

Abstract
Calpain is a calcium-dependent protease that plays a significant role in synaptic plasticity, cell motility, and neurodegeneration. Two major calpain isoforms are present in brain, with μ-calpain (calpain1) requiring micromolar calcium concentrations for activation and m-calpain (calpain2) needing millimolar concentrations. Recent studies in fibroblasts indicate that epidermal growth factor (EGF) can activate m-calpain independently of calcium via mitogen-activated protein kinase (MAPK)-mediated phosphorylation. In neurons, MAPK is activated by both brain-derived neurotrophic factor (BDNF) and EGF. We therefore examined whether these growth factors could activate m-calpain by MAPK-dependent phosphorylation using cultured primary neurons and HEK–TrkB cells, both of which express BDNF and EGF receptors. Calpain activation was monitored by quantitative analysis of spectrin degradation and by a fluorescence resonance energy transfer (FRET)-based assay, which assessed the truncation of a calpain-specific peptide flanked by the FRET fluorophore pair DABCYL and EDANS. In both cell types, BDNF and EGF rapidly elicited calpain activation, which was completely blocked by MAPK and calpain inhibitors. BDNF stimulated m-calpain but not μ-calpain serine phosphorylation, an effect also blocked by MAPK inhibitors. Remarkably, BDNF- and EGF-induced calpain activation was preferentially localized in dendrites and dendritic spines of hippocampal neurons and was associated with actin polymerization, which was prevented by calpain inhibition. Our results indicate that, in cultured neurons, both BDNF and EGF activate m-calpain by MAPK-mediated phosphorylation. These results strongly support a role for calpain in synaptic plasticity and may explain why m-calpain, although widely expressed in CNS, requires nonphysiological calcium levels for activation.
Introduction
Calpains are ubiquitous intracellular proteases that play critical roles in a diverse range of physiological and pathological conditions in the nervous system, including long-term potentiation (LTP), learning and memory, and neurodegeneration (Liu et al., 2005; Wu and Lynch, 2006). In neurons, both theta-burst stimulation and NMDA receptor stimulation produce calpain activation as evidenced by the accumulation of a selective calpain-mediated spectrin breakdown product (SBDP) (Vanderklish et al., 1995). Calpain-mediated spectrin degradation has been implicated in dendritic spine changes associated with LTP induction (Lynch and Baudry, 1984; Vanderklish et al., 2000). In addition, calpain inhibitors block LTP induction in vitro and in vivo (Staubli et al., 1988; Denny et al., 1990).
It is widely acknowledged that calpain, by partially truncating a variety of cytoskeletal proteins, also plays a role in the regulation of shape and motility in numerous cell types (Franco and Huttenlocher, 2005). Two major calpain isoforms are present in brain: μ-calpain (calpain1) and m-calpain (calpain2). The long-held dogma suggested that calpain activation requires calcium influx followed by autoproteolysis, with μ-calpain being activated by micromolar calcium concentrations and m-calpain by millimolar concentrations (Melloni et al.,1996). This has raised a number of questions regarding the physiological role of m-calpain in brain, because it is not clear under which conditions m-calpain could be activated (Friedrich, 2004). However, recent studies in fibroblasts have revealed that m-calpain can be activated independently of calcium; in particular, epidermal growth factor (EGF) activates m-calpain by phosphorylation catalyzed by mitogen-activated protein kinase (MAPK) (Glading et al., 2001, 2004; Shiraha et al., 2002). Brain-derived neurotrophic factor (BDNF) also activates the MAPK pathway, and both BDNF and MAPK play critical roles in synaptic plasticity and in learning and memory (Patterson et al., 2001; Ying et al., 2002; Koponen et al., 2004). We therefore tested the hypothesis that BDNF could activate m-calpain via MAPK-mediated phosphorylation in neurons. Because EGF activates MAPK in various cell types, we also examined whether EGF-dependent activation of MAPK could similarly lead to calpain activation in neurons (Thomas et al., 1997; Niapour and Berger, 2007).
To monitor calpain activation, we used two approaches: (1) Western blotting to measure calpain-generated SBDP (Saido et al., 1994) and (2) fluorescence resonance energy transfer (FRET) kinetics of a synthetic calpain substrate (Vanderklish et al., 2000; Stockholm et al., 2005), consisting of a peptide with an optimized calpain cleavage site coupled to the FRET pair DABCYL and EDANS (Bánóczi et al., 2008). We also determined the effects of BDNF and EGF on μ- and m-calpain phosphorylation by immunoprecipitation (IP) and Western blotting. BDNF has also been shown to stimulate actin polymerization in neurons; therefore, we also tested the potential role of calpain activation in this process. Our results indicate that both BDNF and EGF rapidly activate m-calpain via MAPK-dependent phosphorylation in HEK cells expressing BDNF and EGF receptors and in cultured cortical and hippocampal neurons and provide a novel mechanism by which m-calpain plays a pivotal role in synaptic plasticity.
Materials and Methods
HEK–TrkB cell cultures.
HEK–TrkB cells engineered to express the BDNF receptor TrkB were a generous gift from Dr. Hiroyuki Nawa (Niigata, Japan) and were described previously (Narisawa-Saito et al., 2002). These cells also express endogenous EGF receptors ErbB1; thus, they are good tools to examine BDNF- and EGF-dependent MAPK activation (Thomas and Bradshaw, 1997). Frozen HEK293–TrkB cells were thawed directly into DMEM containing 10% fetal bovine serum (FBS–DMEM) and 400 μg/ml geneticin and cultured at 37°C inside a 5% CO2 incubator; after isolation of geneticin-resistant colonies, cells were expanded and maintained in FBS–DMEM with 200 μg/ml geneticin. For all experiments, HEK–TrkB cells were used 24–36 h after cell passage when they reached 70–80% confluence. Cells were incubated for 4 h with the FRET substrate (330 μm in 100 mm PBS), washed twice with 100 mm PBS to remove excess substrate, and supplemented with fresh medium immediately before being treated with growth factors and inhibitors (see below).
Primary neuronal cultures.
