

Abstract
Cells of the gliding bacterium Flavobacterium johnsoniae move rapidly over surfaces. Mutations in gldN cause a partial defect in gliding. A novel bacteriophage selection strategy was used to aid construction of a strain with a deletion spanning gldN and the closely related gene gldO in an otherwise wild-type F. johnsoniae UW101 background. Bacteriophage transduction was used to move a gldN mutation into F. johnsoniae UW101 to allow phenotypic comparison with the gldNO deletion mutant. Cells of the gldN mutant formed nonspreading colonies on agar but retained some ability to glide in wet mounts. In contrast, cells of the gldNO deletion mutant were completely nonmotile, indicating that cells require GldN, or the GldN-like protein GldO, to glide. Recent results suggest that Porphyromonas gingivalis PorN, which is similar in sequence to GldN, has a role in protein secretion across the outer membrane. Cells of the F. johnsoniae gldNO deletion mutant were defective in localization of the motility protein SprB to the cell surface, suggesting that GldN may be involved in secretion of components of the motility machinery. Cells of the gldNO deletion mutant were also deficient in chitin utilization and were resistant to infection by bacteriophages, phenotypes that may also be related to defects in protein secretion.
Cells of Flavobacterium johnsoniae, and of many other members of the phylum Bacteroidetes, crawl over surfaces at approximately 2 μm/s in a process called gliding motility. F. johnsoniae cells glide on agar, glass, polystyrene, Teflon, and many other surfaces (16, 22). Cells suspended in liquid also bind and propel added particles such as polystyrene latex spheres (23). The mechanism of this form of cell movement is not well understood despite decades of research (15). Genome analyses suggest that F. johnsoniae gliding is genetically unrelated to other well-studied forms of bacterial movement such as bacterial flagellar motility, type IV pilus-mediated twitching motility, myxobacterial gliding motility, and mycoplasma gliding motility (10, 20, 21). Genes and proteins required for F. johnsoniae motility have been identified (1-3, 7-9, 17, 18). GldA, GldF, and GldG appear to form an ATP-binding cassette transporter that is required for gliding (1, 7). Eight other Gld proteins (GldB, GldD, GldH, GldI, GldJ, GldK, GldL, and GldM) are also required for movement (2, 3, 8, 9, 17, 18). Many of these are unique to members of the phylum Bacteroidetes. Disruption of the genes encoding any of these 11 proteins results in complete loss of motility. The mutants form nonspreading colonies, and individual cells exhibit no movement on agar, glass, Teflon, and other surfaces tested. The Gld proteins are associated with the cell envelope and presumably constitute the gliding motor, but none of them appear to be exposed on the cell surface. Mutations in sprA and sprB, which encode cell surface proteins, result in partial motility defects. Cells form nonspreading colonies, but some of the cells exhibit limited movement in wet mounts. SprA is required for efficient attachment to glass (22), and SprB appears to be a mobile adhesin that is propelled along the cell surface by the gliding motor and thus transmits the force generated by the motor to the surface over which cells crawl (10, 21). The surface localization of SprA and SprB and the phenotypes of sprA and sprB mutants suggest that the gliding motor is at least partially functional in these mutants but that force is inefficiently transmitted to the substratum. Analysis of the F. johnsoniae genome revealed the presence of multiple paralogs of sprB, which may explain the residual motility of sprB mutants (20).
gldN lies downstream of gldL and gldM, and the three genes constitute an operon (2). Cells with transposon insertions in gldN form nonspreading colonies that are indistinguishable from those of other gld mutants. However, unlike other gld mutants, gldN mutants exhibit some residual ability to glide in wet mounts (2). One possible explanation for this phenotype is that GldN may have a peripheral and nonessential role in gliding. Alternatively, GldN may perform a critical function in gliding, but in its absence another cellular protein may compensate for the missing GldN function. F. johnsoniae has a gldN paralog, gldO, that is located downstream of gldN but is transcribed independently (2). The GldN and GldO proteins are 85% identical over their entire lengths, making GldO a prime candidate for a protein that might compensate for lack of GldN.
Recent results suggest that some of the F. johnsoniae Gld and Spr proteins, including GldN, may be components of a novel bacteroidete protein translocation apparatus referred to as the Por secretion system (PorSS) (28). This conclusion emerged from studies of gingipain protease secretion by the distantly related nonmotile bacteroidete Porphyromonas gingivalis. P. gingivalis is a human periodontal pathogen, and gingipain proteases are important virulence factors. Gingipains have signal peptides that allow export across the cytoplasmic membrane via the Sec machinery, but they rely on components of the PorSS for secretion across the outer membrane (27-29). P. gingivalis cells with mutations in genes homologous to F. johnsoniae gldK, gldL, gldM, gldN, and sprA are defective in gingipain secretion across the outer membrane (28). F. johnsoniae has a homologue to another P. gingivalis gene required for gingipain secretion, porT. Disruption of the F. johnsoniae porT homologue (referred to as sprT) results in motility defects and defects in surface localization of SprB (28).
This study was designed to identify possible roles for GldN in motility and to determine whether GldN and GldO are partially redundant components of the motility apparatus. The results demonstrate that F. johnsoniae GldN has an important function in motility and that GldO can replace GldN in this role. They suggest that GldN is needed for efficient secretion of the cell surface motility protein SprB, which may explain some of the motility defects of the gldN mutants.
MATERIALS AND METHODS
Bacterial strains, bacteriophages, plasmids, and growth conditions.
F. johnsoniae ATCC 17061 strain UW101 was the wild-type strain used in this study (17, 20). F. johnsoniae MM101 is a direct descendant of F. johnsoniae ATCC 17061. Strain MM101 has a partial defect in chitin utilization, as previously described (17). The 39 spontaneous and chemically induced motile nonspreading mutants of F. johnsoniae UW101 were obtained from J. Pate and are designated UW102-1, -2, -3, -18, -24, -37, -43, -45, -46, -50, -51, -67, -73, -88, -91, -93, -95, -99, -103, -106, -128, -135, -136, -142, -143, -148, -149, -150, -155, -156, -158, -168, -171, -172, -176, -298, -301, -344, and -345 (4, 24, 32). F. johnsoniae strains were grown in Casitone-yeast extract (CYE) medium at 30°C, as previously described (19). To observe colony spreading, F. johnsoniae was grown on PY2 agar medium (1) at 25°C. Motility medium (MM) was used to observe movement of individual cells in wet mounts (14). The bacteriophages active against F. johnsoniae that were used in this study were φCj1, φCj13, φCj23, φCj28, φCj29, φCj42, φCj48, and φCj54 (4, 25, 32). Plasmids and primers used in this study are listed in Table 1. The plasmids used for complementation were all derived from pCP1 and have copy numbers of approximately 10 in F. johnsoniae (1, 11, 19). Antibiotics were used at the following concentrations when needed: ampicillin, 100 μg/ml; cefoxitin, 100 μg/ml; erythromycin, 100 μg/ml; kanamycin, 35 μg/ml; and tetracycline, 20 μg/ml.
TABLE 1.
