

Abstract
Assimilation of acetyl coenzyme A (acetyl-CoA) is an essential process in many bacteria that proceeds via the glyoxylate cycle or the ethylmalonyl-CoA pathway. In both assimilation strategies, one of the final products is malate that is formed by the condensation of acetyl-CoA with glyoxylate. In the glyoxylate cycle this reaction is catalyzed by malate synthase, whereas in the ethylmalonyl-CoA pathway the reaction is separated into two proteins: malyl-CoA lyase, a well-known enzyme catalyzing the Claisen condensation of acetyl-CoA with glyoxylate and yielding malyl-CoA, and an unidentified malyl-CoA thioesterase that hydrolyzes malyl-CoA into malate and CoA. In this study the roles of Mcl1 and Mcl2, two malyl-CoA lyase homologs in Rhodobacter sphaeroides, were investigated by gene inactivation and biochemical studies. Mcl1 is a true (3S)-malyl-CoA lyase operating in the ethylmalonyl-CoA pathway. Notably, Mcl1 is a promiscuous enzyme and catalyzes not only the condensation of acetyl-CoA and glyoxylate but also the cleavage of β-methylmalyl-CoA into glyoxylate and propionyl-CoA during acetyl-CoA assimilation. In contrast, Mcl2 was shown to be the sought (3S)-malyl-CoA thioesterase in the ethylmalonyl-CoA pathway, which specifically hydrolyzes (3S)-malyl-CoA but does not use β-methylmalyl-CoA or catalyze a lyase or condensation reaction. The identification of Mcl2 as thioesterase extends the enzyme functions of malyl-CoA lyase homologs that have been known only as “Claisen condensation” enzymes so far. Mcl1 and Mcl2 are both related to malate synthase, an enzyme which catalyzes both a Claisen condensation and thioester hydrolysis reaction.
Many organic compounds are initially metabolized to acetyl coenzyme A (acetyl-CoA), at which point they enter the central carbon metabolism. Examples of such growth substrates are C1 and C2 compounds (e.g., methanol and ethanol), fatty acids, waxes, esters, alkenes, or (poly)hydroxyalkanoates. The synthesis of all cell constituents from acetyl-CoA requires a specialized pathway for the conversion of this central C2 unit into other biosynthetic precursor metabolites. This (anaplerotic) process is referred to as acetyl-CoA assimilation, and two very different strategies have been described, i.e., the glyoxylate cycle and the ethylmalonyl-CoA pathway (12, 21) (Fig. 1).
FIG. 1.

Pathways for acetyl-CoA assimilation. (A) Glyoxylate cycle. The key enzymes are isocitrate lyase and malate synthase. (B) Ethylmalonyl-CoA pathway. The unique enzymes of the pathway are crotonyl-CoA carboxylase/reductase, ethylmalonyl-CoA/methylmalonyl-CoA epimerase, (2R)-ethylmalonyl-CoA mutase, (2S)-methylsuccinyl-CoA dehydrogenase, mesaconyl-CoA hydratase, (3S)-malyl-CoA/β-methylmalonyl-CoA lyase, and (3S)-malyl-CoA thioesterase. The enzymes involved in the (apparent) malate synthase reaction(s) are boxed for each pathway.
The glyoxylate cycle for acetyl-CoA assimilation is in fact a modified citric acid cycle that converts two molecules of acetyl-CoA to the citric acid cycle intermediate malate (Fig. 1A) (21). In a first reaction sequence, one molecule of acetyl-CoA is converted into glyoxylate due to the combined action of the initial enzymes of the citric acid cycle and isocitrate lyase, the key enzyme of this assimilation strategy. Isocitrate lyase cleaves the citric cycle intermediate isocitrate into succinate and glyoxylate (22). The glyoxylate formed is then condensed in a second step with another molecule of acetyl-CoA to yield malate and free CoA. Because the two decarboxylation reactions of the citric acid cycle are circumvented by this acetyl-CoA assimilation strategy, the glyoxylate cycle is also referred to as the “glyoxylate bypass” or “glyoxylate shunt.”
The ethylmalonyl-CoA pathway for acetyl-CoA assimilation replaces the glyoxylate cycle in bacteria that lack isocitrate lyase (1, 12). In this linear pathway, three molecules of acetyl-CoA, one molecule of CO2, and one molecule of bicarbonate are converted to the citric acid cycle intermediates succinyl-CoA and malate (Fig. 1B). The ethylmalonyl-CoA pathway requires at least seven unique enzymes. Crotonyl-CoA carboxylase/reductase, ethylmalonyl-CoA mutase, and methylsuccinyl-CoA dehydrogenase are considered key enzymes of the pathway, and all three enzymes have been characterized from Rhodobacter sphaeroides (12-14).
Although these two acetyl-CoA strategies differ with respect to their reaction sequence, intermediates and overall balance, the glyoxylate cycle and the ethylmalonyl-CoA pathway both require the condensation of acetyl-CoA and glyoxylate to form malate (Fig. 1, boxed). In the glyoxylate cycle, this reaction is catalyzed by malate synthase, whereas in the ethylmalonyl-CoA pathway malate synthase is catalyzed by two separate enzymes, malyl-CoA lyase and malyl-CoA thioesterase (7, 26).
Malyl-CoA lyases catalyze the reversible condensation of acetyl-CoA and glyoxylate into malyl-CoA and have been purified from Methylobacterium extorquens, Chloroflexus aurantiacus, Aminobacter aminovorans, and Rhodobacter capsulatus; the corresponding genes were identified as mclA (M. extorquens), mcl (C. aurantiacus), and mcl1 (R. capsulatus) (5, 16, 17, 19, 26). Remarkably, these proteins are promiscuous enzymes that also catalyze the (reversible) cleavage of β-methylmalyl-CoA into glyoxylate and propionyl-CoA, and it has been suggested that these enzymes catalyze both reactions in vivo (16, 19, 26). However, in contrast to malyl-CoA lyase, the malyl-CoA thioesterase catalyzing the highly exergonic hydrolysis of the CoA-thioester into malate and free CoA has not been identified so far, and the nature of the enzyme has remained enigmatic (7, 26).
For R. sphaeroides, a malyl-CoA lyase homolog has been shown to be upregulated during growth on acetate, and it was proposed that this protein (Mcl1) catalyzes the cleavage of β-methylmalyl-CoA, as well as the condensation of acetyl-CoA and glyoxylate in the ethylmalonyl-CoA pathway (1). Interestingly, R. sphaeroides encodes a second malyl-CoA lyase homolog with 34% amino acid sequence identity to Mcl1. This protein, named Mcl2, was also shown to be upregulated during growth of R. sphaeroides on acetate, but a function could not be assigned so far (1). We therefore addressed the function of both malyl-CoA lyase homologs by gene inactivation and biochemical studies of recombinant Mcl1 and Mcl2. Based on our findings, we confirm here the function of Mcl1 in R. sphaeroides as (3S)-malyl-CoA/β-methylmalyl-CoA lyase and identify its paralog Mcl2 as the long-sought (3S)-malyl-CoA thioesterase.
MATERIALS AND METHODS
Materials.
Chemicals were obtained from Amresco (Solon, OH), Sigma-Aldrich (Deisenhofen, Germany, or St. Louis, MO), Research Products Inc. (Mt. Prospect, IL), Fluka (Neu-Ulm, Germany), Merck (Darmstadt, Germany), Serva (Heidelberg, Germany), Fisher (Fair Lawn, NJ), or Roth (Karlsruhe, Germany). Biochemicals were from EMB (Gibbstown, NJ), ISC Bioexpress (Kaysville, UT), Roche Diagnostics (Mannheim, Germany), Applichem (Darmstadt, Germany), or Gerbu (Craiberg, Germany). Materials for cloning and expression were purchased from MBI Fermentas (St. Leon-Rot, Germany), New England Biolabs (Frankfurt, Germany, or Ipswich, MA), Stratagene (La Jolla, CA), Novagen (Schwalbach, Germany), Genaxxon Bioscience GmbH (Biberach, Germany), MWG Biotech AG (Ebersberg, Germany), or Qiagen (Hilden, Germany, or Valencia, CA). Materials and equipment for protein purification were obtained from GE Healthcare (Freiburg, Germany) or Millipore (Eschborn, Germany). Nucleotide sequencing was done at the Plant Microbe Genetics Facility at Ohio State University.
Bacteria and growth conditions.