Whole cerebral neocortex of embryonic day 18–19 rats (Sprague Dawley) or hippocampi of embryonic day 18 mice (BALB/c) were digested with trypsin before mechanical dissociation and plating onto poly-d-lysine-coated dishes at low (100–300 cells/mm2) density for imaging experiments or at medium density (600–800 cells/mm2) for all other experiments and maintained in Neurobasal medium supplemented with B27 nutrient mixture for 2 weeks.
FRET substrate and in vitro calpain proteolysis assay.
A cell-penetrating FRET substrate [RRRRRRRR-(EDANS)-GQQEVYGMMPRDG-(DABCYL)] consisting of a peptide with an optimized calpain cleavage site and a hepta-arginine motif to facilitate cellular penetration was synthesized by solid-phase methodology on a rink-amide resin by the 9-fluorenylmethoxycarbonyl strategy and coupled to the following FRET pair: FRET acceptor probe, DABCYL (N-{4-[4′(dimethylamino) phenylazo]benzoyloxy} succinimide) (emission, none; absorption, 425 nm), and FRET donor probe, EDANS [5-(2-aminoethylamino)-1-naphtalenesulfonic acid] (emission, 493 nm; absorption, 335 nm) at >90% purity (BioMer Technologies) (Bánóczi et al., 2008). The FRET substrate was dissolved in 100 mm PBS, pH 7.4. In vitro analysis was performed using purified rat m-calpain (0.5 μm; Calbiochem), the FRET substrate (100 μm), and CaCl2 (5 mm) in 50 μl calpain buffer (in mm: 50 Tris, 150 NaCl, and 1 EDTA, pH 7.5) inside a 10 mm UV quartz cuvette at room temperature and using a HORIBA Jobin Yvon FluoroMax-3 spectrofluorometer. Excitation and emission wavelengths were λ = 320 nm and λ = 480 nm, respectively. Alternatively, cell cultures (HEK–TrkB or cortical neurons) pretreated with the FRET probe (330 μm) for 4 h and after washing were subjected to the indicated treatments for various periods of time. After treatments, cells were trypsinized (PBS buffer containing 5 mg/ml trypsin and 2 mg/ml EDTA for 10 min), and cell lysates were processed for spectrofluorometric analysis as described above.
Image acquisition and FRET imaging analysis.
The FRET substrate was dissolved in 100 mm PBS and used in all imaging experiments at a final concentration of 330 μm. FRET substrate uptake by HEK–TrkB cells was confirmed with fluorescence microscopy (LMS5; Carl Zeiss). Hippocampal neurons kept for 14 d in vitro (DIV) were incubated for 4 h with the FRET substrate diluted in prewarmed (37°C) imaging buffer (100 mm NaCl, 3 mm KCl, 10 mm HEPES, 2 mm CaCl2, 2 mm MgCl2, 10 mm glucose, and 2% B27 dissolved in 100 mm PBS) and washed three times with the imaging buffer to remove excess substrate. Substrate uptake by neurons was confirmed and analyzed by confocal microscopy (Nikon Eclipse TE-2000 with D-Eclipse C1 system). Growth factors were directly added from stock solution while fluorescence was monitored. Images were acquired at various time points before and during treatments with constant acquisition parameters. Pseudocolor saturation treatment of captured images was applied to highlight time- and treatment-dependent changes in FRET.
Filamentous actin staining and actin polymerization assay.
Hippocampal neurons preincubated with the FRET substrate were washed and treated with either 50 ng/ml BDNF or 20 ng/ml EGF for the indicated periods of time before fixation in 4% paraformaldehyde for 30 min. Neurons were then washed three times in 100 mm PBS and incubated with phalloidin–Alexa Fluor594 (Invitrogen) for 30 min. Neurons were subsequently washed twice with PBS and imaged via confocal microscopy (Nikon Eclipse TE-2000 with D-Eclipse C1 system). Fluorescence signals of the FRET substrate and that of the phalloidin–Alexa Fluor594 were captured by sequential acquisition to avoid potential fluorescence signal overlap. Images were then superimposed to localize polymerized actin within dendrites via D-Eclipse C1 System (Nikon). Actin polymerization was further quantified by a rhodamine–phalloidin fluorescence enhancement assay described previously by Katanaev and Wymann (1998). Briefly, DIV14 cultured cortical neurons were preincubated at 37°C in the absence or presence of calpain inhibitor III (10 μm) for 20 min; they were then treated with or without 50 ng/ml BDNF for 30 min. Cultures were then washed twice with 100 mm PBS and subsequently fixed in 4% paraformaldehyde and 1% octyl-β-d-glucopyranoside for 15 min at room temperature. This was followed by incubation with 10 nm rhodamine–phalloidin for 30 min at room temperature. Cultures were collected in 1.0 ml of PBS, and normalized fluorescent intensity was recorded in a 10 mm UV cuvette using a spectrophotometer (excitation and emission wavelengths were λ = 552 nm and λ = 580 nm, respectively).
Drug treatments.
HEK–TrkB cells and cultured neurons of DIV14 were treated with the following inhibitors: calpain inhibitor III (10 μm), K252a [9S,10R,12R)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12,-epoxy-1H-diindolo[1,2,3,-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid methyl ester] (0.5 μm; TrkB inhibitor), PD153035 [N-(3-bromophenyl)-6,7-dimethoxyquinazolin-4-amine] (25 μm; ErbB1 inhibitor), PD98059 [2-(2-amino-3-methyoxyphenyl)-4H-1-benzopyran-4-one] (25 μm) and U0126 [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene] (10 μm; both are MAPK pathway inhibitors), BAPTA-AM (50 μm; cell membrane-permeable calcium chelator), and KT5720 [(9S,10R,12R)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylicacid hexyl ester] (10 μm; a PKA inhibitor) for 10–20 min before being further treated with growth factors (BDNF, 50 ng/ml; EGF, 20 ng/ml; Millipore Corporation) for 20 min (for FRET spectrofluorometric analysis) or for 60 min (for Western blotting and immunoprecipitation analysis).