Plasmids and primers used in this study
| Plasmid or primer | Sequence and/or descriptiona | Reference |
|---|---|---|
| Plasmids | ||
| pCP23 | E. coli-F. johnsoniae shuttle plasmid; Apr (Tcr) | 1 |
| pCP29 | E. coli-F. johnsoniae shuttle plasmid; Apr (Cfr Emr) | 11 |
| pDH223 | gldB in pCP11; Apr (Emr) | 9 |
| pET30a | Protein expression vector; Kmr | Novagen |
| pJVB8 | 1.1-kbp fragment between primers 711 and 717, encoding the C-terminal end of GldN, inserted into EcoRI/SalI-digested pET30a; Kmr | This study |
| pJVB9 | 2.3-kbp PCR product between primers 732 and 733 upstream of gldN, inserted into BamHI/SalI-cut pLYL03; Apr (Emr) | This study |
| pLYL03 | Bacteroides-Flavobacterium suicide vector; Apr (Emr) | 13 |
| pMK315 | gldF in pCP29; Apr (Cfr Emr) | 7 |
| pMM213 | gldD in pCP23; Apr Kmr (Tcr) | 8 |
| pMM265 | gldJ in pCP11; Apr (Emr) | 3 |
| pMM313 | gldJ in pCP11; Apr (Emr) | 3 |
| pNap3 | 3.0-kbp PCR product between primers 734 and 735 downstream of gldN, inserted into SalI/SphI-cut pJVB9; for construction of gldNO deletion strain; Apr (Emr) | This study |
| pSA11 | gldA in pCP11; Apr (Emr) | 1 |
| pSN48 | sprA in pCP23; Apr Tcr (Tcr) | 22 |
| pSN60 | sprB in pCP29; Apr Kmr (Cfr Emr) | 21 |
| pSP24 | sprC in pCP23; Apr (Tcr) | 26 |
| pTB79 | gldN in pCP23; Apr (Tcr) | 2 |
| pTB81a | gldL in pCP23, expressed from the pCP23 orf1 promoter; Apr (Tcr) | 2 |
| pTB94a | gldM and the first 781 bp of gldN in pCP23. gldM expressed from the pCP23 orf1 promoter; Apr (Tcr) | 2 |
| pTB97a | gldO in pCP23; Apr Kmr (Tcr) | 2 |
| pTB98 | gldL, gldM, gldN, and gldO in pCP29; Apr Kmr (Cfr Emr) | 2 |
| pTB99 | gldK in pCP23; Apr (Tcr) | 2 |
| Primers | ||
| 398 | 5′TCTTTTAAAGCGTAATGAAAGC3′; primer in gldG | |
| 399 | 5′CTGCAGGAAGTTCTCCCTGC3′; primer in gldG | |
| 609 | 5′TGGGAATCATTTGAAGGTTGG3′; used for amplifying or sequencing chromosomal DNA adjacent to IR1 of HimarEm1 or HimarEm2 | |
| 684 | 5′CAAAGGCTTGTCGTTATCAG3′; primer within gldO | |
| 692 | 5′AAGGATTTCCTGCGATCGC3′; primer within gldM | |
| 694 | 5′CAATGTATTTTCCTGTAGATACAGC3′; primer within gldN | |
| 711 | 5′TTGCAAAGTCGACCACTAATAAGGGCAAAACC3′; used in construction of pJVB8b | |
| 717 | 5′TCTCAGAATTCGATAAGCCTTTAGCTTACG3′; used in construction of pJVB8c | |
| 732 | 5′GTTATGGGATCCGCTTATGGTATGGGAGCGGC3′; used in construction of pNap3d | |
| 733 | 5′TAGAAGGTCGACCTCCAGCGATAGAAACAATAGC3′; used in construction of pNap3b | |
| 734 | 5′AATGATGTCGACTCTGCGCTTCCTATTTCG3′; used in construction of pNap3b | |
| 735 | 5′CATATCGCATGCTTTCATACGATTTGTATCTGTAGCTGC3′; used in construction of pNap3e | |
| 939 | 5′ACAAGCCTCCTGCAATTCTCGAAG3′; primer downstream of gldO |
Antibiotic resistance phenotypes: ampicillin, Apr; cefoxitin, Cfr; erythromycin, Emr, kanamycin, Kmr; tetracycline, Tcr. Unless indicated otherwise, antibiotic resistance phenotypes are those expressed in E. coli. Antibiotic resistance phenotypes listed in parentheses are those expressed in F. johnsoniae but not in E. coli.
The SalI site is underlined.
The EcoRI site is underlined.
The BamHI site is underlined.
The SphI site is underlined.
Construction of a mutant lacking gldN and gldO.
A 2.3-kb region of F. johnsoniae DNA which spans the upstream region of gldN (Fig. 1) was amplified using primer 732 (which introduces a BamHI site), primer 733 (which introduces a SalI site), and Phusion high-fidelity DNA polymerase (New England Biolabs). The fragment was digested with BamHI and SalI and inserted into the suicide vector pLYL03 which had been digested with the same enzymes to generate pJVB9. A 3.0-kb region of F. johnsoniae DNA downstream of gldO was amplified using primer 734 (which introduces a SalI site) and primer 735 (which introduces a SphI site). This fragment was digested with SalI and SphI and inserted into pJVB9 that had been digested with the same enzymes, to generate pNap3 (Fig. 1). Transfer of pNap3 by conjugation into wild-type F. johnsoniae UW101 followed by recombination resulted in strain CJ1624A, which carried wild-type and deleted versions of the region spanning gldN and gldO. The known resistance of gldN mutants to some bacteriophages (2) was used to select for a second recombination event resulting in loss of the plasmid and generation of a deletion spanning gldN and gldO. A 3-ml culture of F. johnsoniae CJ1624A was incubated overnight at 25°C without antibiotic selection to allow for recombination and loss of the integrated plasmid. To 0.1 ml of these cells, 0.1 ml of bacteriophage φCj1 (108 PFU) was added. Cells with bacteriophage were incubated for 15 min at 23°C and plated in 4 ml of CYE top agar overlaying CYE agar medium. Plates were incubated for 2 days at 25°C, and surviving colonies were screened for loss of erythromycin resistance. The resulting strain, CJ1631A, carried a deletion within the region spanning gldN and gldO.
FIG. 1.

Map of the gldNO region and characterization of gldN and gldNO mutations. (A) Map of the gldNO region. Numbers below the map refer to kilobase pairs of sequence. The sites of Himar insertions are indicated by triangles, and primers used for plasmid construction or for verification of strains are listed. The regions of DNA carried by the plasmid used to make the gldNO deletion (pNap3) and the regions of DNA carried by the complementation plasmids pTB79, pTB81a, pTB94, pTB97a, pTB98, and pTB99 are indicated beneath the map. (B) Verification of loss of gldN and gldO in the deletion mutant CJ1631A. PCR using primers 692 and 939, which flank the deletion, amplified a 3.4-kbp product from wild-type cells (UW101) and amplified a 1.5-kbp product from cells of the deletion mutant CJ1631A. PCR using primers 684 and 694, which lie within the deleted region, resulted in a 1.1-kbp product from wild-type cells and no product from cells of the deletion mutant. (C) Verification of transduction of HimarEm2 from F. johnsoniae CJ1304 (derived from F. johnsoniae MM101) into F. johnsoniae UW101. PCR using primer 692, which anneals within gldM, and primer 609, which anneals near the end of HimarEm2, amplified a 1.4-kbp product from the original gldN mutant CJ1304 and from the transductant CJ1743 but not from wild-type F. johnsoniae UW101.
Transduction of gldN carrying a HimarEm2 insertion into F. johnsoniae UW101.
Cells of the gldN mutant F. johnsoniae CJ1304 carrying pTB79 (to restore motility and phage sensitivity) were grown overnight in MM at 25°C. Approximately 108 cells (0.2 ml of overnight growth in MM) were mixed with approximately 105 PFU of φCj54, incubated for 15 min at 23°C, and plated in 4 ml CYE top agar overlay on CYE agar containing erythromycin. After incubation overnight at 25°C, plates with confluent lysis were harvested by scraping the overlay agar into 5 ml of a buffer consisting of 10 mM Tris and 8 mM MgSO4 (pH 7.5) (TM buffer), followed by addition of 0.1 ml of chloroform and incubation for 4 h at 23°C. Agar and debris were removed by centrifugation for 10 min at 3,700 × g, and 0.1 ml of chloroform was added to the supernatant which contained the phage. Phage were stored at 4°C, and phage titers were determined by plating dilutions on lawns of wild-type cells. To avoid damaging effects of chloroform on recipient or host cells, phage were either diluted or incubated in open tubes at 30°C for 30 min to allow evaporation of chloroform prior to use. F. johnsoniae UW101 recipient cells for transduction were grown overnight in 10 ml MM in a 125-ml flask without shaking at 25°C. The cells (approximately 5 × 109) were sedimented by centrifugation for 10 min at 3,700 × g and resuspended in 1 ml TM buffer containing 1 × 108 to 2.5 × 109 PFU (multiplicity of infection of 0.02 to 0.5). Cells were incubated with phage for 15 min at 23°C, plated in 4 ml CYE top agar overlaying CYE agar containing erythromycin, and incubated overnight at 30°C. Erythromycin-resistant transductants were streaked for isolation, and the resulting mutants were confirmed by PCR using primer 692 (which binds within gldM) and primer 609 (which binds near the end of HimarEm2). F. johnsoniae MM101 has a silent mutation within gldG, which lies 1.1 Mbp from gldO. This can be used to distinguish strains derived from F. johnsoniae MM101 from those derived from F. johnsoniae UW101. To verify that the mutation had been transduced from CJ1304 (derived from F. johnsoniae MM101) into F. johnsoniae UW101, we amplified a fragment of gldG with primers 398 and 399 and determined the nucleotide sequence.
Microscopic observations of cell attachment and movement.
Wild-type and mutant cells of F. johnsoniae were examined for attachment to glass using a Petroff-Hausser counting chamber, as previously described (22). Cells were grown overnight in MM at 25°C without shaking. Equal numbers of cells of different strains were introduced into the chambers, and the number of cells that attached to the glass slide after a 2-min incubation was determined. Cells were also examined for movement over glass and Teflon by phase-contrast microscopy at 25°C. Cells in MM were spotted onto a glass microscope slide and were covered with a glass coverslip or with an oxygen-permeable Teflon membrane (Yellow Springs Instrument Co., Yellow Springs, OH), incubated for 1 min, and observed for motility using an Olympus BH-2 phase-contrast microscope with a heated stage set at 25°C. Images were recorded using a Photometrics CoolSNAPcf2 camera and were analyzed using MetaMorph software (Molecular Devices, Downingtown, PA).