R. sphaeroides strain 2.4.1 (DSMZ 158) was grown anaerobically in the light (3,000 lx) at pH 6.8 and 29°C on a defined medium as described previously (1). The sodium salt of each carbon source (10 mM each) was used. For regulatory studies, cells were grown in 2-liter bottles and harvested at an optical density at 578 nm (OD578) of 0.4 to 0.9. For conjugation experiments R. sphaeroides was also grown aerobically in the dark. Escherichia coli strains DH5α, DH5α/λpir, BL21(DE3), BW020767(pRL27) (24), and S17-1/λpir were grown in Luria-Bertani (LB) broth. Antibiotic concentrations were 100 μg ml−1 ampicillin and 20 μg ml−1 kanamycin.
Syntheses.
Acetyl-CoA and propionyl-CoA were synthesized from their acid anhydrides, (3S)-malyl-CoA from l-malylcaprylcysteamine, and erythro-β-methylmalyl-CoA enzymatically from propionyl-CoA and glyoxylate as described elsewhere (18, 19). (3S)-Malyl-CoA contained 80% CoA thioester and 20% free CoA as determined by high-pressure liquid chromatography (HPLC), while β-methylmalyl-CoA contained 10% CoA thioester and 90% free CoA. For determination of the substrate specificity of malyl-CoA thioesterase and apparent Km values of malyl-CoA/β-methylmalyl-CoA lyase, (3S)-malyl-CoA and β-methylmalyl-CoA were synthesized enzymatically and purified by preparative HPLC. For that purpose a reaction mixture containing 36 mM Tris-HCl (pH 7.8) 0.4 mg Mcl1, 4 mM MnCl2, 10 mM glyoxylate, and 4 mM acetyl-CoA (or propionyl-CoA) was incubated at 30°C. After 75 min, 100 μl of 20% formic acid was added to stop the reaction and to avoid hydrolysis of the malyl-CoA (or β-methylmalyl-CoA) formed. The reaction mixture was freeze-dried, and the dry matter was resuspended in 200 μl H2O. Malyl-CoA and β-methylmalyl-CoA were isolated by HPLC as described below, applying 1 μmol of CoA ester (∼50 μl suspension) per run onto the column. The pure CoA esters were freeze-dried and resuspended in 400 μl H2O. Malyl-CoA and β-methylmalyl-CoA, were quantified by determining the absorption of the solution at 260 nm (ɛ = 16.4 mM−1 cm−1) (8, 9) and stored at −20°C until use.
Isolation of mcl1::kan and mcl2::kan mutants.
The mcl1::kan mutant was isolated from a transposon library through a screen for the ability of mutants to grow on minimal medium with succinate but not with acetate. A transposon was transferred to R. sphaeroides by conjugation with E. coli strain BW20767 carrying the transposon delivery plasmid pRL27 (24). The library was constructed and screened as described before (1) except that the mutants were initially grown on minimal medium with succinate only; glucose was omitted. One mutant was isolated, and the site of transposon insertion on the chromosome was identified as described before, except that EcoRI instead of BamHI was used to restrict the chromosomal DNA of the mutant (1). A site-directed mcl2 mutant was constructed by replacing most of mcl2 with a kanamycin cassette. Chromosomal DNA was prepared from R. sphaeroides using standard techniques. Two fragments containing 600- to 700-nucleotide flanking regions on either side of the site of insertion were amplified, the PCR products were isolated and cloned into pUC18, and the nucleotide sequences of both inserts were confirmed to ensure that no errors had been introduced. For the upstream region, forward primer 5′-GCC GAA TTC ATC TAG ACT CGG ACA TGT TCG GGC TTT ACG C-3′ and reverse primer 5′-TGA GGT ACC ATG ACG ATT CTC CTC CAA GCA ACA ATC TTG CG-3′, introducing EcoRI, XbaI, and KpnI sites (underlined), were used. For the downstream region, forward primer 5′-TCC AGG ATC CTC GAG AAC CTG CAT ATC GTG ACC-3′ and reverse primer 5′-ACA CTG CAG TTC TAG ATC AGC CCG TAC ATC ACC AGA AC-3′, introducing BamHI and PstI restriction sites (underlined), were used. PCR was performed using Pfu polymerase (Stratagene) for 30 cycles, including denaturation for 1 min, annealing at 64°C for 1 min, and extension at 72°C for 2 min. A kanamycin resistance cassette was amplified using forward primer 5′-CCG AGG ATC CTA GAA AAA CTC ATC GAG CAT C-3′ and reverse primer 5′-CAT CGG TAC CGA AAG CCA CGT TGT GTC TC-3′, introducing BamHI and KpnI restriction sites (underlined). PCR was performed as described above except that the annealing temperature was 56°C and pUC4KSAC (Promega) was used as a template. The PCR product was isolated, restricted with BamHI and KpnI, and cloned into pUC18 containing the downstream region of mcl2. The insert of the resulting plasmid was excised with KpnI and PstI and cloned into pUC18 containing the upstream region of mcl2, resulting in plasmid pLF5. The plasmid was restricted with XbaI and PstI, and the fragment containing the upstream region, the kanamycin cassette, and the downstream region of mcl2 was ligated into pJQ200mp18 (28) and was named pLF7. This plasmid was transferred into R. sphaeroides by conjugation with E. coli S17-1/λpir carrying pLF7 as described previously (13). The deletion/insertion mutation of the mcl2 gene was confirmed by PCR.
Cloning of the mcl1 and mcl2 genes and production of the proteins in E. coli.
The gene encoding (3S)-malyl-CoA lyase/β-methylmalyl-CoA lyase (mcl1) was amplified from chromosomal DNA by using the forward primer (RSLy_for_01) 5′-CTT GCC CAG AGA CCA TAT GAG CTT CC-3′, introducing an NdeI site (underlined), and reverse primer (RSLy_rev_01) 5′-CCT CCC AAG CTT CCG CAG CTC-3′, introducing a HindIII site (underlined). PCR was performed with a 1:8 mixture of Pfu and TaqS polymerases (Genaxxon, Biberach, Germany) for 25 cycles, including denaturation at 94°C for 1 min, annealing at 60°C for 1 min, and extension at 72°C for 1 min. The PCR product was isolated and cloned into pET16b (Novagen) to obtain pMCL1_RS_JZ_03 for expression of mcl1 and production of an N-terminal deca-His tag fusion protein. The gene encoding (3S)-malyl-CoA thioesterase (mcl2) was amplified from chromosomal DNA using forward primer 5′-TAG ACG CCA TAT GGC GCA TCA GGC T-3′, introducing an NdeI site (underlined), and reverse primer 5′-AGC GGA TCC TTC CTC AGC TTG CCC T-3′, introducing a BamHI site (underlined). PCR was performed with Pfu polymerase (Genaxxon) for 25 cycles as before, except that 61°C was used as the annealing temperature. The PCR product was isolated and cloned into pUC19, and the nucleotide sequence was confirmed to ensure that no errors had been introduced. The insert of the resulting plasmid was excised with NdeI and BamHI and ligated into pET16b (Novagen), resulting in plasmid pJS12mcl2. Competent E. coli BL21(DE3) cells were transformed with pMCL1_RS_JZ_03 or pJS12mcl2 and grown at 37°C in LB medium with 100 μg ml−1 ampicillin to an OD578 of 0.6 to 0.9. After a temperature shock for 15 min at 4°C, expression was induced with 0.5 mM isopropyl-thiogalactopyranoside for 3 to 6 h at 22°C.
Purification of recombinant His-tagged proteins.
All purification steps were performed at 4°C. Frozen cells were suspended in a double volume of 20 mM Tris-HCl (pH 7.8) containing 0.1 mg ml−1 of DNase I. The suspension was passed twice through a chilled French pressure cell at 137 MPa, and the cell lysate was centrifuged (100,000 × g) at 4°C for 1 h. An aliquot of the supernatant (2 ml) was applied at a flow rate of 1 ml min−1 onto a 1-ml Ni-Sepharose Fast Flow column (HisTrap FF; Amersham, Braunschweig, Germany) that had been equilibrated with buffer A (20 mM Tris-HCl, 200 mM KCl, pH 7.8). After application of cell extract, the column was washed with buffer A and with buffer A containing 75 mM imidazole at a flow rate of 1 ml min−1 to elute nonspecifically bound protein. Recombinant Mcl1 (or Mcl2) was eluted with buffer A containing 500 mM imidazole. Enzymes were desalted and concentrated by ultrafiltration with an Amicon YM 10 membrane (Millipore, Eschborn, Germany) and were stored at −20°C in 20 mM Tris-HCl (pH 7.8) and 10 mM MgCl2 in the presence or absence of 50% glycerol. Recombinant mesaconyl-CoA hydratase was a gift from J. Zarzycki (33).
Enzyme assays.