Western blotting.
Cells were lysed in modified Laemli's sample loading buffer (10% glycerol, 2% SDS, and 65 mm Tris). After centrifugation (10,000 × g, 10 min), proteins in supernatants were denatured at 95°C in the presence of 0.1 m 2-mercaptoethanol, and protein samples were separated by SDS-PAGE with 8% polyacrylamide gels. Proteins were transferred to nitrocellulose membranes by electrophoresis. Primary antibodies were diluted with blocking solution and incubated with membranes at 4°C overnight. Antibodies against the following proteins were used at the indicated concentrations: μ- and m-calpain (MAB3104 and AB81023; 1:1000 dilution; Millipore Corporation); phospho-serine (catalog #p5747; 1:1000 dilution; Sigma); spectrin and actin (MAB1622 and MAB1501; 1:1000 and 1:10,000 dilution, respectively; Millipore Corporation), and calpastatin (CSL5-10; 2 μg/ml; Abcam). After incubation with primary antibodies, membranes were washed in TBS containing 0.1% Tween 20 and incubated with peroxidase-conjugated secondary antibodies (1: 10,000; Vector Laboratories), for 1 h, developed with ECL plus solution (ECL kit; GE Healthcare) and exposed to negative film. Exposed films were developed, fixed, scanned, and analyzed quantitatively by densitometry with NIH ImageJ software. In addition, all blots were stripped and reincubated with anti-β-actin as loading control, and results were adjusted against actin signal.
Small interfering RNA treatment in cultured cortical neurons.
Cultured cortical neurons (DIV14) were transfected with 40 μm small interfering RNA (siRNA) diluted in Neurobasal media and 40 μl of HiPerfect. Forty micromolar siRNA was diluted in 900 μl of Neurobasal media, and 40 μl of Hiperfect was diluted in 900 μl of Neurobasal media. siRNA mixtures and Hiperfect mixtures were combined and incubated at room temperature for 15 min. Two milliliters of media were removed from the cells, and siRNA solutions were added to the cells. Cells treated with siRNA or control solutions were incubated for 5 d. The siRNA sequences were as follows: μ-calpain, TACCTCTGTTCAATTGCTCTA; m-calpain, GCGGTCAGATACCTTCATCAA (Qiagen). After 5 d, cells were preincubated with the FRET reagent as described in Materials and Methods for 3 h. They were then treated with vehicle (control), EGF (20 μg/ml), or BDNF (50 μg/ml) for 30 min. Cells were lysed, and fluorescence in the lysates was determined as described above. Aliquots of cell lysates were also processed for Western blots with μ- or m-calpain antibodies.
Immunoprecipitation.
HEK–TrkB cells were treated with 50 ng/ml BDNF or 20 ng/ml EGF for 1 h in the presence or absence of PD98059 (25 μm). Cells were washed twice with HBSS buffer before being harvested and lysed (on ice for 15 min) in cold IP buffer (1% deoxycholate, 10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 5 mm EDTA, 1 mm EGTA, and 1 mm phenylmethylsulfonylfluoride) containing phosphatase inhibitors (10 mm NaF and 1 mm Na3VO4) and protease inhibitor cocktail [2.08 mm 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride, 1.6 μm aprotinin, 40 μm leupeptin, 80 μm bestatin, 30 μm pepstatin A, and 28 μm trans-3-carboxyoxirane-2-carbonyl-l-leucylagmatine] (all from Sigma-Aldrich). Protein concentration was determined, and 0.5–1 mg of protein from total cell lysates was incubated overnight at 4°C on a revolving rotor with anti-m-calpain antibodies (10 μg/IP reaction) in the presence of protein A/protein G-conjugated Sepharose beads (30 μl/IP reaction); total IP reaction volume was adjusted to 1.5–2 ml with HBSS. Sepharose beads were centrifuged and washed three to four times in HBSS buffer, and precipitated proteins were eluted by boiling (two times for 10 min, 95°C) in sample loading buffer (2% SDS, 0.1 m DTT, and 10% 2-mercaptoethanol). Eluted proteins were then processed for Western blotting as indicated previously with anti-phospho-serine antibody.
Statistical analysis.
Statistical analysis was performed using one-way ANOVA, followed by the Bonferroni's test (for multiple factors). Alternatively, Student's t test was applied to data with multiple variables and one factor. Normalized FRET results were obtained from multiple sister cultures, and their means were calculated; means from independently grown cultures were then expressed as means ± SEM (n = 3–6 cultures with a total of 8–16 readings). For Western blotting and immunoprecipitation results, data were derived from duplicate similarly treated sister cultures from four to six independent cultures and analyzed with Student's t test to evaluate statistical significance. p value of <0.05 was selected to indicate statistically significant differences between individual conditions.
Results
FRET substrate provided a reliable and quantitative measure of calpain activation
FRET substrate emission spectrum was determined by spectrofluorometry, as indicated in Materials and Methods. The substrate was excited at 320 nm, and emission spectrum recorded from 381 to 541 nm. As expected, a single emission peak was observed at ∼480 nm (Fig. 1A). The fluorescence intensity of the dual-labeled substrate was measured in a calpain assay solution over 60 min with spectrofluorometry. A clear FRET signal was observed, because donor emission was gradually quenched by the acceptor fluorescent probe. Fluorescence intensity of the FRET substrate was then analyzed in the presence of purified calpain and calcium. A decrease in FRET was observed, with a threefold increase in fluorescence intensity displayed, as expected. This loss of FRET signal is consistent with the cleavage of the FRET substrate, which eliminated quenching of donor fluorescence signal.
Figure 1.