Measurements of chitin digestion.
Chitin utilization on plates was observed as previously described (17) except that cells were cultured in MM overnight at 25°C prior to spotting 2 μl on the MYA-chitin medium. For chitinase activity assays, 3-ml cultures of F. johnsoniae cells were grown in MM at 25°C overnight with gentle mixing by rotation. Cells were pelleted by centrifugation at 21,000 × g for 10 min, and culture supernatants were filtered through a 0.22-μm polyvinylidene difluoride (PVDF) filter (Fisher Scientific). For whole-cell samples, cells were suspended in the original volume of a buffer consisting of 137 mM NaCl, 2.7 mM KCl, 10 mM Na2PO4, and 2 mM KH2PO4 (pH 7.4) (phosphate-buffered saline [PBS]). For cell lysates, cells were washed in PBS and suspended in the original volume of bacterial protein extraction reagent (BPER) (Thermo Fisher Scientific, Waltham, MA). Chitinase activity was determined by a modification of the procedure described by Thompson et al. (31) using the synthetic substrates 4-methylumbelliferyl β-d-N-acetyl-glucosaminide (4-MU-GlcNAc), 4-methylumbelliferyl β-d-N,N′-diacetyl-chitobioside [4-MU-(GlcNAc)2], and 4-methylumbelliferyl β-d-N,N′,N"-triacetylchitotrioside [4-MU-(GlcNAc)3], which were each obtained from Sigma-Aldrich (St. Louis, MO). Specifically, 15 μl of sample (supernatant, whole cells, or lysed cells) was added to 75 μl of Tris-buffered saline (50 mM Tris, 150 mM NaCl, pH 7.5) in a black half-well microtiter plate (Greiner Bio-one, Frickenhausen, Germany). Reactions were started by adding 10 μl of 1.0 mM fluorescent substrate, and plates were incubated at 37°C for 4 h. Enzyme activity was detected using a BioTek Synergy HT microplate reader (BioTek Instruments, Inc., Winooski, VT) at an excitation wavelength of 360 nm and an emission wavelength of 460 nm. Standard curves using authentic 4-methylumbelliferone were used to determine pmol of substrate hydrolyzed. Enzyme assays were performed in duplicate and averages calculated. Means and standard errors were determined from three independent experiments. Chitinase activities in whole cells, cell extracts, and cell-free supernatants are indicated as pmol 4-methylumbelliferone released per μg of total protein in the original cell suspension. Protein concentrations were determined by the bicinchoninic acid (BCA) assay (Thermo Fisher Scientific).
Measurements of bacteriophage sensitivity.
Sensitivity to F. johnsoniae bacteriophages was determined essentially as previously described by spotting 3 μl of phage lysates (109 PFU/ml) onto lawns of cells in CYE overlay agar (9), except that host cells were from overnight cultures in MM at 25°C. The plates were incubated for 24 h at 25°C to observe lysis.
Expression of recombinant GldN and antibody production.
A 1,110-bp fragment encoding the C-terminal 283 amino acids of GldN was amplified using Phusion DNA polymerase (New England Biolabs, Ipswich, MA), primer 711 (which introduces a SalI site), and primer 717 (which introduces an EcoRI site). The product was digested with EcoRI and SalI and inserted into pET30a (Novagen, Madison, WI) which had been digested with the same enzymes, to generate pJVB8. pJVB8 was introduced into Escherichia coli Rosetta2(DE3) (Novagen), which produces seven rare tRNAs required for efficient expression of some F. johnsoniae proteins in E. coli (21, 22). Expression was induced by addition of 1.0 mM IPTG (isopropyl-β-d-thiogalactopyranoside) and incubation for 3 h at 37°C. Cells were collected by centrifugation and disrupted using a French pressure cell, and recombinant GldN was purified by Ni affinity chromatography. Polyclonal antibodies against recombinant GldN were produced and affinity purified using the recombinant protein by Proteintech Group, Inc. (Chicago, IL).
Detection and localization of GldN, GldO, and SprB.
Western blots were performed to detect GldN and GldO in extracts of wild-type and mutant cells of F. johnsoniae. Overnight cultures were grown in MM at 25°C without shaking. Cells were pelleted by centrifugation at 4,400 × g for 10 min and suspended in a buffer consisting of 20 mM sodium phosphate and 10 mM EDTA, pH 7.5. Proteins were separated by SDS-PAGE essentially as described previously (12) and transferred to nitrocellulose membranes using a Trans Blot cell (Bio-Rad, Hercules, CA). Approximately 6 μg protein was loaded per lane. GldN and GldO were detected using antiserum against recombinant GldN, goat anti-rabbit secondary antibody linked to horseradish peroxidase (Bio-Rad), and a SuperSignal West Pico detection kit (Thermo Fisher Scientific) with a Foto/Analyst LuminaryFx Workstation (Fotodyne, Hartland, WI). F. johnsoniae cells grown in MM at 25°C without shaking were fractionated into soluble, inner membrane (Sarkosyl-soluble), and outer membrane (Sarkosyl-insoluble) fractions as described previously (9) except that EDTA-free protease inhibitor cocktail (Thermo Fisher Scientific) was added to cells in 20 mM sodium phosphate-10 mM EDTA (pH 7.5) before lysis in a French pressure cell. Proteins were separated by SDS-PAGE, and Western blotting was performed as described above. To detect SprB, cells were lysed using a French pressure cell as described above. Proteins (25 μg) were separated on 3 to 8% Criterion XT Tris-acetate acrylamide gels (Bio-Rad) before transfer to nitrocellulose and detection as described above except that antisera against SprB were used (21).
Detection of surface-localized SprB.
Movement of surface localized SprB was detected as previously described (21). Cells were grown overnight at 25°C in MM without shaking. Purified anti-SprB (1 μl of a 1:10 dilution of a 300-mg/liter stock), 0.5-μm-diameter protein G-coated polystyrene spheres (1 μl of a 0.1% stock preparation; Spherotech Inc., Libertyville, IL), and bovine serum albumin (BSA) (1 μl of a 1% solution) were added to 7 μl of cells (approximately 5 × 108 cells per ml) in MM. The cells were spotted on a glass slide, covered with a glass coverslip, and examined using an Olympus BH2 phase-contrast microscope with a heated stage at 25°C. Images were recorded with a Photometrics CoolSNAPcf2 camera and analyzed using MetaMorph software. Samples were examined 1 min after spotting, and images were captured for 30 s.
Wild-type and mutant cells were also examined by immunofluorescence microscopy to identify cell surface-localized SprB. Cells were grown overnight in MM at 25°C. Twenty microliters of cells was diluted in 130 μl of MM and fixed with 1% formaldehyde for 15 min. Cells were collected on 0.4-μm Isopore membrane filters (Millipore, Billerica, MA) by filtration. Cells were washed three times with 200 μl of PBS and were blocked with 0.1% BSA in PBS for 30 min. After removal of the blocking solution by filtration, cells were exposed to 200 μl of a 1:200 dilution of purified anti-SprB (21) in PBS with 0.1% BSA for 90 min. Cells were washed five times with 200 μl of PBS and exposed to 200 μl of F(ab′) fragment of goat anti-rabbit IgG conjugated to Alexa-488 (0.4 μg/ml; Invitrogen, Carlsbad, CA) in PBS plus 0.1% BSA. Cells were incubated for 60 min in the dark, the liquid was removed, and cells were washed five times with PBS. During the final PBS wash, 1 μl of InSpeck 0.3% relative intensity fluorescence beads (Invitrogen-Molecular Probes, Eugene, OR) was added as a control. The final wash was removed by filtration, the filters were mounted on glass slides with 6 μl of VectaShield with DAPI (4′,6′-diamidino-2-phenylindole) (Vector Laboratories Inc., Burlingame, CA), coverslips were applied, and samples were observed using a Nikon Eclipse 50i microscope. Images were captured with a Photometrics CoolSNAPES camera with exposure times of 700 to 1200 ms (DAPI) and 500 ms (Alexa-488).
RESULTS
Construction of a gldNO deletion mutant and of an isogenic gldN mutant.