All enzymes assays were performed at 30°C. One unit corresponds to one micromole of substrate converted per minute. Protein concentrations were determined by the method of Bradford (6) using bovine serum albumin as a standard. For measuring activities in R. sphaeroides cell extracts, 400 to 600 mg frozen cells was resuspended in 0.6 ml 100 mM Tris-HCl (pH 7.8) containing 0.1 mg ml−1 DNase I. After addition of 1 g glass beads (diameter, 0.1 to 0.25 mm), the cell solution was treated in a mixer mill (type MM2; Retsch, Haare, Germany) for 9 min at 30 Hz. Cell debris and glass beads were removed by centrifugation at 14,000 × g for 10 min at 4°C. The protein content of the cell extract was 2 to 10 mg ml−1.
(i) (Apparent) malate synthase.
Malate synthase activity was measured by the method of Srere et al. (30) by following the glyoxylate- and acetyl-CoA-dependent release of CoA in a discontinuous assay. The reaction mixture (0.2 ml) contained 200 mM potassium morpholinepropanesulfonic acid (MOPS-KOH) (pH 7.5), 10 mM MnCl2, 0.5 mM acetyl-CoA, 10 mM glyoxylate, and cell extract protein. At various time points, samples (30 μl) were withdrawn and mixed with 80 μl 5 mM 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) in 200 mM MOPS-KOH (pH 7.5) and 15 mM EDTA, and the absorbance was measured at 412 nm (ɛ412 = 13.6 mM−1 cm−1) (8).
(ii) Cleavage of (3S)-malyl-CoA and β-methylmalyl-CoA (Mcl1).
The cleavage of (3S)-malyl-CoA into acetyl-CoA and glyoxylate was monitored spectrophotometrically by following the formation of the glyoxylate phenylhydrazone derivative at 324 nm. The standard assay mixture (0.3 ml) contained 200 mM MOPS-KOH (pH 7.5), 5 to 10 mM MnCl2, 3.5 mM phenylhydrazinium chloride, 0.5 mM (3S)-malyl-CoA, and cell extract protein (or 0.8 to 3.2 μg Mcl1). The cleavage of β-methylmalyl-CoA was measured as described above, using β-methylmalyl-CoA instead of (3S)-malyl-CoA. The influence of chelating inhibitors on Mcl1 was determined by addition of 10 mM EDTA to the reaction mixture. The influence of divalent cations on Mcl1 activity was measured by preincubating an Mcl1 aliquot with EDTA (10 mM) before adding the EDTA-treated protein to the assay mixture containing MgCl2, CaCl2, CoCl2, or NiCl2 (10 mM each) instead of MnCl2. Km values for the pure protein were determined by varying the concentration of either (3S)-malyl-CoA (0.02 to 0.5 mM) or β-methylmalyl-CoA (0.02 to 0.5 mM) while keeping the concentrations of the other substrates constant.
(iii) Formation of (3S)-malyl-CoA and β-methylmalyl-CoA (Mcl1).
The formation of (3S)-malyl-CoA from acetyl-CoA and glyoxylate was measured by an HPLC-based assay. The reaction mixture (0.3 ml) contained 200 mM MOPS-KOH (pH 7.5), 10 mM MnCl2, 1.0 mM acetyl-CoA, 10 mM glyoxylate, and 0.8 to 3.2 μg Mcl1. The formation of β-methylmalyl-CoA was measured by using propionyl-CoA instead of acetyl-CoA. Samples of 50 μl were taken after 0 to 40 min of incubation at 30°C, and the reaction was stopped by addition of 5 μl 20% formic acid. Water (30 μl) was added, and protein was removed by centrifugation. The samples were analyzed for CoA derivatives by HPLC as described below, and the amount of product formed was calculated from the peak area. Km values for the pure protein were determined by varying the concentration of either acetyl-CoA (0.06 to 1 mM), propionyl-CoA (0.06 to 1 mM), or glyoxylate (1.2 to 10 mM) while keeping the concentrations of the other substrates constant. The formation of β-methylmalyl-CoA from propionyl-CoA and glyoxylate was also monitored spectrophotometrically at 290 nm by coupling the reaction to the formation of mesaconyl-CoA with recombinant mesaconyl-CoA hydratase, which dehydrates β-methylmalyl-CoA (33). For that purpose, the reaction mixture was essentially the same as described above but also contained recombinant mesaconyl-CoA hydratase from R. sphaeroides in excess (33). The increase of absorption at 290 nm due to the formation of mesaconyl-(C1)-CoA from β-methylmalyl-CoA was followed spectrophotometrically (ɛ = 3.1 mM−1 cm−1, determined experimentally).
(iv) Hydrolysis of (3S)-malyl-CoA (Mcl2).
Release of CoA from (3S)-malyl-CoA (or other CoA esters) was monitored in a discontinuous assay as described above (malate synthase assay), replacing acetyl-CoA and glyoxylate with (3S)-malyl-CoA. The reaction mixture (0.2 ml) contained 200 mM MOPS-KOH (pH 7.5), 10 mM MnCl2, 0.1 to 0.2 mM (3S)-malyl-CoA, and cell extract protein or 0.01 to 0.1 μg Mcl2. At various time points, samples (30 μl) were withdrawn and mixed with 80 μl 5 mM DTNB in 200 mM MOPS-KOH (pH 7.5) and 15 mM EDTA, and the absorbance was measured at 412 nm (ɛ412 = 13.6 mM−1 cm−1) (8). The influence of chelating inhibitors on Mcl2 activity was determined by addition of 10 mM EDTA to the reaction mixture. The influence of divalent cations on Mcl2 was measured by preincubating an Mcl2 aliquot with EDTA (10 mM) before adding the EDTA-treated protein to the assay mixture containing MgCl2, CaCl2, CoCl2, or NiCl2 (10 mM each) instead of MnCl2. Km values for the pure protein were determined by varying the concentration of (3S)-malyl-CoA (0.02 to 0.20 mM) while keeping the concentrations of the other substrates constant. Substrate specificity of Mcl2 was tested using 100 mM Tris-HCl (pH 7.6) instead of MOPS-KOH and replacing (3S)-malyl-CoA with 0.1 to 0.2 mM β-methylmalyl-CoA, acetyl-CoA, propionyl-CoA, crotonyl-CoA, succinyl-CoA, or 3-hydroxybutyryl-CoA.
HPLC.
A reversed-phase column (LiChrospher 100, end-capped, 5 μm, 125 by 4 mm; Merck, Darmstadt, Germany) was developed for 30 min with a linear gradient from 2 to 10% acetonitrile in 40 mM potassium phosphate buffer (pH 4.2). Reaction products and standard compounds were detected by UV absorbance with a Waters 996 photodiode array detector (Waters, Eschborn, Germany). Retention times were 8.9 min for (3S)-malyl-CoA, 10.4 min for free CoA, 13.7 min for β-methylmalyl-CoA, 17.1 min for acetyl-CoA, and 24.7 min for propionyl-CoA (UV absorbance at 260 nm).
Protein analysis techniques.
Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) was performed as described by Laemmli (23) using 12% gels. Proteins were stained with Coomassie blue according to the method of Zehr et al. (34). The native molecular mass was determined by fast protein liquid chromatography, using a 24-ml Superdex 200 (Amersham Biosciences) gel filtration column calibrated with ferritin (440 kDa), aldolase (158 kDa), bovine serum albumin (67 kDa), ovalbumin (45 kDa), and RNase A (14 kDa). Protein samples (0.2 ml) were applied onto the column, equilibrated with 10 mM MOPS-KOH (pH 7.5) and 150 mM KCl, and eluted with the same buffer with a flow rate of 0.4 ml min−1.
RESULTS
Isolation and characterization of the R. sphaeroides mcl1::kan mutant.
A mutant of R. sphaeroides that grew photoheterotrophically on minimal medium with succinate but showed no growth with acetate as the sole carbon source was isolated from a transposon library. The site of transposon insertion in this mutant was identified at positions 355146 to 355155 on chromosome 1 of R. sphaeroides (accession number NC_007493). The corresponding gene (accession number ABA77918) encodes a protein with 88% sequence identity to (3S)-malyl-CoA/β-methylmalyl-CoA lyase from Rhodobacter capsulatus (Mcl1, accession number ACI22682), which operates in acetate assimilation (26), and the gene in R. sphaeroides was named accordingly (accession number GU320612). Unlike the case for other acetate-negative mutants of R. sphaeroides (1, 13, 14), growth of the mcl1::kan mutant on acetate was not rescued by the addition of glyoxylate (Fig. 2A). Instead, the slow increase in optical density over time for the mutant in the presence of glyoxylate and acetate was comparable to that of the wild type with glyoxylate alone (Fig. 2C). This phenotype is consistent with the function of Mcl1 as malyl-CoA lyase, which catalyzes the condensation of acetyl-CoA and glyoxylate during acetyl-CoA assimilation. As expected, the mcl1::kan mutant grew with growth rates comparable to those of the wild type on substrates downstream of the proposed metabolic block, i.e., malate and propionate plus bicarbonate (Fig. 2A).
