [Google Scholar]
- 2.Ying W. (2008) Antioxid. Redox Signal. 10, 179–206 [DOI] [PubMed] [Google Scholar]
- 3.Schuber F., Lund F. E. (2004) Curr. Mol. Med. 4, 249–261 [DOI] [PubMed] [Google Scholar]
- 4.Lee H. C., Munshi C., Graeff R. (1999) Mol. Cell Biochem. 193, 89–98 [DOI] [PubMed] [Google Scholar]
- 5.Holbourn K. P., Shone C. C., Acharya K. R. (2006) FEBS J. 273, 4579–4593 [DOI] [PubMed] [Google Scholar]
- 6.Stevens D. L., Salmi D. B., McIndoo E. R., Bryant A. E. (2000) J. Infect. Dis. 182, 1117–1128 [DOI] [PubMed] [Google Scholar]
- 7.Karasawa T., Takasawa S., Yamakawa K., Yonekura H., Okamoto H., Nakamura S. (1995) FEMS Microbiol. Lett. 130, 201–204 [DOI] [PubMed] [Google Scholar]
- 8.Grushoff P. S., Shany S., Bernheimer A. W. (1975) J. Bacteriol. 122, 599–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ajdic D., McShan W. M., Savic D. J., Gerlach D., Ferretti J. J. (2000) FEMS Microbiol. Lett. 191, 235–241 [DOI] [PubMed] [Google Scholar]
- 10.Bricker A. L., Cywes C., Ashbaugh C. D., Wessels M. R. (2002) Mol. Microbiol. 44, 257–269 [DOI] [PubMed] [Google Scholar]
- 11.Madden J. C., Ruiz N., Caparon M. (2001) Cell 104, 143–152 [DOI] [PubMed] [Google Scholar]
- 12.Cunningham M. W. (2000) Clin. Microbiol. Rev. 13, 470–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bricker A. L., Carey V. J., Wessels M. R. (2005) Infect. Immun. 73, 6562–6566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michos A., Gryllos I., Håkansson A., Srivastava A., Kokkotou E., Wessels M. R. (2006) J. Biol. Chem. 281, 8216–8223 [DOI] [PubMed] [Google Scholar]
- 15.Kimoto H., Fujii Y., Hirano S., Yokota Y., Taketo A. (2006) J. Biol. Chem. 281, 9181–9189 [DOI] [PubMed] [Google Scholar]
- 16.Meehl M. A., Pinkner J. S., Anderson P. J., Hultgren S. J., Caparon M. G. (2005) PLoS Pathog. 1, e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh J., Caparon M. G. (2006) Mol. Microbiol. 62, 1203–1214 [DOI] [PubMed] [Google Scholar]
- 18.Lei B., Mackie S., Lukomski S., Musser J. M. (2000) Infect. Immun. 68, 6807–6818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuber F., Travo P. (1976) Eur. J. Biochem. 65, 247–255 [DOI] [PubMed] [Google Scholar]
- 20.Kaplan N. O. (1955) Methods Enzymol. 2, 551–557 [Google Scholar]
- 21.Oppenheimer N. (1994) Mol. Cell Biochem. 138, 245–251 [DOI] [PubMed] [Google Scholar]
- 22.Love M. L., Szebenyi D. M., Kriksunov I. A., Thiel D. J., Munshi C., Graeff R., Lee H. C., Hao Q. (2004) Structure 12, 477–486 [DOI] [PubMed] [Google Scholar]
- 23.Domenighini M., Magagnoli C., Pizza M., Rappuoli R. (1994) Mol. Microbiol. 14, 41–50 [DOI] [PubMed] [Google Scholar]
- 24.Han S., Tainer J. A. (2002) Int. J. Med. Microbiol. 291, 523–529 [DOI] [PubMed] [Google Scholar]
- 25.Kushner S. (1978) in Genetic Engineering (Boyer H. W., Nicosia S. eds) p. 173, Elsevier/North Holland Biomedical Press, New York [Google Scholar]
- 26.Caparon M. G., Scott J. R. (1991) Methods Enzymol. 204, 556–586 [DOI] [PubMed] [Google Scholar]
- 27.Liu Q., Kriksunov I. A., Gerdes K., Munshi C., Lee H. C., Hao Q. (2005) Structure 13, 1331–1339 [DOI] [PubMed] [Google Scholar]
- 28.Segel I. H. (1975) Enzyme Kinetics: Behaviour and Analysis of Rapid Equilibrium and Steady-state Enzyme Systems, pp. 544–577, John Wiley and Sons, Inc., New York [Google Scholar]
- 29.Graeff R., Lee H. C. (2002) Biochem. J. 361, 379–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lund F. E., Moutin M. J., Muller-Steffner H., Schuber F. (2005) Anal. Biochem. 346, 336–338 [DOI] [PubMed] [Google Scholar]
- 31.Tarnus C., Muller H. M., Schuber F. (1988) Bioorgan. Chem. 16, 38–51 [Google Scholar]
- 32.Berthelier V., Tixier J. M., Muller-Steffner H., Schuber F., Deterre P. (1998) Biochem. J. 330, 1383–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kandel J., Collier R. J., Chung D. W. (1974) J. Biol. Chem. 249, 2088–2097 [PubMed] [Google Scholar]
- 34.Oppenheimer N. J., Handlon A. L. (1992) in The Enzymes (Sigman D. S. ed) pp. 453–505, Academic Press, New York [Google Scholar]
- 35.Gerlach D., Ozegowski J. H., Gunther E., Vettermann S., Kohler W. (1996) FEMS Microbiol. Lett. 136, 71–78 [DOI] [PubMed] [Google Scholar]
- 36.Tatsuno I., Sawai J., Okamoto A., Matsumoto M., Minami M., Isaka M., Ohta M., Hasegawa T. (2007) Microbiology 153, 4253–4260 [DOI] [PubMed] [Google Scholar]
- 37.Sauve A. A., Munshi C., Lee H. C., Schramm V. L. (1998) Biochemistry 37, 13239–13249 [DOI] [PubMed] [Google Scholar]
- 38.Armstrong S., Merrill A. R. (2004) Biochemistry 43, 183–194 [DOI] [PubMed] [Google Scholar]
- 39.Muller-Steffner H., Muzard M., Oppenheimer N., Schuber F. (1994) Biochem. Biophys. Res. Commun. 204, 1279–1285 [DOI] [PubMed] [Google Scholar]
- 40.Sun J., Barbieri J. T. (2003) J. Biol. Chem. 278, 32794–32800 [DOI] [PubMed] [Google Scholar]
- 41.Fink S. L., Cookson B. T. (2005) Infect. Immun. 73, 1907–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davis W. B. (1980) Antimicrob. Agents Chemother. 17, 663–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mather I. H., Knight M. (1972) Biochem. J. 129, 141–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Everse J., Griffin J. B., Kaplan N. O. (1975) Arch. Biochem. Biophys. 169, 714–723 [DOI] [PubMed] [Google Scholar]
- 45.Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 46.Everse J., Everse K. E., Kaplan N. O. (1975) Arch. Biochem. Biophys. 169, 702–713 [DOI] [PubMed] [Google Scholar]