FRET analysis of calpain activation in vitro and in HEK–TrkB cells. A, Emission spectrum analysis of the FRET substrate incubated in the absence or presence of calpain. As expected, the peak of emission was at ∼480 nm. In the presence of activated calpain, a clear decrease in FRET was observed. B, FRET substrate (arrows) was internalized into HEK–TrkB cells (bottom). Top shows phase-contrast image of the same field. C, Kinetic of calpain activation by BDNF and EGF in HEK–TrkB cells. BDNF (50 ng/ml) or EGF (20 ng/ml) was added in cells preloaded with the FRET substrate, and fluorescence at 480 nm recorded at various times. Results are expressed as percentage of T0 and are means ± SEM of three experiments. D, BDNF-elicited calpain activation in HEK–TrkB cells is suppressed by MAPK inhibition or TrkB receptor inhibition. Fluorescence was measured 20 min after BDNF (50 ng/ml) addition in HEK–TrkB cells preloaded with the FRET substrate and pretreated with U0126 (10 μm) or K252a (0.5 μm). Results are expressed as normalized fluorescence (percentage of untreated control values) and are means ± SEM of six experiments with a total of 8–18 readings per condition. **p < 0.01 compared with control; ‡p < 0.01 compared with BDNF control samples. E, EGF-elicited calpain activation in HEK–TrkB cells is inhibited by MAP kinase inhibition or ErbB1 receptor inhibition. Fluorescence was measured 20 min after EGF (100 ng/ml) addition in HEK–TrkB cells preloaded with the FRET substrate and pretreated with PD98059 (25 μm) or PD153035 (0.5 μm). Results are expressed as normalized fluorescence (percentage of untreated control values) and are means ± SEM of six experiments with a total of 8–18 readings per condition. *p < 0.05 compared with control; ‡p < 0.01 compared with EGF control samples. F, BDNF- and EGF-induced calpain activation is blocked by calpain inhibitor III. Fluorescence was measured 20 min after BDNF (50 ng/ml) or EGF (100 ng/ml) addition in HEK–TrkB cells preloaded with the FRET substrate and pretreated with calpain inhibitor III (10 μm). Results are expressed as normalized fluorescence (percentage of untreated control values) and are means ± SEM of four experiments with three to four readings/experiment. **p < 0.01 compared with control; ‡p < 0.01 compared with BDNF and EGF control samples.
FRET analysis of BDNF- and EGF-dependent calpain activation in HEK–TrkB cells
HEK–TrkB cells incubated with the FRET substrate were first imaged to confirm internalization of the FRET substrate (Fig. 1B), followed by spectrofluorometric analysis of calpain activity. Time course of calpain activation was then determined in BDNF- and EGF-treated samples. Normalized fluorescence values were calculated by subtracting fluorescence values at time 0 (T0, i.e., before initiation of growth factor treatment) from fluorescence values recorded every 2 min for 20–50 min after stimulation with BDNF or EGF. Averaged mean values of growth factor-stimulated increases in fluorescence signal intensity were then calculated and expressed as percentage of the fluorescence values at T0 (Fig. 1C). The results indicated a rapid (within 5 min of BDNF or EGF addition) loss of FRET, because calpain activation resulted in FRET substrate cleavage. Calpain activation reached maximal levels by 20–30 min of treatment with both BDNF (346 ± 5%) and EGF (235 ± 4%). Compared with EGF, BDNF-induced calpain activation produced a larger increase in fluorescence intensity at all recorded time points, possibly reflecting stronger activation of calpain downstream from the overexpressed TrkB receptors in these cells. Reactions were monitored for up to 50 min and did not show additional increases in fluorescence. Accordingly, we chose the 20 min time point for all remaining experiments.
To confirm that BDNF- and EGF-dependent cleavage of the FRET substrate was mediated by calpain, HEK–TrkB cells were incubated with calpain inhibitor III (10 μm; added 20 min before growth factor treatment). Calpain inhibitor III completely eliminated BDNF- and EGF-dependent changes in fluorescence of the FRET substrate (Fig. 1F). Specificity of calpain activation by BDNF and EGF in HEK–TrkB cells was also examined using inhibitors of BDNF and EGF receptors, TrkB and ErbB1 (K252a and PD153035, respectively). Inhibitors were added 20–30 min before application of growth factors. In the presence of K252a or PD153035, BDNF or EGF treatments had no effect on FRET signals (Fig. 1D,E).
BDNF- and EGF-elicited calpain activation in HEK–TrkB cells is mediated by MAPK
MAPK pathway is rapidly activated (within minutes) by several signaling pathways, including BDNF–TrkB and EGF–ErbB1. The similar activation profiles of calpain after BDNF and EGF treatments (Fig. 1C) suggested convergence on a common downstream effector activated by both BDNF–TrkB and EGF–ErbB1 pathways. Accordingly, we examined whether the MAPK pathway was involved in BDNF- and EGF-dependent calpain activation using MAPK inhibitors. HEK–TrkB cells were incubated with the FRET substrate and treated with inhibitors of the MAPK pathway, U0126 or PD98059, before adding BDNF (50 ng/ml) or EGF (20 ng/ml) for 20 min. No change in FRET was observed, because both BDNF- and EGF-mediated FRET was abolished by pretreatment with MAPK pathway inhibitors (Fig. 1D,E). Similar results were obtained with both inhibitors (data not shown for BDNF + PD98059 and for EGF + U0126).
BDNF and EGF-induced calpain activation and actin polymerization in cultured neurons
Similar experiments as those described above were performed using primary neuronal cultures. Primary hippocampal or cortical neurons were pretreated with the FRET substrate for 3 h. Confocal microscopy was used to confirm internalization of the FRET substrate into cultured hippocampal neurons. Neurons were then treated with BDNF or EGF, and images were captured at different time points (0, 2, and 10 min for BDNF and 0 and 10 s and 2 min for EGF) (Fig. 2A,B). Fluorescence signal intensity increased rapidly within dendrites during growth factor treatment. Interestingly, BDNF treatment induced slower increases than EGF, because EGF produced calpain activation as early as 10 s after application.
Figure 2.