FIG. 2.
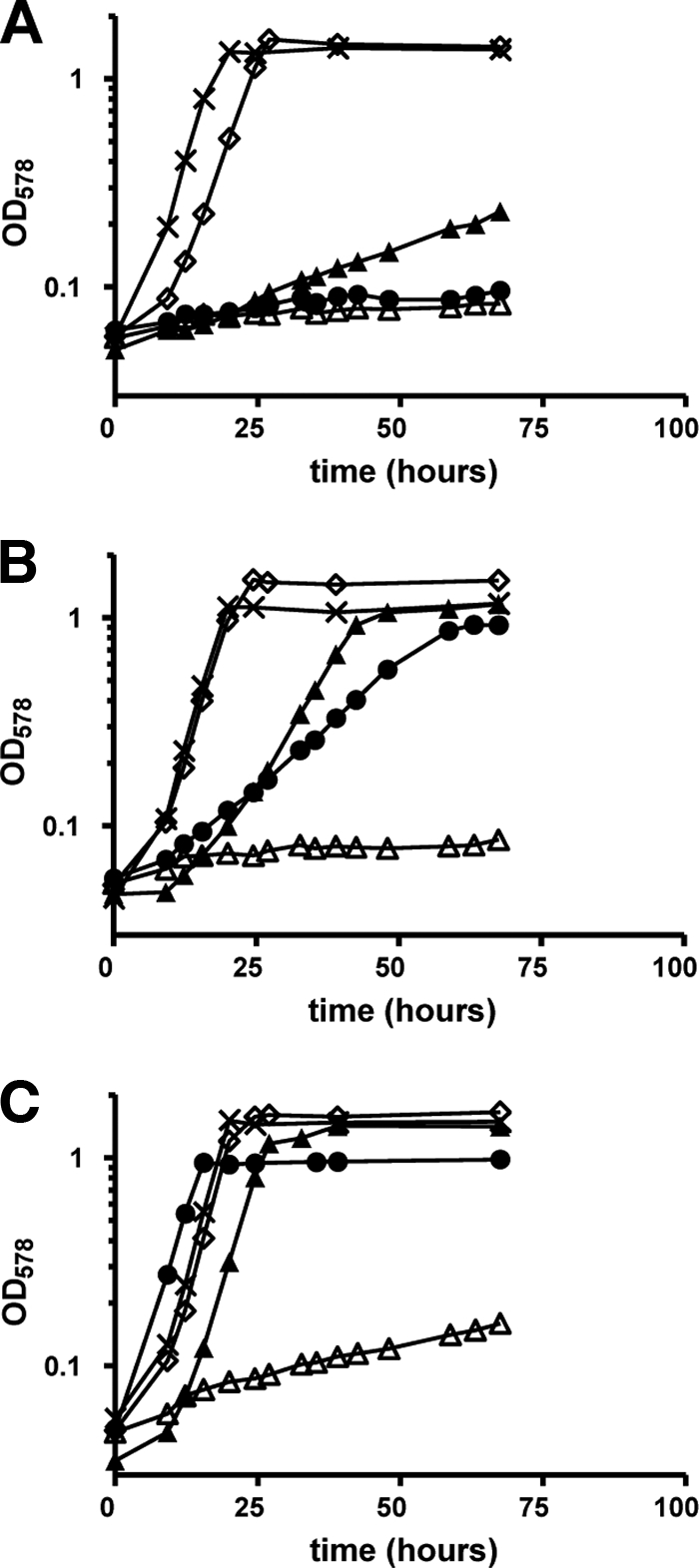
Growth curves of the R. sphaeroides mcl1::kan (A) and mcl2::kan (B) mutants compared to the wild type (C). R. sphaeroides was pregrown on minimal medium with succinate and transferred to minimal medium containing: acetate (•), glyoxylate (▵), acetate plus glyoxylate (▴), propionate plus bicarbonate (⋄), or malate (×).
To confirm the role of Mcl1 as malyl-CoA lyase, glyoxylate formation from (3S)-malyl-CoA was measured in cell extracts of wild-type R. sphaeroides and the mcl1::kan mutant. Malyl-CoA lyase activity in cell extract of wild-type cells grown with acetate was 220 nmol min−1 mg−1. This activity was downregulated 10-fold in cells grown on succinate (20 nmol min−1 mg−1). Interestingly, when cells were grown on succinate plus acetate, malyl-CoA lyase activity was upregulated to levels seen with acetate alone (210 nmol min−1 mg−1). In contrast to that, (3S)-malyl-CoA lyase activity was not detected in cell extracts of the mcl1::kan mutant grown on succinate plus acetate (0.01 nmol min−1 mg−1), supporting the proposed role of Mcl1 as (3S)-malyl-CoA lyase.
Heterologous production of Mcl1 from R. sphaeroides and characterization of the recombinant protein.
To study its biochemical properties, mcl1 from R. sphaeroides was expressed in Escherichia coli and the N-terminally histidine-tagged protein with a calculated molecular mass of 36.8 kDa was purified by one affinity chromatographic step (Fig. 3A). As expected, recombinant Mcl1 catalyzed the reversible cleavage of (3S)-malyl-CoA into acetyl-CoA and glyoxylate in the presence of divalent cations such as Mn2+ or Mg2+. The reaction followed Michaelis-Menten kinetics, with a maximum specific activity of 4.1 U mg−1 and an apparent Km value of 0.02 mM for (3S)-malyl-CoA.
FIG. 3.

Heterologous production and purification of recombinant (3S)-malyl-CoA/β-methylmalyl-CoA lyase and (3S)-malyl-CoA thioesterase (12.5% SDS). (A) (3S)-Malyl-CoA/β-methylmalyl-CoA lyase (Mcl1). Lanes: 1, 20 μg of E. coli cell extract protein before induction; 2, 20 μg of E. coli cell extract protein after 3 h of induction; 3, 8 μg of protein from the elution step (Ni2+-chelating column). (B) (3S)-malyl-CoA thioesterase (Mcl2). Lanes: 1, 20 μg of E. coli cell extract protein before induction; 2, 20 μg of E. coli cell extract protein after 3 h of induction; 3, 8 μg of protein from the elution step (Ni2+-chelating column). The molecular masses are indicated.
The closely related Mcl1 proteins from R. capsulatus and C. aurantiacus are promiscuous enzymes that catalyze not only the (reversible) cleavage of (3S)-malyl-CoA but also the (reversible) cleavage of β-methylmalyl-CoA into propionyl-CoA and glyoxylate (19, 26). When tested for this enzymatic activity, recombinant Mcl1 from R. sphaeroides was likewise able to split β-methylmalyl-CoA into propionyl-CoA and glyoxylate. The kinetic properties of this reversible cleavage reaction were comparable to those of malyl-CoA cleavage, with a specific activity of 4.5 U mg−1 and an apparent Km value of 0.01 mM for β-methylmalyl-CoA (Table 1).
TABLE 1.