Fragments upstream of gldN and downstream of gldO were cloned into the bacteroidete suicide vector pLYL03 to generate pNap3 (Fig. 1). Introduction of pNap3 into F. johnsoniae UW101 and selection for erythromycin resistance resulted in integration of the vector into the genome by a single recombination event. Methods to select for loss of vector DNA by a second recombination event have not been reported for F. johnsoniae. To facilitate isolation of the gldNO deletion, we developed a selection strategy that relies on the known resistance of F. johnsoniae motility mutants to bacteriophage infection. gldN mutants are resistant to infection by φCj1, φCj13, φCj23, and φCj29 (2). Exposure of F. johnsoniae UW101 carrying pNap3 integrated into its genome to φCj1 resulted in the isolation of phage-resistant, erythromycin-sensitive colonies. These were analyzed by PCR using primer pairs 692/939 and 694/684 (Fig. 1B) and by sequencing the amplified products. Each of seven phage-resistant erythromycin-sensitive colonies tested carried identical deletions within the region spanning gldN and gldO, and one of these (F. johnsoniae CJ1631A) was selected for further study. This approach to deletion construction is not suitable for all genes, but it is a general method for constructing unmarked deletions within F. johnsoniae motility genes since in all cases examined, disruption of motility genes results in resistance to one or more bacteriophages (1-3, 7-9, 17, 18, 21, 22).
Strains with HimarEm insertions in gldN have previously been isolated (2). The original HimarEm2 gldN mutant, CJ1304, was constructed in F. johnsoniae MM101, which is derived from F. johnsoniae ATCC 17061 UW101 but has an unidentified mutation resulting in a partial deficiency in chitin utilization (17). In order to facilitate comparisons between the effects of this gldN HimarEm2 mutation (in the chitin utilization-deficient background) and the gldNO deletion (in the chitin utilization-proficient background), a method was developed to transduce the gldN HimarEm2 mutation from F. johnsoniae CJ1304 into F. johnsoniae UW101 using φCj54. Bacteriophage φCj54 was grown on F. johnsoniae CJ1304 carrying pTB79, and the lysate was used to transduce the transposon, conferring erythromycin resistance, and adjacent chromosomal DNA into F. johnsoniae UW101, resulting in the gldN mutant F. johnsoniae CJ1743. The presence of the transposon in gldN was confirmed by PCR (Fig. 1C). To demonstrate that the mutation had been introduced into F. johnsoniae UW101, we took advantage of the fact that F. johnsoniae MM101 has a silent C-T mutation at position 114 (numbered from the first nucleotide of the start codon of gldG). This mutation is located 1.1 Mbp from gldN and gldO, making cotransduction unlikely. Amplification and sequencing of gldG verified that the transposon had been transduced into F. johnsoniae UW101. φCj54 was used for transduction in this study, but we also demonstrated that φCj1, φCj13, φCj23, φCj28, φCj29, φCj42, and φCj48 can also be used to transduce markers between strains of F. johnsoniae (data not shown). Phage transduction is a useful addition to the genetic tools available for F. johnsoniae and should facilitate strain construction.
Either GldN or GldO is sufficient to allow formation of spreading colonies.
Motility phenotypes of wild-type F. johnsoniae UW101 and of isogenic strains carrying either a transposon insertion in gldN (CJ1743) or a deletion spanning gldN and gldO (CJ1631A) were examined. Wild-type cells formed spreading colonies, whereas cells of the gldN mutant CJ1743 formed nonspreading colonies (Fig. 2A and E). Introduction of pTB79, which expresses gldN, into CJ1743 resulted in complementation and formation of spreading colonies (Fig. 2F). Colony spreading was partially restored to CJ1743 by introduction of pTB97a, which carries gldO, suggesting that increased levels of GldO can partially compensate for the defect in gldN (Fig. 2G). The gldNO deletion mutant CJ1631A formed nonspreading colonies (Fig. 2I) that were indistinguishable from those of CJ1743. The ability to form spreading colonies was restored by introduction of pTB79 (which carries gldN) and was partially restored by introduction of pTB97a (which carries gldO) (Fig. 2J and K). Introduction of pTB98 (which carries gldL, gldM, gldN, and gldO) into wild-type cells or into cells of the gldN or gldNO mutants resulted in the formation of small colonies that exhibited some spreading (Fig. 2D, H, and L). The presence of pTB98 resulted in a decreased growth rate, presumably because of deleterious effects of moderate overexpression of gldL, gldM, gldN, and gldO together. A similar effect was previously reported for cells expressing plasmid-borne gldK and gldL together (2).
FIG. 2.

Photomicrographs of F. johnsoniae colonies. Colonies were incubated at 25°C on PY2 agar medium for 48 h, and photomicrographs were taken with a Photometrics CoolSNAPcf2 camera mounted on an Olympus IMT-2 phase-contrast microscope. (A) Wild-type (WT) F. johnsoniae UW101 with control vector pCP23. (B) Wild-type F. johnsoniae UW101 with pTB79, which carries gldN. (C) Wild-type F. johnsoniae UW101 with pTB97a, which carries gldO. (D) Wild-type F. johnsoniae UW101 with pTB98, which carries gldL, gldM, gldN, and gldO. (E) gldN mutant CJ1743 with pCP23. (F) gldN mutant CJ1743 with pTB79. (G) gldN mutant CJ1743 with pTB97a. (H) gldN mutant CJ1743 with pTB98. (I) gldNO deletion mutant CJ1631A with pCP23. (J) gldNO deletion mutant CJ1631A with pTB79. (K) gldNO deletion mutant CJ1631A with pTB97a. (L) gldNO deletion mutant CJ1631A with pTB98. Bars indicate 0.5 mm.
Disruption of gldN and gldO eliminates motility and results in decreased attachment to surfaces.
Wild-type and mutant cells were examined for attachment to glass coverslips in a Petroff-Hauser counting chamber. Wild-type cells readily attached to glass, whereas few cells of the gldN or gldNO mutant strains attached (Table 2). Complementation of the mutants with pTB79, which carries gldN, or pTB97a, which carries gldO, restored attachment to glass to near-wild-type levels.
TABLE 2.
Effect of mutations in gldN and gldO on attachment of cells to glass coverslips
| Strain | Description | Avg (SD) no. of cells attached to 0.03-mm2 region of glass coverslipa |
|---|---|---|
| UW101 with control plasmid pCP23 | Wild type | 171.6 (34.1) |
| CJ1743 with pCP23 | gldN mutant | 13.8 (6.6) |
| CJ1743 with pTB79 | gldN mutant complemented with gldN | 121.5 (30.5) |
| CJ1743 with pTB97a | gldN mutant complemented with gldO | 133.3 (31.5) |
| CJ1631A with pCP23 | gldNO deletion mutant | 0.4 (0.7) |
| CJ1631A with pTB79 | gldNO deletion mutant complemented with gldN | 140.4 (62.4) |
| CJ1631A with pTB97a | gldNO deletion mutant complemented with gldO | 96.6 (20.6) |
Approximately 2 × 106 cells in 2.5 μl of MM were introduced into a Petroff-Hausser counting chamber and incubated for 2 min at 25°C. Samples were observed using an Olympus BH-2 phase-contrast microscope, and cells attached to a 0.03-mm2 region of the cover glass were counted. Numbers in parentheses are standard deviations calculated from 12 measurements.
Wild-type cells moved rapidly over glass surfaces in wet mounts (see Movie S1 in the supplemental material). Cells of the gldN mutant CJ1743 were severely deficient in motility, but a few cells exhibited some ability to glide (see Movie S2 in the supplemental material). In contrast, cells of CJ1631A carrying a deletion spanning gldN and gldO were completely nonmotile (see Movie S3 in the supplemental material). Even the rare cells that attached to the glass failed to exhibit any movements. Introduction of pTB79, which spans gldN, and pTB97a, which spans gldO, into the mutants resulted in restoration of motility (see Movies S4 and S5 in the supplemental material), indicating that the presence of either GldN or GldO supports gliding. Essentially identical results were obtained in separate experiments using Teflon membranes instead of glass coverslips, indicating that the attachment and motility defects of gldN and gldNO mutants were not specific to glass surfaces (data not shown).
Identification and localization of GldN and GldO.
Antiserum to GldN were used to detect GldN and GldO in cell extracts. GldN, which migrated with an apparent molecular mass of approximately 36 kDa, was detected in extracts of wild-type cells (Fig. 3, lane 1) but was absent from extracts of the gldN mutant CJ1743 and of the gldNO deletion mutant CJ1631A (Fig. 3, lanes 2 and 5). Introduction of gldN on pTB79 restored production of GldN. Antiserum raised against GldN also recognized GldO. GldO, which migrated with an apparent molecular mass of approximately 38 kDa, was not usually apparent in extracts of wild-type cells but was detected in cells of the gldN mutant CJ1743 (Fig. 3 lane 2) or in cells expressing GldO from pTB97a or pTB98 (Fig. 3 lanes 4, 7, and 8).