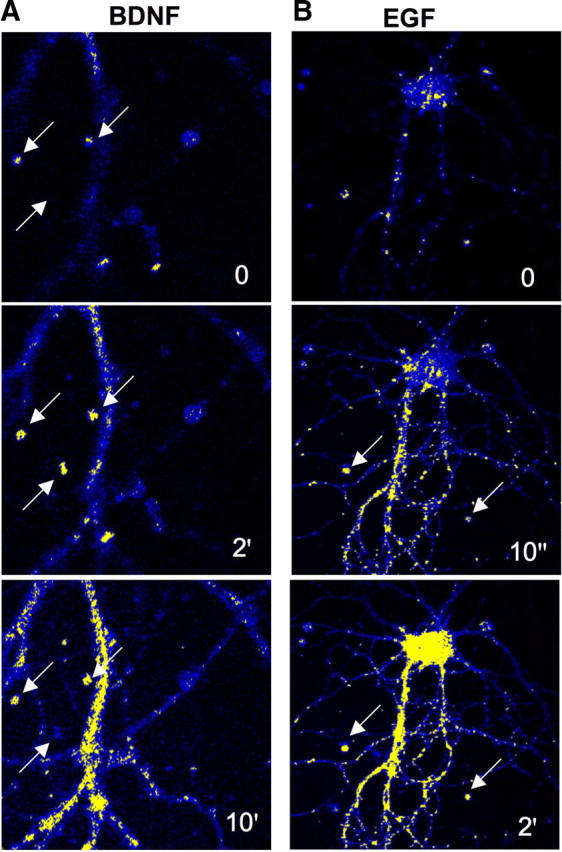
FRET analysis of calpain activation in primary hippocampal neuronal cultures. Cultured hippocampal neurons were preloaded with the FRET substrate as described in Materials and Methods. A, B, FRET analysis of calpain activation by BDNF (50 ng/ml; A) or EGF (20 ng/ml; B) at the indicated time points. Arrows indicate spine-like structures.
BDNF treatment of cultured neurons increases actin polymerization in dendritic spines (Lin et al., 2005; Lynch et al., 2007). Accordingly, polymerized actin [filamentous actin (F-actin)] can be used as a marker of dendritic spines. To better localize calpain activation within neurons, hippocampal neurons incubated with the FRET substrate and treated with EGF or BDNF for 5 min were then fixed and stained with phalloidin–Alexa Fluor594 to label F-actin. Calpain activation revealed with FRET was colocalized with polymerized actin, and numerous structures double labeled with FRET and phalloidin had the appearance of dendritic spines (Fig. 3A).
Figure 3.
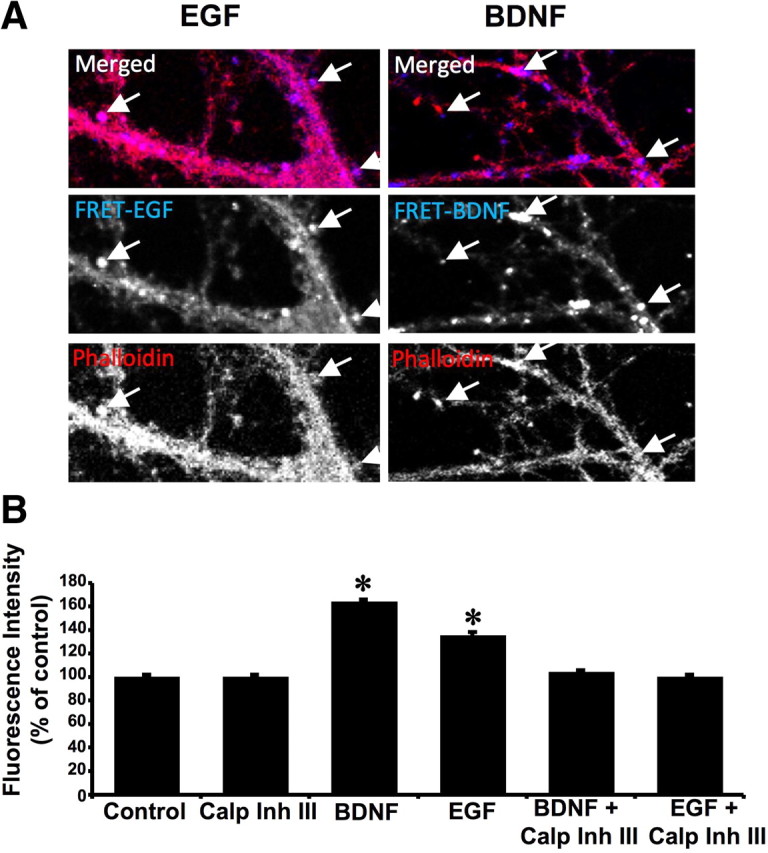
Calpain activation is localized in dendritic spine-like structures and stimulates actin polymerization. A, Hippocampal neurons were preloaded with the FRET substrate and treated with BDNF (50 ng/ml) for 10 min or EGF (20 ng/ml) for 2 min. They were then fixed, stained with phalloidin–Alexa Fluor594, and processed for confocal microscopy. Decrease in FRET signal (increased fluorescence) was observed in dendrites as well as in dendritic spine-like structures labeled with phalloidin–Alexa Fluor594 (arrows). B, Hippocampal neuronal cultures were pretreated with calpain inhibitor III (10 μm) for 20 min before adding vehicle or BDNF (50 ng/ml) for 20 min. At the end of incubation, actin polymerization was analyzed as described under Materials and Methods with the rhodamine–phalloidin fluorescence enhancement assay. Results of the rhodamine– phalloidin fluorescence were normalized (by subtracting control fluorescence signals) and represent means ± SEM of five experiments. *p < 0.001 (ANOVA, followed by Bonferroni's test).
Because calpain has been shown previously to be involved in actin polymerization in various cell types (Flevaris et al., 2007), we tested the possibility that calpain was causally involved in BDNF-induced actin polymerization by evaluating the effects of calpain inhibitor III on actin polymerization assayed with the rhodamine–phalloidin fluorescence enhancement method of Katanaev and Wymann (1998). Cultured hippocampal neurons were pretreated with calpain inhibitor III (10 μm) for 20 min before adding BDNF (50 ng/ml) for 30 min. BDNF treatment resulted in a 60% increase in actin polymerization, and this effect was completely prevented by the calpain inhibitor (Fig. 3B). Calpain inhibitor III alone did not modify actin polymerization.