Molecular and catalytic properties of recombinant (3S)-malyl-CoA/β-methylmalyl-CoA lyase and (3S)-malyl-CoA thioesterase from R. sphaeroides
| Property | (3S)-Malyl-CoA/β-methylmalyl-CoA lyase (Mcl1) | (3S)-Malyl-CoA thioesterase (Mcl2) |
|---|---|---|
| Reaction(s) catalyzed | Acetyl-CoA + glyoxylate ⇆ (3S)-malyl-CoA Propionyl-CoA + glyoxylate ⇆ β-methylmalyl-CoA | (3S)-Malyl-CoA → (3S) malate + CoA |
| Sp act, U mg−1 (reaction) | 14 (condensation of acetyl-CoA + glyoxylate), 4.1 [cleavage of (3S)-malyl-CoA], 20 (condensation of propionyl-CoA + glyoxylate), 4.5 (cleavage of β-methylmalyl-CoA) | 200 (thioester cleavage) |
| Apparent Km value(s), mM (substrate) | 0.1 (acetyl-CoA), 3.1 (glyoxylate), 0.02 [(3S)-malyl-CoA], 0.2 (propionyl-CoA), 4.1 (glyoxylate), 0.01 (β-methylmalyl-CoA) | 0.09 [(3S)-malyl-CoA] |
| Turnover no. per subunit kcat, s−1 (reaction) | 8.6 (condensation of acetyl-CoA + glyoxylate), 2.5 [cleavage of (3S)-malyl-CoA], 12 (condensation of propionyl-CoA + glyoxylate), 2.8 (cleavage of β-methylmalyl-CoA) | 110 (thioester cleavage) |
| Catalytic efficiency per subunit kcat/Km, M−1 s−1 (reaction) | 2.8 × 103 (condensation of acetyl-CoA + glyoxylate), 1.3 × 105 [cleavage of (3S)-malyl-CoA], 3.0 × 103 (condensation of propionyl-CoA + glyoxylate), 2.8 × 105 (cleavage of β-methylmalyl-CoA) | 1.2 × 106 (thioester cleavage) |
| Influence (sp act, %) of cations/inhibitor at 10 mM each | Mn2+, 100; Mg2+, 73; Ca2+, 14; Co2+, 13; Ni2+, 6; EDTA, 1 | Mg2+, 100; Mn2+, 40; Ca2+, 19; Co2+, 3; Ni2+, 7; EDTA, 1 |
| Native molecular mass (kDa) | 200 | 80 |
| Subunit molecular mass (kDa) | 36.8 | 33.4 |
| Suggested composition | α6 | α2 or α3 |
When tested in the direction of condensation, Mcl1 from R. sphaeroides catalyzed the condensation of glyoxylate with acetyl-CoA and with propionyl-CoA at comparable rates. At saturating substrate concentrations, these condensation rates were about 3- to 4-fold higher than the rates of the (“reverse”) cleavage reactions, with apparent Km values of 3.1 mM for glyoxylate and 0.07 mM for acetyl-CoA; the Km values for the condensation reaction to form β-methylmalyl-CoA were 4.1 mM for glyoxylate and 0.2 mM for propionyl-CoA (Table 1). The ratios of the catalytic efficiencies for the forward and reverse reactions, however, are consistent with the fact that the equilibrium of the reaction catalyzed by Mcl1 clearly is on the side of glyoxylate and acetyl-CoA or glyoxylate and propionyl-CoA, as previously determined (Table 1) (17). Like Mcl1 from R. capsulatus, the enzyme from R. sphaeroides was dependent on the presence of divalent cations. Incubation with the metal chelator EDTA inhibited enzymatic activity reversibly. Tests for different cations with EDTA-preincubated enzyme showed that Mn2+ served best compared to Mg2+, Ca2+, Co2+, and Ni2+ (Table 1). The native molecular mass determined by gel filtration was 200 kDa, suggesting a homohexameric composition. This has also been reported for other malyl-CoA lyases (16, 19, 26).
In summary, these results indicate that Mcl1 from R. sphaeroides acts as a (3S)-malyl-CoA/β-methylmalyl-CoA lyase in the ethylmalonyl-CoA pathway that catalyzes two consecutive steps: (i) the cleavage of β-methylmalyl-CoA into propionyl-CoA and glyoxylate and (ii) the further condensation of glyoxylate with acetyl-CoA into (3S)-malyl-CoA.
Construction, isolation, and characterization of the R. sphaeroides mcl2::kan mutant.
Mcl1 was upregulated during growth on acetate, as was another protein with 34% amino acid sequence identity to Mcl1 (1). Because of its significant amino acid sequence identity to Mcl1, this malyl-CoA lyase homolog was named Mcl2 (accession number ABA80153) (1, 26). Although the genomic context of the gene encoding Mcl2 strongly suggested that it functions in the ethylmalonyl-CoA pathway (1), the role of Mcl2 in acetyl-CoA assimilation was not obvious. In particular, the fact that Mcl1 is a promiscuous enzyme, which is in principle able to catalyze the reversible cleavage of both malyl-CoA and β-methylmalyl-CoA, raised the question of which function this second malyl-CoA homolog (Mcl2) might in fact serve.
To elucidate the role of Mcl2, a site-directed mutant was constructed, with almost 800 nucleotides of the mcl2 gene replaced by a kanamycin resistance cassette. The R. sphaeroides mutant mcl2::kan grew normally on malate or propionate plus bicarbonate, whereas growth on acetate or acetate plus glyoxylate was impaired (Fig. 2B), confirming the role of Mcl2 in acetyl-CoA assimilation. However, (3S)-malyl-CoA lyase activity was not affected in cell extracts of the mutant, but the apparent malate synthase activity was greatly decreased, consistent with the growth phenotype of the mcl2::kan mutant with acetate plus glyoxylate (Table 2; Fig. 2B). The decrease in apparent malate synthase activity in cell extracts of the mcl2::kan mutant was therefore not due to a decrease in (3S)-malyl-CoA lyase activity but rather was caused by a decrease in (3S)-malyl-CoA thioesterase activity (Table 2).
TABLE 2.
Enzyme activities in cell extracts of wild-type and mutant R. sphaeroides
| Strain | Growth substrate | Mean generation time (h) ± SD | Mean sp act (μmol min−1 mg−1) ± SD |
||||
|---|---|---|---|---|---|---|---|
| Malyl-CoA lyase | Apparent malate synthase | Apparent malate synthase + Mcl1 | Apparent malate synthase + Mcl2 | Malyl-CoA thioesterase | |||
| Wild type | Acetate | 3.5 ± 0.0 | 0.22 | 0.36 ± 0.09 | 0.56 ± 0.05 | 1.84 ± 0.17 | 0.43 ± 0.01 |
| Succinate-acetate | 3.2 ± 0.0 | 0.21 | 0.29 ± 0.05 | 0.44a | 1.54 ± 0.05 | 0.34 ± 0.05 | |
| Succinate | 3.5 ± 0.0 | 0.02 | 0.05 ± 0.04 | 0.06a | 0.17 ± 0.02 | 0.11 ± 0.07 | |
| mcl1::kan mutant | Succinate-acetate | 3.2 ± 0.0 | <0.02 | 0.02 ± 0.01 | 0.22 ± 0.1 | 0.06 ± 0.01 | 0.17 ± 0.01 |
| Succinate | 3.2 ± 0.0 | <0.02 | 0.03 ± 0.01 | 0.14a | 0.03 ± 0.02 | 0.14 ± 0.05 | |
| Δmcl2::kan mutant | Acetate | 17.9 ± 0.0 | 0.21 | 0.07 ± 0.02 | 0.04a | 1.39 ± 0.12 | 0.02 ± 0.02 |
| Succinate-acetate | 3.1 ± 0.0 | 0.18 | 0.06 ± 0.03 | 0.05a | 1.15 ± 0.22 | 0.08 ± 0.04 | |
| Succinate | 3.3 ± 0.0 | 0.02 | 0.03 ± 0.03 | 0.03a | 0.10 ± 0.02 | 0.06 ± 0.03 | |
Activity was determined using one cell extract only.
Heterologous production of Mcl2 from R. sphaeroides and characterization of the recombinant protein.
To gain further insights into the function of Mcl2, the corresponding gene of R. sphaeroides was expressed as an N-terminally histidine-tagged protein in E. coli. The recombinant protein (calculated molecular mass, 33.4 kDa) was purified to homogeneity from cell extracts of E. coli by affinity chromatography, as shown by electrophoresis (Fig. 3B). Notably, in contrast to Mcl1, Mcl2 was able to catalyze neither the cleavage of (3S)-malyl-CoA into glyoxylate and acetyl-CoA nor the cleavage of β-methylmalyl-CoA into glyoxylate and propionyl-CoA. Instead, Mcl2 catalyzed the hydrolysis of (3S)-malyl-CoA to malate and free CoA, with a specific activity of 200 U mg−1 and an apparent Km value of 0.09 mM for (3S)-malyl-CoA in the presence of divalent cations (Mg2+ or Mn2+). The recombinant enzyme was highly specific for (3S)-malyl-CoA and did not use β-methylmalyl-CoA (Fig. 4) or other CoA-esters tested as substrates (acetyl-CoA, propionyl-CoA, crotonyl-CoA, succinyl-CoA, and 3-hydroxybutyryl-CoA), suggesting that Mcl2 is a bona fide (3S)-malyl-CoA thioesterase. Incubation with divalent cations (Mg2+ or Mn2+; 10 mM) stimulated enzymatic activity of the recombinant protein, whereas addition of EDTA inhibited the reaction reversibly, indicating that divalent cations are essential for enzymatic function. Similar findings have been reported for a malyl-CoA thioesterase activity in cell extracts of M. extorquens (7). When EDTA-preincubated enzyme was tested for activity with different cations, Mg2+ (10 mM) served best in thioester hydrolysis compared to Mn2+, Ca2+, Co2+, and Ni2+ (Table 1). According to gel filtration analysis, the native molecular mass of the enzyme was 80 kDa, which indicates a homodimeric or homotrimeric composition.