FIG. 3.

Immunodetection of GldN and GldO. Whole-cell extracts were examined for GldN and GldO by Western blot analysis. Lane 1, wild-type F. johnsoniae with control vector pCP23. Lane 2, gldN mutant CJ1743 with pCP23. Lane 3, gldN mutant CJ1743 with pTB79, which carries gldN. Lane 4, gldN mutant CJ1743 with pTB97a, which carries gldO. Lane 5, gldNO deletion mutant CJ1631A with pCP23. Lane 6, gldNO deletion mutant CJ1631A with pTB79, which carries gldN. Lane 7, gldNO deletion mutant CJ1631A with pTB97a, which carries gldO. Lane 8, gldNO deletion mutant CJ1631A with pTB98, which carries gldL, gldM, gldN, and gldO.
GldN and GldO were found in both the soluble and particulate fractions of cell extracts (Fig. 4). Addition of 100 and 500 mM NaCl had no effect on this distribution, suggesting that localization to the particulate fraction was not the result of electrostatic interactions (Fig. 4A and data not shown). GldN and GldO both have predicted cleavable signal peptides, but they do not have obvious features of membrane proteins such as additional predicted hydrophobic alpha-helical segments as expected for cytoplasmic membrane proteins or extensive beta-sheet structure as expected for outer membrane proteins. Programs used to predict protein localization did not allow prediction of localization with confidence. The subcellular localization predictor Cello v.2.5 (34, 35) predicted that both proteins were likely to localize to either the periplasm or outer membrane, and PSORTb (6) gave no prediction for either GldN or GldO. Sarkosyl has been demonstrated to solubilize cytoplasmic membrane proteins but not outer membrane proteins of F. johnsoniae (9). Exposure to Sarkosyl resulted in solubilization of most of the GldN and GldO that were present in the particulate (membrane) fraction (Fig. 4B), suggesting that the proteins were associated with the cytoplasmic membrane. However, it is possible that exposure to Sarkosyl disrupted a protein complex containing GldN and GldO or that these proteins were loosely associated with the outer membrane and were solubilized by the detergent.
FIG. 4.

Localization of GldN and GldO. (A) Fractionation of GldN between soluble and particulate fractions. Cells of wild-type F. johnsoniae UW101 were disrupted and separated into soluble and membrane fractions. Equal amounts of each fraction based on the starting material were separated by SDS-PAGE, and GldN was detected by Western blot analysis. NaCl was added to some extracts to determine whether the increased ionic strength would alter the localization of GldN. Lanes 1, 4, and 7, whole cells. Lanes 2, 5, and 8, soluble fraction. Lanes 3, 6, and 9, particulate fraction. Lanes 1, 2, and 3, no salt added. Lanes 4, 5, and 6, 100 mM NaCl. Lanes 7, 8, and 9, 500 mM NaCl. (B) Fractionation of GldN and GldO. Wild-type and mutant cells were disrupted and separated into soluble and membrane fractions. Membranes were fractionated further by differential solubilization in Sarkosyl, and proteins were detected by Western blot analysis. Equal amounts of each fraction based on the starting material were loaded in each lane. Lanes 1 to 4, wild-type F. johnsoniae with control vector pCP23. Lanes 5 to 8, gldN mutant CJ1743 with pCP23. Lanes 9 to 12, gldNO deletion mutant CJ1631A with pTB97a, which carries gldO. Lanes 1, 5, and 9, whole-cell extracts. Lanes 2, 6, 10, soluble (cytoplasmic and periplasmic) fractions. Lanes 3, 7, and 11, Sarkosyl-soluble (cytoplasmic membrane) fractions. Lanes 4, 8, and 12, Sarkosyl insoluble (outer membrane) fractions.
SprB is improperly localized in cells of the gldN and gldNO mutants.
Comparative genome analysis revealed that the distantly related nonmotile bacteroidete P. gingivalis has homologues to some of the F. johnsoniae motility genes, including gldN (2, 20). P. gingivalis is a common cause of gum disease, and secretion of gingipain proteases is important for pathogenesis. Recent results suggest that the P. gingivalis “motility” proteins, such as the GldN homologue PorN, are required for secretion of gingipain protease virulence factors rather than for cell movement (28). F. johnsoniae GldN may also be involved in protein secretion, and an inability to secrete cell surface motility proteins could account for some of the motility defects of gldN mutants. Antibodies were used to examine the localization of SprB in wild-type cells, in cells with a transposon insertion in gldN, and in cells carrying a deletion spanning gldN and gldO. Each of these strains produced SprB protein (Fig. 5). Protein G-coated polystyrene spheres carrying antibodies against SprB were used to detect the presence of surface-exposed SprB on live cells. As previously reported (21), antibody-coated spheres attached specifically to wild-type cells expressing SprB and were rapidly propelled along the surfaces of the cells (Table 3; see Movie S6 in the supplemental material). Also as previously reported (21), such spheres failed to attach to sprB mutant cells, and Protein-G coated spheres without antibodies failed to bind to wild-type cells (Table 3; see Movie S7 in the supplemental material). Antibody-coated spheres failed to bind to cells of the gldNO deletion mutant CJ1631A (Table 3; see Movie S8 in the supplemental material), indicating that SprB was not exposed on the surface of these cells. Complementation of the gldNO mutant with pTB79, which carries gldN, or with pTB97a, which carries gldO, restored surface localization of SprB (Table 3; see Movie S9 in the supplemental material). Cells of the gldN mutant CJ1743 appeared to have some surface-localized SprB, since some cells bound to the antibody-coated spheres. Surprisingly, although these cells bound some spheres, very few (fewer than 1% of those with spheres attached) were observed to move them. In contrast, examination of 100 wild-type cells with antibody-coated spheres attached revealed that 90 of these cells rapidly propelled the spheres during a 30-s observation. Cells of the gldN mutant CJ1743 that had been complemented with pTB79 or pTB97a behaved similarly to wild-type cells and rapidly propelled attached spheres. The inability of gldN mutant cells to propel spheres that were attached to surface-localized SprB may indicate that GldN is directly involved in cell movement, in addition to its role in surface localization of SprB.
FIG. 5.
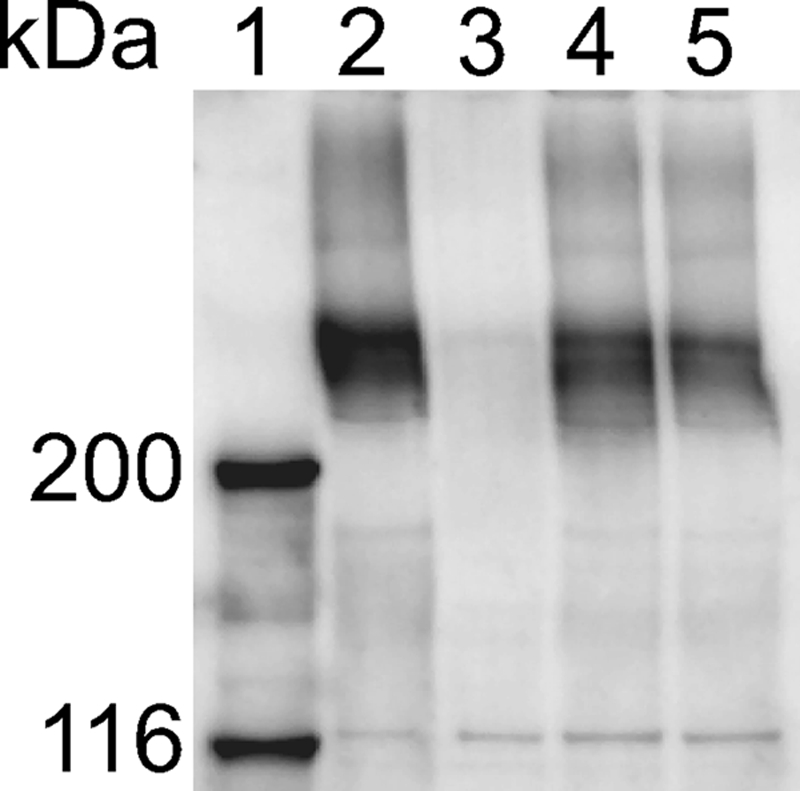
Western blot analysis of SprB in cells of wild-type and mutant F. johnsoniae strains. Cells were disrupted using a French pressure cell, and samples were boiled in SDS-PAGE loading buffer. Proteins (25 μg per lane) were separated by electrophoresis, and SprB was detected using anti-SprB antibody. Lane 1, molecular weight markers. Lane 2, wild-type F. johnsoniae carrying control plasmid pCP23. Lane 3, sprB mutant FJ156 carrying pCP23. Lane 4, gldN mutant CJ1743 carrying pCP23. Lane 5, gldNO deletion mutant CJ1631A carrying pCP23.