The striking difference in time course for calpain activation between EGF and BDNF suggested to us that another level of regulation was involved in their effects. It had been shown previously that, in addition to the positive activation of calpain by MAP kinase, calpain could be negatively regulated by PKA-mediated phosphorylation of serine369/threonine370 residues. We therefore tested the effects of a PKA inhibitor, KT5720, on EGF- and BDNF-mediated calpain activation (Fig. 4). Although PKA inhibition did not affect EGF-mediated calpain activation, it markedly increased the rate of BDNF-mediated calpain activation. In fact, in the presence of KT5720, the time course for calpain activation was identical for BDNF and EGF. A similar enhancement of the effect of BDNF on calpain activation was observed with another PKA inhibitor, PKI 14–22 (data not shown).
Figure 4.

Effect of a PKA inhibitor, KT5720, on EGF- and BDNF-mediated calpain activation. Cortical neurons were preloaded with the FRET substrate and treated with BDNF (A; 50 ng/ml) or EGF (B; 20 ng/ml) for the indicated periods of time. They were pretreated without or with the PKA inhibitor (PKI) KT5720 (10 μm) for 20 min before BDNF or EGF treatment. At the indicated times, cells were trypsinized (PBS buffer containing 5 mg/ml trypsin and 2 mg/ml EDTA for 10 min), and cell lysates were processed for spectrofluorometric analysis as described in Materials and Methods. Fluorescence intensity was expressed as percentage of values measured at time 0, and results are means ± SEM of six experiments from independent cell dishes. KT5720 alone had no effect on fluorescence intensity (data not shown).
BDNF- and EGF-induced calpain activation in cultured neurons is also mediated by MAPK and is independent of intracellular calcium
DIV14 neurons were incubated with BDNF in the presence or absence of K252a, U0126, or calpain inhibitor III. Similarly, neurons were also treated with EGF in the presence or absence of PD153035, PD98059, or calpain inhibitor III (Fig. 5A,B). As observed in HEK–TrkB cells, BDNF- and EGF-induced calpain activation was blocked by inhibitors of the MAPK pathway. Calpain inhibitor III also completely eliminated BDNF- and EGF-dependent decrease in FRET (Fig. 5C). To further clarify the involvement of μ- or m-calpain in EGF- and BDNF-mediated calpain activation, we performed siRNA experiments to downregulate specifically these two calpain isoforms in cultured neurons (Fig. 6). Although treatment with siRNA against μ-calpain had not effect on either EGF- or BDNF-mediated calpain activation, treatment with siRNA against m-calpain markedly reduced EGF- and BDNF-mediated calpain activation. We also verified the specificity of the siRNA and found that treatment with siRNA against μ-calpain decreased levels of μ- but not m-calpain, and conversely treatment with siRNA against m-calpain decreased levels of m- but not μ-calpain.
Figure 5.
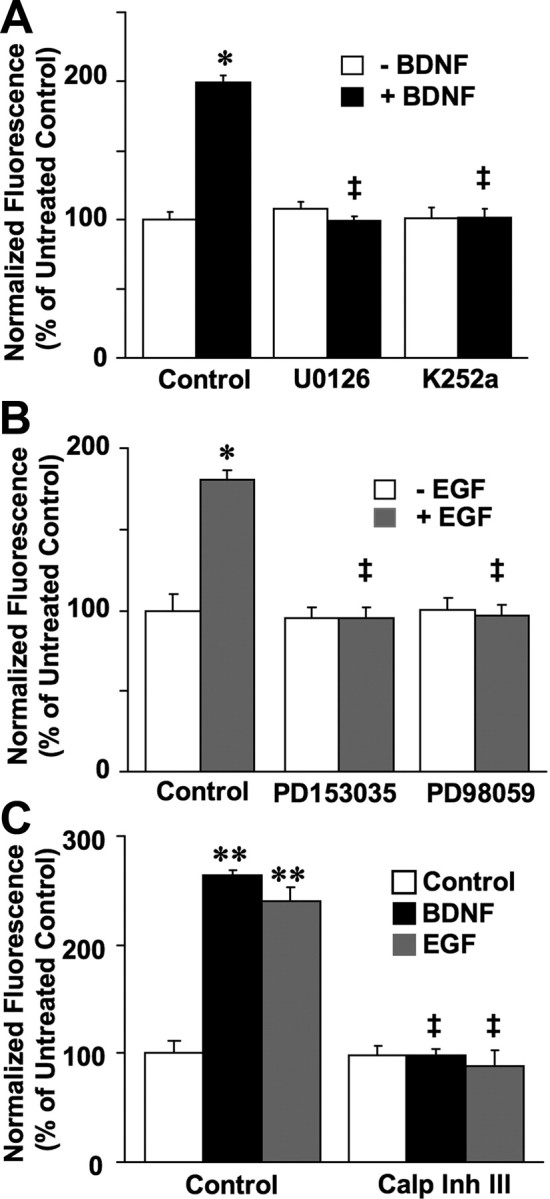
Calpain activation induced by BDNF and EGF in cortical neurons depends on MAPK. A, BDNF-elicited calpain activation in cortical neurons is blocked by inhibitors of MAP kinase and TrkB receptor. Fluorescence was measured 20 min after BDNF (50 ng/ml) addition to cortical neurons preloaded with the FRET substrate and pretreated with U0126 (10 μm) or K252a (0.5 μm). Results are expressed as normalized fluorescence (percentage of untreated control values) and are means ± SEM of five experiments with a total of 6–14 readings per condition. *p < 0.05 compared with control; ‡p < 0.01 compared with BDNF control samples. B, EGF-elicited calpain activation in cortical neurons is inhibited by MAP kinase inhibition or ErbB1receptor inhibition. Fluorescence was measured 20 min after EGF (20 ng/ml) addition to cortical neurons preloaded with the FRET substrate and pretreated with PD98059 (25 μm) or PD153035 (0.5 μm). Results are expressed as normalized fluorescence (percentage of untreated control values) and are means ± SEM of six experiments. *p < 0.05 compared with control; ‡p < 0.01 compared with EGF control samples. C, BDNF- and EGF-induced calpain activation in cortical neurons is blocked by calpain inhibitor III. Fluorescence was measured 20 min after BDNF (50 ng/ml) or EGF (20 ng/ml) addition to cortical neurons preloaded with the FRET substrate and pretreated with calpain inhibitor III (10 μm). Results are expressed as normalized fluorescence (percentage of untreated control values) and are means ± SEM of six to eight experiments. **p < 0.01 compared with control; ‡p < 0.01 compared with BDNF or EGF control samples.