FIG. 4.

Substrate specificity of (3S)-malyl-CoA thioesterase. Recombinant (3S)-malyl-CoA thioesterase (Mcl2) was incubated with (3S)-malyl-CoA (♦) or β-methylmalyl-CoA (○). The reaction mixture (0.2 ml) contained 100 mM Tris-HCl (pH 7.6), 5 mM MgCl2, and 0.1 mM (malyl-CoA) CoA ester or 0.15 mM (β-methylmalyl-CoA) CoA ester, respectively. Twenty micrograms of Mcl2 (malyl-CoA) or 180 μg of Mcl2 (β-methylmalyl-CoA) was added to start the reaction. Samples were withdrawn at various time points, and the amount of free CoA formed was quantified by DTNB (see Materials and Methods). As a control, residual (3S)-malyl-CoA and β-methylmalyl-CoA were chemically hydrolyzed by addition of KOH after 5 and 8 min, respectively.
Physiological roles of Mcl1 and Mcl2 in R. sphaeroides.
The functional assignment of Mcl1 as malyl-CoA lyase and its paralog Mcl2 as malyl-CoA thioesterase was examined by cell extract measurements of wild-type R. sphaeroides and the mcl1::kan and mcl2::kan mutant strains. For that purpose, acetyl-CoA- and glyoxylate-dependent formation of malate (and free CoA) in various cell extracts was monitored by detection of the free CoA released (30). This CoA-releasing activity from acetyl-CoA plus glyoxylate relies on the combined action of malyl-CoA lyase and malyl-CoA thioesterase and is also referred to as “apparent malate synthase” activity (7, 26). To determine (3S)-malyl-CoA thioesterase activity separately, the reaction was started with (3S)-malyl-CoA, and the formation of free CoA was detected (Table 2).
The “apparent malate synthase” was present in wild-type R. sphaeroides grown on acetate and acetate plus succinate, with specific rates of 430 and 340 nmol min−1 mg−1, respectively. This activity was downregulated in wild-type cells grown on succinate as the sole carbon source. In either case, the “apparent malate synthase” activities were comparable to the thioesterase activities in these extracts, suggesting that the thioesterase is rate determining for the overall “apparent malate synthase” activity (Table 2). This was confirmed by adding recombinant Mcl2 in excess to extracts of wild-type cells grown on acetate or acetate plus succinate, which led to a significant stimulation of “apparent malate synthase” activity (Table 2). In contrast, addition of Mcl1 to these cell extracts did not stimulate “apparent malate synthase” activity (Table 2). This indicates that the rate of conversion of acetyl-CoA and glyoxylate into malate is limited in R. sphaeroides by the thioesterase and not by the malyl-CoA lyase activity. Malyl-CoA lyase is also regulated in response to the growth substrate, with malyl-CoA lyase and “apparent malate synthase plus excess of Mcl2” activities being about 10-fold decreased in cells grown on succinate versus acetate or acetate plus succinate (Table 2).
In contrast to the case for the wild type, “apparent malate synthase” activity was very low in cell extracts of the R. sphaeroides mcl1::kan and mcl2::kan mutants when grown on acetate or acetate plus succinate. Although malyl-CoA lyase activities were nondetectable in cell extracts of the mcl1::kan mutant, some “apparent malate synthase” activity remained (Table 2). This activity may be due to a true malate synthase that is encoded by the R. sphaeroides genome. In the case of the mcl2::kan mutant, malyl-CoA thioesterase activity was not completely abolished in any cell extract. Residual activity might be due to hydrolysis of malyl-CoA catalyzed by an ambiguous enzyme present in cell extracts (as observed previously [7]). A possible candidate is citrate synthase; the enzyme from pig liver has been previously shown to catalyze the hydrolysis of (3S)-malyl-CoA (11). This, however, was not further investigated. A residual thioesterase and therefore malate synthase activity explains the slow growth of the mcl2::kan mutant with acetate plus glyoxylate as carbon substrates (Fig. 2B). Addition of excess Mcl1 to cell extracts of the mcl2::kan mutant did not increase “apparent malate synthase” activity, indicating that Mcl1 has no thioesterase activity as demonstrated with the purified recombinant enzyme.
In summary, the cell extract measurements confirmed that Mcl1 and Mcl2 together catalyze the “apparent malate synthase” activity in R. sphaeroides during acetyl-CoA assimilation via the ethylmalonyl-CoA pathway. Whereas Mcl1 is the (3S)-malyl-CoA(/β-methylmalyl-CoA) lyase catalyzing the Claisen condensation of acetyl-CoA with glyoxylate, Mcl2 is the sought thioesterase that catalyzes the subsequent hydrolysis of the (3S)-malyl-CoA thioester into malate and free CoA.
DISCUSSION
(3S)-Malyl-CoA thioesterase.
We have described a novel thioesterase which functions in the ethylmalonyl-CoA pathway for acetyl-CoA assimilation. (3S)-Malyl-CoA thioesterase (Mcl2, accession number GU320613) catalyzes the hydrolysis of (3S)-malyl-CoA. Release of CoA from (3S)-malyl-CoA pulls the entire reaction sequence of the ethylmalonyl-CoA pathway toward the formation of citric cycle intermediates (Fig. 1B). Notably, the enzyme is a paralog to (3S)-malyl-CoA lyase/β-methylmalyl-CoA lyase (amino acid sequence identity of 34%), which catalyzes a very different type of reaction (Claisen condensation versus thioester hydrolysis) and also differs in substrate specificity.
“Apparent malate synthase” in the ethylmalonyl-CoA pathway as a reaction sequence catalyzed by two paralogous enzymes.
The “apparent malate synthase” of R. sphaeroides operating during acetyl-CoA assimilation via the ethylmalonyl-CoA pathway could be attributed to the combined action of (3S)-malyl-CoA lyase (Mcl1) and (3S)-malyl-CoA thioesterase (Mcl2). Malyl-CoA lyase (Mcl1) from R. sphaeroides is similar to other well-characterized malyl-CoA lyases, which all catalyze the reversible Claisen condensation of a CoA thioester with glyoxylate (7, 17, 19, 26). These proteins are typically promiscuous enzymes that accept both acetyl-CoA and also propionyl-CoA with comparable kinetic parameters. In the ethylmalonyl-CoA pathway, this promiscuousness is necessary, because the cleavage of β-methylmalyl-CoA as well as the formation of (3S)-malyl-CoA is essential during assimilation of acetyl-CoA. In contrast to that, (3S)-malyl-CoA thioesterase (Mcl2), as reported here, is highly specific for malyl-CoA and does not hydrolyze β-methylmalyl-CoA or any other substrate tested so far. A promiscuousness of malyl-CoA thioesterase similar to that of malyl-CoA/β-methylmalyl-CoA lyase would result in the (wasteful) hydrolysis of β-methylmalyl-CoA (or structurally related CoA esters), which strongly decreases the efficiency of acetyl-CoA assimilation via the ethylmalonyl-CoA pathway. Notably, (3S)-malyl-CoA thioesterase is the first malyl-CoA lyase homolog that does not catalyze a Claisen condensation, and Mcl2 therefore serves as the prototype of malyl-CoA lyase(-like) proteins with a new enzymatic function. These findings are particularly remarkable because malyl-CoA lyase and malyl-CoA thioesterase share 34% amino acid sequence identity.
Roles of malyl-CoA/β-methylmalyl-CoA lyase and malyl-CoA thioesterase in other pathways.
Besides the ethylmalonyl-CoA pathway (Fig. 5A), malyl-CoA/β-methylmalyl-CoA lyase has also been reported to operate in the bicyclic 3-hydroxypropionate/malyl-CoA cycle for autotrophic CO2 fixation of C. aurantiacus, where it links the first (“CO2 fixation”) cycle with the second (“glyoxylate assimilation”) cycle (Fig. 5B). Malyl-CoA is an important intermediate in the 3-hydroxypropionate cycle of C. aurantiacus and is formed by two carboxylation steps from acetyl-CoA. Malyl-CoA is then split into acetyl-CoA and glyoxylate. Acetyl-CoA as the primary CO2 acceptor is used again in the first cycle for CO2 fixation, whereas glyoxylate is condensed with one molecule of propionyl-CoA for the assimilation of the primary fixation product (glyoxylate) into cell material (18, 19, 32, 33). Thus, in the 3-hydroxypropionate cycle, malyl-CoA/β-methylmalyl-CoA lyase operates in the opposite direction from the ethylmalonyl-CoA pathway. An (irreversible) hydrolysis of malyl-CoA is not required here, and an Mcl2 homolog is not present in the genome.