TABLE 3.
Effect of mutations in gldN and gldO on binding of protein G-coated polystyrene spheres carrying antibodies against SprB
| Strain | Description | Antibody added | Avg (SD) % of cells with spheres attacheda |
|---|---|---|---|
| UW101 with control plasmid pCP23 | Wild type | None | 0.3 (0.6) |
| UW101 with pCP23 | Wild type | Anti-SprB | 51.7 (4.0) |
| FJ156 | sprB mutant | Anti-SprB | 1.0 (1.0) |
| CJ1743 with pCP23 | gldN mutant | Anti-SprB | 21.0 (2.6) |
| CJ1743 with pTB79 | gldN mutant complemented with gldN | Anti-SprB | 45.7 (6.1) |
| CJ1743 with pTB97a | gldN mutant complemented with gldO | Anti-SprB | 46.7 (5.9) |
| CJ1631A with pCP23 | gldNO deletion mutant | Anti-SprB | 0.0 (0.0) |
| CJ1631A with pTB79 | gldNO deletion mutant complemented with gldN | Anti-SprB | 48.0 (4.6) |
| CJ1631A with pTB97a | gldNO deletion mutant complemented with gldO | Anti-SprB | 58.0 (2.6) |
Purified anti-SprB and 0.5-μm-diameter protein G-coated polystyrene spheres were added to cells as described in Materials and Methods. Samples were spotted on a glass slide, covered with a glass coverslip, incubated for 1 min at 25°C, and examined using a phase-contrast microscope. Images were recorded for 30 s, and 100 randomly selected cells were examined for the presence of attached spheres during this period. Numbers in parentheses are standard deviations calculated from three measurements.
Analysis of surface exposure of SprB by immunofluorescence microscopy of fixed cells confirmed that gldN and gldNO mutant cells are defective in surface localization of SprB (Fig. 6). Cells of F. johnsoniae with mutations in another gene required for efficient motility, sprT, also fail to efficiently assemble SprB on the cell surface, suggesting a defect in protein secretion (28). The decreased levels of SprB, and perhaps of other motility proteins, on the cell surface may explain some of the motility defects exhibited by cells of the gldN and gldNO deletion mutants.
FIG. 6.

Detection of surface-localized SprB protein by immunofluorescence microscopy. Cells of wild-type and mutant F. johnsoniae were exposed to DAPI and to anti-SprB antibodies followed by secondary antibodies conjugated to Alexa-488. The fluorescent spheres are InSpeck relative intensity control fluorescence beads. WT, wild-type F. johnsoniae UW101. sprB refers to the HimarEm2 sprB mutant FJ156. gldN refers to the HimarEm2 gldN mutant CJ1743. gldNO refers to the gldNO deletion mutant CJ1631A. pCP23 is a control vector, pTB79 carries gldN, and pTB97a carries gldO. The bar indicates 10 μm.
Cells of the gldNO mutant fail to digest chitin.
F. johnsoniae rapidly digests chitin (17, 30). Genome analysis identified five potential chitinases predicted to cut the long chitin polymers and four potential β-N-acetyl-glucosaminidases predicted to release N-acetyl-glucosamine or chitobiose from the oligomers. Wild-type cells of F. johnsoniae rapidly digested colloidal chitin, cells of the gldN mutant CJ1743 digested chitin more slowly, and cells of the gldNO mutant CJ1631A were completely deficient in digestion of colloidal chitin (Fig. 7). In each case, complementation of the mutants with plasmids carrying gldN or gldO restored the ability to digest chitin. In order to determine the reason for the defect in colloidal chitin utilization of the gldN and gldNO mutants we examined chitinase activities in intact cells, cell extracts, and cell-free supernatants using the synthetic substrates 4-MU-GlcNAc, 4-MU-(GlcNAc)2, and 4-MU-(GlcNAc)3. Release of methylumbelliferone from 4-MU-GlcNAc is typically the result of β-N-acetyl-glucosaminidases, release of methylumbelliferone from 4-MU-(GlcNAc)3 is typically the result of chitinases, and release of methylumbelliferone from 4-MU-(GlcNAc)2 may be the result of either activity. Intact cells and cell extracts released 4-methylumbelliferone from each of the substrates, and there were no significant differences between the activities associated with wild-type and mutant cells (Fig. 8). Cell-free supernatants from wild-type cells failed to digest 4-MU-GlcNAc but had substantial activities against 4-MU-(GlcNAc)2 and 4-MU-(GlcNAc)3. This suggests that the F. johnsoniae β-N-acetyl-glucosaminidases are primarily cell bound, as predicted from bioinformatics analyses (20), but that cells secrete one or more chitinases. Cell-free supernatants from cells of the gldN mutant contained much lower levels of chitinase activities, and supernatants of cells of the gldNO deletion mutant contained no detectable chitinase activity (Fig. 8). Complementation with plasmids carrying gldN or gldO restored the soluble extracellular chitinase activities to wild-type levels. These results suggest that one or more chitinases may be secreted via the proposed PorSS.
FIG. 7.

Mutations in gldN and gldO result in defects in chitin utilization. Approximately 106 cells of F. johnsoniae were spotted on MYA-chitin medium and incubated at 25°C for 4 days. (A) Wild-type F. johnsoniae UW101 with control vector pCP23. (B) gldN mutant CJ1743 with pCP23. (C) gldN mutant CJ1743 with pTB79, which carries gldN. (D) gldN mutant CJ1743 with pTB97a, which carries gldO. (E) gldNO deletion mutant CJ1631A with pCP23. (F) gldNO deletion mutant CJ1631A with pTB79. (G) gldNO deletion mutant CJ1631A with pTB97a.
FIG. 8.

Chitinase and β-N-acetyl-glucosaminidase activities. Chitinase and β-N-acetyl-glucosaminidase activities of intact cells, cell lysates, and culture supernatants of F. johnsoniae strains were determined using the synthetic substrates 4-MU-GlcNAc, 4-MU-(GlcNAc)2, and 4-MU-(GlcNAc)3. Equal amounts of each sample, based on the protein content of the cell suspension, were incubated with 10 nmol of synthetic substrate for 4 h at 37°C, and the amount of 4-MU released was determined by measuring fluorescence emission at 460 nm following excitation at 360 nm. Yellow, wild-type F. johnsoniae UW101 carrying control vector pCP23. Red, gldN mutant CJ1743 with pCP23. Dark blue, gldN mutant CJ1743 with pTB79, which carries gldN. Orange, gldN mutant CJ1743 with pTB97a, which carries gldO. Green, gldNO deletion mutant CJ1631A with pCP23. Pink, gldNO deletion mutant CJ1631A with pTB79. Light blue, gldNO deletion mutant CJ1631A with pTB97a. Error bars indicate standard errors.
Bacteriophage resistance of gldNO mutants.
Sensitivity to F. johnsoniae bacteriophages was determined as previously described (9) by spotting 5 μl of phage lysates (109 PFU/ml) onto lawns of cells in CYE overlay agar. The plates were incubated for 24 h at 25°C to observe lysis (Fig. 9). Wild-type F. johnsoniae showed complete lysis by all of the bacteriophages. The gldN mutant exhibited resistance to most of the bacteriophages but was partially susceptible to φCj28 and φCj54 (Fig. 9B). In contrast, the gldNO mutant, F. johnsoniae CJ1631A, was resistant to infection by each of the phages (Fig. 9E). Introduction of the plasmids pTB79, which spans gldN, and pTB97a, which spans gldO, into the gldN mutant or the gldNO deletion mutant resulted in restoration of sensitivity to each of the bacteriophages.
FIG. 9.

Mutations in gldN and gldO result in resistance to bacteriophages. Bacteriophages (5 μl of lysates containing approximately 109 phage/ml) were spotted onto lawns of cells in CYE overlay agar. The plates were incubated at 25°C for 24 h to observe lysis. Bacteriophages were spotted in the following order from left to right, as indicated also by the numbers in panel A: top row, φCj1, φCj13, and φCj23; middle row, φCj28, φCj29, and φCj42; bottom row, φCj48 and φCj54. (A) Wild-type F. johnsoniae UW101 with control vector pCP23. (B) gldN mutant CJ1743 with pCP23. (C) gldN mutant CJ1743 with pTB79, which carries gldN. (D) gldN mutant CJ1743 with pTB97a, which carries gldO. (E) gldNO deletion mutant CJ1631A with pCP23. (F) gldNO deletion mutant CJ1631A with pTB79. (G) gldNO deletion mutant CJ1631A with pTB97a.