Figure 6.

Effect of siRNA against μ- or m-calpain on calpain activity in cultured cortical neurons. Cultured cortical neurons (DIV14) were transfected with 40 μm siRNA diluted in Neurobasal media and 40 μl of HiPerfect, as described in Materials and Methods. After 5 d, cells were preincubated with the FRET reagent for 3 h. They were then treated with vehicle (control), EGF (20 μg/ml), or BDNF (50 μg/ml) for 30 min. Cells were lysed, and fluorescence in the lysates was determined (A). Aliquots of cell lysates were also processed for Western blots with m-calpain (B) or μ-calpain (C) antibodies (actin antibodies were used as loading control). Fluorescence intensity results are expressed as percentage of values found in control-treated nontransfected neurons and are means ± SEM of three experiments. **p < 0.01 compared with the respective control values. Levels of m- and μ-calpain were normalized and expressed as percentage of values found in control-treated nontransfected neurons and are means ± SEM of nine experiments. **p < 0.01 compared with control values.
Primary neuronal cultures preloaded with the FRET substrate were also treated with BAPTA-AM, a cell membrane-permeable and intracellular calcium chelator, before application of either BDNF or EGF. Both confocal image analysis and spectrofluorometric analysis indicated that BDNF- and EGF-induced changes in FRET were not altered by BAPTA-AM, indicating that BDNF- and EGF-mediated calpain activation occurs independently of calcium (Fig. 7).
Figure 7.

BDNF- and EGF-mediated calpain activation is calcium independent. Top, Representative images of BDNF-induced (50 ng/ml) or EGF-induced (20 ng/ml) changes in FRET in hippocampal neurons preincubated with BAPTA-AM (BAPTA) 10 min after addition of BDNF or EGF (for details, see Materials and Methods). Calpain activation is still observed in dendrites (arrows). Bottom, Primary cortical neurons preloaded with the FRET substrate were incubated with BAPTA-AM (50 μm) for 20 min before application of BDNF (50 ng/ml) or EGF (20 ng/ml) for 20 min. Normalized FRET signal was measured using spectrofluorometry. Results are expressed as normalized fluorescence and are means ± SEM of four experiments with a total of 7–14 readings per condition. *p < 0.05 compared with control.
BDNF-induced calpain activation was also monitored by Western blot analysis of SBDP, the truncated form of α-spectrin that is specifically generated by calpain-dependent cleavage (Saido et al., 1994). Neuronal cultures were incubated with BDNF or EGF for 60 min in the presence or absence of the MAPK pathway inhibitor PD98059. BDNF or EGF treatment of neurons increased SBDP accumulation, an effect that was blocked by pretreatment with PD98059 (Fig. 8). Treatment with PD98059 alone slightly reduced SBDP accumulation compared with control, suggesting that phosphorylation reactions might participate in basal calpain-dependent spectrin degradation. As expected, calpain inhibitor III eliminated BDNF-dependent increases in SBDP (data not shown). In addition, K252a treatment eliminated BDNF-dependent increase in SBDP accumulation, suggesting the involvement of BDNF receptors (data not shown).
Figure 8.

BDNF and EGF stimulate calpain-mediated spectrin degradation in cultured cortical neurons. Cortical neurons were pretreated with vehicle or the MAP kinase inhibitor PD98059 (25 μm) for 15 min before being treated with BDNF (50 ng/ml) or EGF (20 ng/ml) for 60 min. Cell lysates were processed for Western blotting with spectrin antibodies and actin antibodies (as a loading control). Levels of the 145–150 kDa calpain-specific breakdown products were quantified and expressed as percentage of values found in control cultures. Results are means ± SEM of six independent experiments. *p < 0.01 compared with control; ‡p < 0.05 compared with BDNF- or EGF-treated samples.
MAPK-dependent m-calpain phosphorylation by BDNF in cultured neurons and in HEK–TrkB cells
Immunoprecipitation experiments were performed to examine whether BDNF-induced calpain activation was mediated by MAPK-dependent serine phosphorylation of μ- or m-calpain. Cultured cortical neurons were treated with BDNF in the presence or absence of a MAPK inhibitor as indicated previously. Cells were lysed in immunoprecipitation buffer, and cell lysates were immunoprecipitated with anti-μ- or anti-m-calpain antibodies. Pull-down products were then analyzed by Western blot using anti-phospho-serine as well as anti-μ-calpain and anti-m-calpain antibodies as indicated in Materials and Methods. BDNF treatment increased serine phosphorylation of immunoprecipitated m-calpain (Fig. 9B). However, the low levels of μ-calpain did not allow determining potential changes in μ-calpain phosphorylation. Treatment with PD98059 alone resulted in decreased levels of serine-phosphorylated m-calpain, suggesting that m-calpain was constitutively phosphorylated by MAP kinase (Fig. 9).
Figure 9.