FIG. 5.

Roles of malyl-CoA/β-methylmalyl-CoA lyase and malyl-CoA thioesterase in different pathways. The reactions of malyl-CoA/β-methylmalyl-CoA lyase and malyl-CoA thioesterase in different pathways are highlighted in red and blue, respectively. (A) Acetate assimilation via the ethylmalonyl-CoA pathway (EMPW) in Rhodobacter sphaeroides (1, 12-14). (B) CO2 fixation via the 3-hydroxypropionate cycle in the phototrophic bacterium Chloroflexus aurantiacus (modified from reference 33). Here, malyl-CoA/β-methylmalyl-CoA lyase also catalyzes the hydrolysis of citramalyl-CoA into acetyl-CoA and pyruvate, the final CO2 assimilation product (32). (C) C1 assimilation via the serine cycle and reactions of the ethylmalonyl-CoA pathway (EMPW) in Methylobacterium extorquens AM1 (modified from references 12 and 27). (D) C2 assimilation via the ethylmalonyl-CoA pathway in M. extorquens (modified from reference 4).
The methylotroph M. extorquens uses the ethylmalonyl-CoA pathway for the assimilation of C1 compounds (e.g., methanol and methylamine) (12, 27), and its genome encodes both malyl-CoA/β-methylmalyl-CoA lyase and malyl-CoA thioesterase homologs. During methylotrophic growth, malyl-CoA/β-methylmalyl-CoA lyase is required (i) to generate acetyl-CoA from the cleavage of malyl-CoA, which is the product of the serine cycle for C1 fixation (reviewed in reference 4), and later (ii) for the cleavage of β-methylmalyl-CoA to yield propionyl-CoA, which is assimilated into cell material (Fig. 5C). Glyoxylate formed in either case is supplied to the serine cycle to serve as an acceptor for C1 fixation. The presence of malyl-CoA thioesterase activity would be detrimental under these conditions, as it would, together with the malate thiokinase, result in a futile cycle of malate activation and hydrolysis of malyl-CoA. For growth on C2 compounds (e.g., ethanol and ethylamine), the situation changes and “apparent malate synthase” activity is actually required to release malate as an assimilation product from malyl-CoA that is formed during the assimilation of acetyl-CoA (as discussed in reference 4) (Fig. 5D). This is consistent with earlier mutant studies which had shown that a malyl-CoA thioesterase activity is essential for growth on the C2 substrate ethanol but not for growth on the C1 compound methanol (7, 31).
These examples show that in fact some pathways exist that do not necessarily rely on the combined action of Mcl1 and Mcl2 like “apparent malate synthase” but rather rely on the enzymatic function of only one of the two homologs. One might speculate that some bacteria (or routes) require only malyl-CoA thioesterase and not malyl-CoA lyase. This might be the case, for example, for a number of fully sequenced Anaeromyxobacter species and also Rhodopseudomonas palustris, which seem to encode only an Mcl2 homolog and not the Mcl1 counterpart, as inferred from genomic analysis. However, the true enzymatic function (or substrates) of these Mcl2 homologs as well as their physiological role remains to be demonstrated.
Structural and mechanistic considerations for malyl-CoA(/β-methylmalyl-CoA) lyase, malyl-CoA thioesterase, and malate synthase.
(3S)-Malyl-CoA/β-methylmalyl-CoA lyase and (3S)-malyl-CoA thioesterase are both members of the malate synthase superfamily but are only distantly related to malate synthase on the protein level (26). Malate synthase catalyzes the overall conversion of acetyl-CoA and glyoxylate into malate and free CoA without the formation of a free intermediate; initially acetyl-CoA is condensed with glyoxylate in a Claisen-like mechanism to form malyl-CoA. Malyl-CoA remains (noncovalently) bound to the same active site and in a second step is hydrolyzed to malate and free CoA (20). In principle these two reaction steps are also conserved in the “apparent malate synthase” described here, but in this case two proteins participate: Mcl1 exclusively catalyzes the Claisen condensation and Mcl2 hydrolyzes the CoA thioester.
Two malate synthase isoforms have been described and named A (for acetate assimilation) and G (for glycolate assimilation). They differ mainly by the presence of an additional domain in the G isoform that may have a regulatory function (25). The crystal structures of malate synthase G (MSG) from Mycobacterium tuberculosis and E. coli, of malate synthase A (MSA) from E. coli and Bacillus anthracis, and of a malyl-CoA lyase-like protein with unknown function from M. tuberculosis have been determined (2, 3, 15, 20, 25, 29). They all contain a conserved α8β8 TIM (triose phosphate isomerase fold) barrel that harbors the active site.
Based on structural considerations, a partial alignment of these sequences is possible, even with amino sequence identities of below 20% (Fig. 6). Two residues that are involved in binding of the catalytically essential divalent cation are strictly conserved between these five proteins (Fig. 6, highlighted in red); so are amino acids that coordinate two water ligands to the divalent cation (Fig. 6, highlighted in blue). The coordination of the divalent cation is completed by the substrate/product molecule which also interacts with an arginine residue in the protein; this residue is also conserved for all five proteins (Fig. 6, highlighted in yellow). Alignment of sequences beyond the α8β8 TIM barrel is more difficult, but the essential aspartate residue D631 of E. coli MSG (D447 of E. coli MSA), which is thought to act as the catalytic base, likely corresponds to glutamate residue E262 of Mcl2 and aspartate residue D299 of Mcl1.
FIG. 6.

Amino acid sequence alignment of Mcl-like proteins. (3S)-Malyl-CoA/β-methylmalyl-CoA lyase (Mcl1) and (3S)-malyl-CoA thioesterase (Mcl2) from R. sphaeroides were aligned with a malyl-CoA lyase-like protein from Mycobacterium tuberculosis (accession number 56554142) (M.t.) and partially with malate synthase G from Escherichia coli (accession number 152149159) (MSG) and malate synthase A from E. coli (accession number 212374959) (MSA). Colored boxes indicate residues that are predicted to be part of the active site based on the structure of the enzyme from M. tuberculosis and E. coli. Red boxes highlight residues that may be involved in binding of the divalent cation, whereas blue boxes indicate residues that may be involved in coordinating two water ligands to this cation. Yellow boxes indicate a conserved arginine that is proposed to interact with the substrate/product and a predicted catalytic base at the active site.
Because all residues appear to be conserved, it is not obvious which residue(s) is specifically involved in proton abstraction of the acyl-CoA substrate to prepare it for the Claisen condensation with glyoxylate (Mcl1) or in the activation of a water molecule for cleavage of the thioester bond (Mcl2). This is complicated by the fact that although malate synthases catalyze both reactions (Claisen condensation and thioester hydrolysis) at the same active site, malyl-CoA is a very poor substrate for these enzymes (10). Because of that, a crystal structure of this intermediate bound to the enzyme that may help to identify essential amino acids for one of the two reaction steps has not be obtained so far. It is therefore not possible, based on the current data, to predict from the primary structure whether a given malyl-CoA lyase-like enzyme catalyzes a Claisen condensation (Mcl1), a thioester hydrolysis (Mcl2), or both reactions (E. coli MSG or MSA). Likewise, the molecular basis of a narrow (Mcl2, malate synthase) or broad (Mcl1) substrate specificity of these enzymes of the malate synthase superfamily remains to be determined.
Concluding remarks.
The malate synthase superfamily comprises a very large group of enzymes from all three domains of life. For most of the well over 1,000 proteins in the database with greater than 20% sequence identity (over the entire protein) to Mcl1 from R. sphaeroides, the reactions that they catalyze are unknown. The assignment of Mcl2 from R. sphaeroides as bona fide (3S)-malyl-CoA thioesterase, as reported here, therefore greatly extents the possible role of malyl-CoA lyase-like enzymes in nature.
Acknowledgments
We thank W. Metcalf (University of Illinois) for providing the transposon delivery plasmid pRL27. We thank Damir Stazic, Jasmin Schleimer, Jan Zarzycki, Kathrin Schneider, and Andreas Präg for technical assistance and H. vanDijken for helpful comments on the manuscript.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (AL 677/1-1) and the National Science Foundation (MCB0842892) and by a start-up fund from the Ohio State University.
Footnotes
Published ahead of print on 4 January 2010.