Many spontaneous or chemically induced motile nonspreading mutants have mutations in gldN or in other gld genes.
Pate and colleagues isolated numerous spontaneous and chemically induced nonspreading mutants, and 85 of these are available for analysis (4, 32). Forty-six of these mutants are completely nonmotile and have mutations in known gld genes, as previously reported (1-3, 7-9, 17, 18). Individual cells of the remaining 39 nonspreading mutants retain some ability to glide in wet mounts and were referred to as “motile nonspreading” mutants (4). The motility phenotypes of these mutants are similar to those of cells with transposon insertions in sprA, sprB, or gldN (2, 21, 22). Plasmids carrying each of the known gld and spr genes were introduced into these mutants. Five of the mutants were complemented by pSN48, which spans sprA; four were complemented by pSN60, which spans sprB; one was complemented by pSP24, which carries sprC; and eight were complemented by pTB79, which carries gldN (Table 4). Further complementation analysis suggested that 18 of the remaining 21 spontaneous and chemically induced motile nonspreading mutants had mutations in known gld genes (gldA, gldB, gldD, gldF, gldJ, gldK, gldL, and gldM) (Table 4). Disruption of any of these gld genes results in complete loss of motility, so the point mutations likely result in production of partially functional Gld proteins and thus cause less drastic effects on motility. These gld mutants with partial function may be useful in elucidating the roles of the individual Gld proteins in motility and in protein secretion. Most of the gld and spr genes were originally identified by transposon mutagenesis. The fact that 82 of 85 independently isolated spontaneous and chemically induced nonspreading mutants were complemented with plasmids carrying the 15 known spr and gld genes suggests that few motility genes may remain to be identified.
TABLE 4.
Complementation of spontaneous and chemically induced motile nonspreading mutants by plasmids
| Straina | Complementing plasmid | Predicted site of mutation |
|---|---|---|
| UW102-1 | Not complemented | Unknownd |
| UW102-2 | pMK315 (gldF) | gldF |
| UW102-3 | pSN48 (sprA) | sprA |
| UW102-18 | pTB79 (gldN) | gldN |
| UW102-24 | pSN60 (sprB) | sprB |
| UW102-37 | pMM313 (gldJ) | gldJ |
| UW102-43 | pSN48 (sprA) | sprA |
| UW102-45b | pSN60 (sprB) | sprB |
| UW102-46 | Not complemented | Unknown |
| UW102-50b | pTB79 (gldN) | gldN |
| UW102-51b | pTB79 (gldN) | gldN |
| UW102-67 | pTB79 (gldN) | gldN |
| UW102-73b | pTB99 (gldK) | gldK |
| UW102-88 | pTB79 (gldN) | gldN |
| UW102-91b | pSP24 (sprC) | sprC |
| UW102-93 | pTB79 (gldN) | gldN |
| UW102-95b | pMM265 (gldJ) | gldJ |
| UW102-99b | pDH223 (gldB) | gldB |
| UW102-103 | pDH223 (gldB) | gldB |
| UW102-106b | pSN48 (sprA) | sprA |
| UW102-128 | pSN60 (sprB) | sprB |
| UW102-135 | pTB79 (gldN) | gldN |
| UW102-136 | pMM213 (gldD) | gldD |
| UW102-142 | pSN48 (sprA) | sprA |
| UW102-143 | pTB99 (gldK) | gldK |
| UW102-148 | pTB79 (gldN) | gldN |
| UW102-149b | pTB99 (gldK) | gldK |
| UW102-150 | pSN48 (sprA) | sprA |
| UW102-155b | pMK315 (gldF) | gldF |
| UW102-156b | pTB99 (gldK) | gldK |
| UW102-158 | pTB81a (gldL) | gldL |
| UW102-168 | pSA11 (gldA) | gldA |
| UW102-171 | pTB99 (gldK) | gldK |
| UW102-172 | pMM313 (gldJ) | gldJ |
| UW102-176 | pTB94a (gldM) | gldM |
| UW102-298 | pSN60 (sprB) | sprB |
| UW102-301b | pMM265 (gldJ) | gldJ |
| UW102-344 | pTB81a (gldL)c | gldL |
| UW102-345b | Not complemented | Unknown |
The motile nonspreading mutants were previously described (4, 24, 32). They form nonspreading colonies, but cells retain some ability to glide in wet mounts.
These strains were described as completely nonmotile in the original publications (4, 24, 32), but more recent observations indicate that a few cells exhibit some motility in wet mounts.
Partial complementation.
The strain was not complemented by plasmids carrying gldA, gldB, gldD, gldF, gldG, gldH, gldI, gldJ, gldK, gldL, gdlM, gldN, gldO, sprA, sprB, sprC, sprD, sprE, or sprT.
DISCUSSION
The results described above demonstrate that GldN, or the GldN-like protein GldO, is essential for F. johnsoniae gliding motility. GldN and GldO appear to be partially redundant components of the motility machinery. Other gliding bacteroidetes for which complete genome sequences are available, such as Flavobacterium psychrophilum (5), and Cytophaga hutchinsonii (33), have single copies of gldN, so the presence of semiredundant GldN-like proteins may be a somewhat unusual feature of F. johnsoniae. We do not know why F. johnsoniae has two GldN-like proteins. GldN and GldO may be produced under different conditions and may allow optimal movement over different surfaces. GldN and GldO may function in protein secretion as part of the F. johnsoniae PorSS. They are required for efficient surface localization of the SprB adhesin, which is thought to be one of the outermost components of the motility machinery. However, mislocalization of SprB cannot explain all of the attachment and motility defects of the gldNO deletion mutant. Unlike cells of the gldNO mutant, cells of sprB mutants retain significant ability to attach to and glide on glass (21). F. johnsoniae has several paralogs of sprB (20) that may encode alternative adhesins for attachment to and movement over different surfaces. GldN and GldO may be required for secretion of these proteins in addition to secretion of SprB, which might account for the complete loss of gliding motility exhibited by the gldNO deletion mutant. It is possible that GldN and GldO provide optimal secretion of different cell surface adhesins required for optimal movement over different surfaces. This could provide an explanation for the presence of the seemingly redundant gldN and gldO genes in F. johnsoniae.
It has been known for decades that motility mutants of F. johnsoniae are often deficient in utilization of chitin and display resistance to bacteriophages, but the underlying reasons for these pleiotropic effects were not understood (4). The identification of cell surface motility proteins such as SprB (21) and of an apparent protein secretion system associated with gliding motility (28) suggest explanations for these phenotypes. Cells with mutations in sprB are resistant to some bacteriophages (21), suggesting the possibility that the mobile cell surface adhesin SprB serves as a receptor for these phages. Since gldNO mutants fail to efficiently assemble SprB on the cell surface, such cells would be expected to be resistant to any bacteriophages that require SprB for attachment or entry into the cell. However, gldNO mutants are resistant to all bacteriophages tested, whereas sprB mutants are resistant to only a subset of bacteriophages (21). Many sprB-like genes are present in the F. johnsoniae genome (20). These may encode semiredundant cell surface components of the motility machinery that may be secreted by the PorSS and serve as receptors for some of the bacteriophages that infect cells lacking SprB. The inability of gldNO mutants to secrete any of these receptors to the cell surface could explain the complete resistance to all bacteriophages. Other motility mutations that directly or indirectly disrupt the PorSS would also likely result in phage resistance. Our results also suggest an explanation for the chitin utilization defect of motility mutants such as those deficient in gldN and gldO. The presence of extracellular chitinase secreted by wild-type cells and the absence of secreted chitinase in the gldNO mutant suggest that the PorSS is involved in chitinase secretion as well as being involved in assembly of the motility apparatus. Further analyses may identify additional functions of this secretion system.
The available evidence supports a model of F. johnsoniae motility in which some of the Gld proteins constitute the “motor” embedded in the cell envelope that propels SprB and related SprB-like adhesins along the cell surface, resulting in cell movement (10, 21). GldN appears to be involved in assembly of the motility apparatus, since it is needed for localization of SprB to the cell surface. Based on the work presented here and on studies conducted with P. gingivalis (28), the F. johnsoniae protein secretion machinery involved in assembly of SprB on the cell surface is likely to involve GldK, GldL, GldM, GldN, SprA, SprE, SprT, and perhaps several other proteins. If these proteins are dedicated to protein secretion, then the remaining motility proteins (GldA, GldB, GldD, GldF, GldG, GldH, GldI, and GldJ) might be more directly involved in cell movement. However, the separation between motility and secretion functions may not be complete. The apparent failure of gldN mutant cells to propel the small amount of SprB that does make it to the surface suggests the possibility that GldN may have other roles in motility besides localization of SprB and related proteins to the cell surface. Further studies are needed to determine the range of functions performed by GldN and to determine which of the motility proteins are involved in assembly of the motility apparatus, which are involved in function of the apparatus, and which, if any, are involved in both processes.