REFERENCES
- 1.Alber, B. E., R. Spanheimer, C. Ebenau-Jehle, and G. Fuchs. 2006. Study of an alternate glyoxylate cycle for acetate assimilation by Rhodobacter sphaeroides. Mol. Microbiol. 61:297-309. [DOI] [PubMed] [Google Scholar]
- 2.Anstrom, D. M., K. Kallio, and S. J. Remington. 2003. Structure of the Escherichia coli malate synthase G:acetyl-coenzyme A abortive ternary complex at 1.95 Å resolution. Protein Sci. 12:1822-1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anstrom, D. M., and S. J. Remington. 2007. The product complex of M. tuberculosis malate synthase revisited. Protein Sci. 15:2002-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anthony, C. 1982. The biochemistry of methylotrophs. Academic Press, London, United Kingdom.
- 5.Arps, P. J., G. F. Fulton, E. C. Minnich, and M. E. Lidstrom. 1993. Genetics of serine pathway enzymes in Methylobacterium extorquens AM1: phosphoenolpyruvate carboxylase and malyl coenzyme A lyase. J. Bacteriol. 175:3776-3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradford, M. 1976. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 7.Cox, R. B., and J. R. Quayle. 1976. Synthesis and hydrolysis of malyl-coenzyme A by Pseudomonas AM1: an apparent malate synthase activity. J. Gen. Microbiol. 95:121-133. [DOI] [PubMed] [Google Scholar]
- 8.Dawson, R. M. C., D. C. Elliot, W. H. Elliot, and K. M. Jones. 1986. Data for biochemical research, 3rd ed., Clarendon Press, Oxford, United Kingdom.
- 9.Decker, K. 1959. Die aktivierte Essigsäure. Das Coenzym A und seine Acylderivate im Stoffwechsel der Zelle, p. 84-89. F. Enke, Stuttgart, Germany.
- 10.Eggerer, H., and A. Klette. 1967. Über das Katalyzeprinzip der Malat-Synthase. Eur. J. Biochem. 1:447-475. [DOI] [PubMed] [Google Scholar]
- 11.Eggerer, H., U. Remberger, and C. H. Grünewälder. 1964. Zum Mechanismus der biologischen Umwandlung von Citronensäure. V. Citrat-synthase, eine Hydrolase für Malyl-coenzym A. Biochem. Z. 339:436-453. [PubMed] [Google Scholar]
- 12.Erb, T. J., I. A. Berg, V. Brecht, M. Müller, G. Fuchs, and B. E. Alber. 2007. Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: the ethylmalonyl-CoA pathway. Proc. Natl. Acad. Sci. U. S. A. 104:10631-10636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erb, T. J., J. Rétey, G. Fuchs, and B. E. Alber. 2008. Ethylmalonyl-CoA mutase from Rhodobacter sphaeroides defines a new subclade of coenzyme B12-dependent acyl-CoA mutases. J. Biol. Chem. 283:32283-32293. [DOI] [PubMed] [Google Scholar]
- 13a.Erb, T. J., V. Brecht, G. Fuchs, M. Müller, and B. E. Alber. 2009. Carboxylation mechanism and stereochemistry of crotonyl-CoA carboxylase/reductase, a carboxylating enoyl thioester reductase. Proc. Natl. Acad. Sci. U. S. A. 106:8871-8876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erb, T. J., G. Fuchs, and B. E. Alber. 2009. (2S)-Methylsuccinyl-CoA dehydrogenase closes the ethylmalonyl-CoA pathway for acetate assimilation. Mol. Microbiol. 73:992-1008. [DOI] [PubMed] [Google Scholar]
- 15.Goulding, C. W., P. M. Bowers, B. Segelke, T. Lekin, C.-Y. Kim, T. C. Terwilliger, and D. Eisenberg. 2007. The structure and computational analysis of Mycobacterium tuberculosis protein CitE suggest a novel enzymatic function. J. Mol. Biol. 365:275-283. [DOI] [PubMed] [Google Scholar]
- 16.Hacking, A. J., and J. R. Quayle. 1974. Purification of malyl-coenzyme A lyase from Pseudomonas AM1. Biochem. J. 139:339-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hersh, L. B. 1973. Malate adenosine triphosphate lyase. Sepaeration of the reaction into malate thiokinase and malyl coenzyme A lyase. J. Biol. Chem. 248:7295-7303. [PubMed] [Google Scholar]
- 18.Herter, S., J. Farfsing, N. Gad'on, C. Rieder, W. Eisenreich, A. Bacher, and G. Fuchs. 2001. Autotrophic CO2 fixation by Chloroflexus aurantiacus: study of glyoxylate formation and assimilation via the 3-hydroxypropionate cycle. J. Bacteriol. 183:4305-4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herter, S., A. Busch, and G. Fuchs. 2002. l-Malyl-coenzyme A lyase/β-methylmalyl-coenzyme A lyase from Chloroflexus aurantiacus, a bifunctional enzyme involved in autotrophic CO2-fixation. J. Bacteriol. 184:5999-6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howard, B. R., J. A. Endrizzi, and S. J. Remington. 2000. Crystal structure of Escherichia coli malate synthase G complexed with magnesium and glyoxylate at 2.0 Å resolution: mechanistic implications. Biochemistry 39:3156-3168. [DOI] [PubMed] [Google Scholar]
- 21.Kornberg, H. L., and H. A. Krebs. 1957. Synthesis of cell constituents from C2-units by a modified tricarboxylic acid cycle. Nature 179:988-991. [DOI] [PubMed] [Google Scholar]
- 22.Kornberg, H. L., and J. Lascelles. 1960. The formation of isocitratase by the athiorhodaceae. J. Gen. Microbiol. 23:511-51728. [DOI] [PubMed] [Google Scholar]
- 23.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 24.Larsen, R. A., M. M. Wilson, A. M. Guss, and W. W. Metcalf. 2002. Genetic analysis of pigment biosynthesis in Xanthobacter autotrophicus Py2 using a new, highly efficient transposon mutagenesis system that is functional in a wide variety of bacteria. Arch. Microbiol. 178:193-201. [DOI] [PubMed] [Google Scholar]
- 25.Lohman, J. R., A. C. Olson, and S. J. Remington. 2008. Atomic resolution structures of Escherichia coli and Bacillus anthracis malate synthase A: comparison with isoform G and implications for structure-based drug discovery. Protein Sci. 17:1935-1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meister, M., S. Saum, B. E. Alber, and G. Fuchs. 2005. l-Malyl-coenzyme A/β-methylmalonyl-coenzyme A lyase is involved in acetate assimilation of the isocitrate lyase-negative bacterium Rhodobacter capsulatus. J. Bacteriol. 187:1415-1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peyraud, R., P. Kiefer, P. Christen, S. Massou, J. C. Portais, and J. A. Vorholt. 2009. Demonstration of the ethylmalonyl-CoA pathway by using 13C metabolomics. Proc. Nat. Acad. Sci. U. S. A. 106:4846-4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quandt, J., and M. F. Hynes. 1993. Versatile suicide vectors which allow direct selection for gene replacement in Gram-negative bacteria. Gene 127:15-21. [DOI] [PubMed] [Google Scholar]
- 29.Smith, C. V., C.-C. Huang, A. Miczak, D. G. Russell, J. C. Sacchettini, and K. Höner zu Bentrup. 2003. Biochemical and structural studies of malate synthase from Mycobacterium tuberculosis. J. Biol. Chem. 278:1735-1743. [DOI] [PubMed] [Google Scholar]
- 30.Srere, P. A., H. Brazil, and L. Gonen. 1963. The citrate condensing enzyme of pigeon breast muscle and moth flight muscle. Acta Chem. Scand. 17:129-134. [Google Scholar]
- 31.Taylor, I. J., and C. Anthony. 1976. A biochemical basis for obligate methylotrophy: properties of a mutant of Pseudomonas AM1 lacking 2-oxoglutarate dehydrogenase. J. Gen. Microbiol. 93:259-265. [DOI] [PubMed] [Google Scholar]
- 32.Zarzycki, J., V. Brecht, M. Müller, and G. Fuchs. 2009. Identifying the missing steps of the autotrophic 3-hydroxypropionate CO2 fixation cycle in Chloroflexus aurantiacus. Proc. Natl. Acad. Sci. U. S. A. 106:21317-21322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zarzycki, J., A. Schlichting, N. Strychalsky, M. Müller, B. E. Alber, and G. Fuchs. 2008. Mesaconyl-coenzyme A hydratase, a new enzyme of two central carbon metabolic pathways in bacteria. J. Bacteriol. 190:1366-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zehr, B. D., T. J. Savin, and R. E. Hall. 1989. A one-step, low-background Coomassie staining procedure for polyacrylamide gels. Anal. Biochem. 182:157-159. [DOI] [PubMed] [Google Scholar]