Supplementary Material
Acknowledgments
This research was supported by grant MCB-0641366 from the National Science Foundation.
Footnotes
Published ahead of print on 28 December 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Agarwal, S., D. W. Hunnicutt, and M. J. McBride. 1997. Cloning and characterization of the Flavobacterium johnsoniae (Cytophaga johnsonae) gliding motility gene, gldA. Proc. Natl. Acad. Sci. U. S. A. 94:12139-12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braun, T. F., M. K. Khubbar, D. A. Saffarini, and M. J. McBride. 2005. Flavobacterium johnsoniae gliding motility genes identified by mariner mutagenesis. J. Bacteriol. 187:6943-6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braun, T. F., and M. J. McBride. 2005. Flavobacterium johnsoniae GldJ is a lipoprotein that is required for gliding motility. J. Bacteriol. 187:2628-2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang, L. Y. E., J. L. Pate, and R. J. Betzig. 1984. Isolation and characterization of nonspreading mutants of the gliding bacterium Cytophaga johnsonae. J. Bacteriol. 159:26-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duchaud, E., M. Boussaha, V. Loux, J. F. Bernardet, C. Michel, B. Kerouault, S. Mondot, P. Nicolas, R. Bossy, C. Caron, P. Bessières, J. F. Gibrat, S. Claverol, F. Dumetz, M. L. Hénaff, and A. Benmansour. 2007. Complete genome sequence of the fish pathogen Flavobacterium psychrophilum. Nat. Biotechnol. 25:763-769. [DOI] [PubMed] [Google Scholar]
- 6.Gardy, J. L., M. R. Laird, F. Chen, S. Rey, C. J. Walsh, M. Ester, and F. S. L. Brinkman. 2005. PSORTb v. 2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics 21:617-623. [DOI] [PubMed] [Google Scholar]
- 7.Hunnicutt, D. W., M. J. Kempf, and M. J. McBride. 2002. Mutations in Flavobacterium johnsoniae gldF and gldG disrupt gliding motility and interfere with membrane localization of GldA. J. Bacteriol. 184:2370-2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hunnicutt, D. W., and M. J. McBride. 2001. Cloning and characterization of the Flavobacterium johnsoniae gliding motility genes gldD and gldE. J. Bacteriol. 183:4167-4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunnicutt, D. W., and M. J. McBride. 2000. Cloning and characterization of the Flavobacterium johnsoniae gliding motility genes, gldB and gldC. J. Bacteriol. 182:911-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jarrell, K. F., and M. J. McBride. 2008. The surprisingly diverse ways that prokaryotes move. Nat. Rev. Microbiol. 6:466-476. [DOI] [PubMed] [Google Scholar]
- 11.Kempf, M. J., and M. J. McBride. 2000. Transposon insertions in the Flavobacterium johnsoniae ftsX gene disrupt gliding motility and cell division. J. Bacteriol. 182:1671-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 13.Li, L.-Y., N. B. Shoemaker, and A. A. Salyers. 1995. Location and characterization of the transfer region of a Bacteroides conjugative transposon and regulation of the transfer genes. J. Bacteriol. 177:4992-4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu, J., M. J. McBride, and S. Subramaniam. 2007. Cell-surface filaments of the gliding bacterium Flavobacterium johnsoniae revealed by cryo-electron tomography. J. Bacteriol. 189:7503-7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McBride, M. J. 2001. Bacterial gliding motility: multiple mechanisms for cell movement over surfaces. Annu. Rev. Microbiol. 55:49-75. [DOI] [PubMed] [Google Scholar]
- 16.McBride, M. J. 2004. Cytophaga-flavobacterium gliding motility. J. Mol. Microbiol. Biotechnol. 7:63-71. [DOI] [PubMed] [Google Scholar]
- 17.McBride, M. J., and T. F. Braun. 2004. GldI is a lipoprotein that is required for Flavobacterium johnsoniae gliding motility and chitin utilization. J. Bacteriol. 186:2295-2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McBride, M. J., T. F. Braun, and J. L. Brust. 2003. Flavobacterium johnsoniae GldH is a lipoprotein that is required for gliding motility and chitin utilization. J. Bacteriol. 185:6648-6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McBride, M. J., and M. J. Kempf. 1996. Development of techniques for the genetic manipulation of the gliding bacterium Cytophaga johnsonae. J. Bacteriol. 178:583-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McBride, M. J., G. Xie, E. C. Martens, A. Lapidus, B. Henrissat, R. G. Rhodes, E. Goltsman, W. Wang, J. Xu, D. W. Hunnicutt, A. M. Staroscik, T. R. Hoover, Y. Q. Cheng, and J. L. Stein. 2009. Novel features of the polysaccharide-digesting gliding bacterium Flavobacterium johnsoniae as revealed by genome sequence analysis. Appl. Environ. Microbiol. 75:6864-6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson, S. S., S. Bollampalli, and M. J. McBride. 2008. SprB is a cell surface component of the Flavobacterium johnsoniae gliding motility machinery. J. Bacteriol. 190:2851-2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nelson, S. S., P. P. Glocka, S. Agarwal, D. P. Grimm, and M. J. McBride. 2007. Flavobacterium johnsoniae SprA is a cell-surface protein involved in gliding motility. J. Bacteriol. 189:7145-7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pate, J. L., and L.-Y. E. Chang. 1979. Evidence that gliding motility in prokaryotic cells is driven by rotary assemblies in the cell envelopes. Curr. Microbiol. 2:59-64. [Google Scholar]
- 24.Pate, J. L., and D. M. De Jong. 1990. Use of nonmotile mutants to identify a set of membrane proteins related to gliding motility in Cytophaga johnsonae. J. Bacteriol. 172:3117-3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pate, J. L., S. J. Petzold, and L.-Y. E. Chang. 1979. Phages for the gliding bacterium Cytophaga johnsonae that infect only motile cells. Curr. Microbiol. 2:257-262. [Google Scholar]
- 26.Pochiraju, S. 2009. Role of cell surface and cytoplasmic membrane proteins in Flavobacterium johnsoniae gliding motility. M.S. thesis. University of Wisconsin-Milwaukee, Milwaukee, WI.
- 27.Saiki, K., and K. Konishi. 2007. Identification of a Porphyromonas gingivalis novel protein Sov required for the secretion of gingipains. Microbiol. Immunol. 51:483-491. [DOI] [PubMed] [Google Scholar]
- 28.Sato, K., M. Naito, H. Yukitake, H. Hirakawa, M. Shoji, M. J. McBride, R. G. Rhodes, and K. Nakayama. 2010. A protein secretion system linked to bacteroidete gliding motility and pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 107:276-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato, K., E. Sakai, P. D. Veith, M. Shoji, Y. Kikuchi, H. Yukitake, N. Ohara, M. Naito, K. Okamoto, E. C. Reynolds, and K. Nakayama. 2005. Identification of a new membrane-associated protein that influences transport/maturation of gingipains and adhesins of Porphyromonas gingivalis J. Biol. Chem. 280:8668-8677. [DOI] [PubMed] [Google Scholar]
- 30.Stanier, R. Y. 1947. Studies on non-fruiting myxobacteria. I. Cytophaga johnsonae, N. Sp., A chitin-decomposing myxobacterium. J. Bacteriol. 53:297-315. [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson, S. E., M. Smith, M. C. Wilkinson, and K. Peek. 2001. Identification and characterization of a chitinase antigen from Pseudomonas aeruginosa strain 385. Appl. Environ. Microbiol. 67:4001-4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolkin, R. H., and J. L. Pate. 1985. Selection for nonadherent or nonhydrophobic mutants co-selects for nonspreading mutants of Cytophaga johnsonae and other gliding bacteria. J. Gen. Microbiol. 131:737-750. [Google Scholar]
- 33.Xie, G., D. C. Bruce, J. F. Challacombe, O. Chertkov, J. C. Detter, P. Gilna, C. S. Han, S. Lucas, M. Misra, G. L. Myers, P. Richardson, R. Tapia, N. Thayer, L. S. Thompson, T. S. Brettin, B. Henrissat, D. B. Wilson, and M. J. McBride. 2007. Genome sequence of the cellulolytic gliding bacterium Cytophaga hutchinsonii. Appl. Environ. Microbiol. 73:3536-3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu, C. S., Y. C. Chen, C. H. Lu, and J. K. Hwang. 2006. Prediction of protein subcellular localization. Proteins 64:643-651. [DOI] [PubMed] [Google Scholar]
- 35.Yu, C. S., C. J. Lin, and J. K. Hwang. 2004. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 13:1402-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.