BDNF stimulates MAP kinase-mediated m-calpain serine phosphorylation in HEK–TrkB cells and cortical neurons. A, HEK–TrkB cells were pretreated with vehicle or the MAP kinase inhibitor PD98059 (25 μm) for 15 min before being treated with BDNF (50 ng/ml) for 60 min. Cell lysates were processed for immunoprecipitation with m-calpain or μ-calpain antibodies, and immunoprecipitated proteins were immunoblotted with anti-m-calpain, anti-μ-calpain, or anti-phospho-serine antibodies. Levels of serine-phosphorylated m-calpain and serine phosphorylated μ-calpain were quantified, and results were expressed as percentage of control. Results are means ± SEM of four experiments. *p < 0.01 compared with control; †p < 0.05 compared with BDNF-treated samples. B, Cortical neurons were pretreated with vehicle or the MAP kinase inhibitor PD98059 (25 μm) for 15 min before being treated with BDNF (50 ng/ml) for 60 min. Cell lysates were processed for immunoprecipitation with m-calpain antibodies, and immunoprecipitated proteins were immunoblotted with anti-m-calpain or anti-phospho-serine antibodies. Levels of serine-phosphorylated m-calpain were quantified, and results were expressed as percentage of control. Results are means ± SEM of four experiments. *p < 0.01 compared with control; †p < 0.05 compared with BDNF-treated samples.
Similar experiments were performed in HEK–TrkB cells. BDNF treatment resulted in increased serine phosphorylation of m- but not μ-calpain (Fig. 9A). Cotreatment of HEK–TrkB cells with PD98059 eliminated BDNF-mediated increase in serine phosphorylation of m-calpain, indicating that it was mediated by MAPK activation downstream from BDNF receptors. Interestingly, these results also indicated that BDNF- or EGF-mediated calpain activation was not associated with calpain autoproteolysis.
Calpain activation is regulated by an endogenous calpain inhibitor, calpastatin. To eliminate the possibility that calpain activation elicited by BDNF treatment could be attributable to decreased levels of endogenous calpastatin, we used Western blots with anti-calpastatin antibodies to evaluate the effects of BDNF treatment on calpastatin levels in cultured neurons. BDNF treatment of cultured cortical neurons for 60 min did not significantly modify the levels of calpastatin (105 ± 5% of control values; means ± SEM of three experiments) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Discussion
The major finding of our study is that m-calpain is rapidly activated by MAPK-mediated phosphorylation, and this mode of calpain regulation is involved in BDNF and EGF signal transduction in neurons: both BDNF and EGF rapidly activate calpain in HEK–TrkB cells as well as in cultured cortical and hippocampal neurons. Also, BDNF- and EGF-induced calpain activation requires MAPK-mediated m-calpain phosphorylation and is not associated with autoproteolysis. We used two separate assays to monitor calpain activation. The first one consisted in the widely used measure of spectrin degradation, because calpain-mediated spectrin truncation generates a 145–150 kDa fragment, which is readily evidenced in Western blots with spectrin antibodies. The second one was a FRET-based analysis with a substrate consisting of a calpain-specific peptide, flanked by two fluorophores exhibiting FRET and a hepta-arginine sequence to facilitate the substrate penetration across cell membranes (Bánóczi et al., 2008). The advantage of the second approach is that, in combination with confocal analysis, it provides precise spatiotemporal information regarding calpain activation within cells. In particular, our results clearly indicated that BDNF and EGF induce calpain activation not only in dendrites but also in dendritic spine-like structures in cultured hippocampal neurons. This result confirms previous studies indicating that calpain is localized in dendritic spines (Perlmutter et al., 1988; Vanderklish et al., 2000) and provides additional support for our hypothesis that calpain activation in dendrites and dendritic spines plays a critical role in synaptic plasticity (Lynch and Baudry, 1984; Baudry and Lynch, 2001).
Our studies provide several lines of evidence indicating that BDNF- and EGF-elicited calpain activation is attributable to MAPK-mediated phosphorylation and is calcium independent. Pharmacological treatments with MAPK pathway inhibitors completely eliminated BDNF- and EGF-induced calpain activation. In addition, immunoprecipitation experiments indicated that, after BDNF treatment of HEK–TrkB cells and cultured neurons, m-calpain but not μ-calpain was phosphorylated at a serine residue, an effect that was completely eliminated by an MAPK pathway inhibitor. Although the immunoprecipitation experiment cannot distinguish between Ser50 and Ser369, results obtained with the PKA inhibitors strongly suggest that it is phosphorylation of Ser50 that is stimulated by BDNF and EGF, because phosphorylation of Ser369 inhibits calpain activation. Our results therefore confirm previous studies showing that MAPK is implicated in m-calpain activation in non-neuronal cells (Niapour and Berger, 2007) and that this occurs downstream from the EGF–ErbB1 pathway (Glading et al., 2004). In their experiments, Glading et al. (2004) clearly showed that m-calpain was phosphorylated at serine 50 and was activated independently of calcium. They also showed that this mechanism plays a critical role in the regulation of cell migration and motility (Glading et al., 2004). Although m-calpain can also be phosphorylated at some threonine residues (Glading et al., 2004), we could not detect any changes in threonine phosphorylation of m-calpain after BDNF treatment of cortical neuronal cultures (data not shown). It has also been shown that m-calpain can also be phosphorylated at serine 369 by PKA, resulting in calpain inhibition; our results with the PKA inhibitor KT5720 clearly indicate that, as in fibroblasts, neuronal calpain is also negatively regulated by PKA, because the rate of calpain activation by BDNF was markedly increased in the presence of KT5720. This result suggests that, in neurons, BDNF stimulates not only the MAP kinase pathway but also the PKA pathway.
The effects of both BDNF and EGF on calpain activation in cultured neurons were still observed in the presence of BAPTA-AM at a concentration generally used to chelate all available calcium. These results indicate that BDNF and EGF activate calpain independently of calcium. In addition, results with siRNA against μ- and m-calpain further strengthen the conclusion that it is activation of m-calpain but not μ-calpain that is responsible for the effects of EGF and BDNF in cultured neurons. Furthermore, we verified that BDNF treatment of cultured neurons did not modify the levels of calpastatin, the endogenous calpain inhibitor, thus excluding the possibility that BDNF activation of calpain was attributable to a decrease in calpastatin levels. Finally, we also provided evidence that calpain activation participates in BDNF-mediated actin polymerization, because the effect of BDNF on actin polymerization was completely blocked by a calpain inhibitor.
Our results therefore provide interesting links between several elements that have been postulated previously to participate in synaptic plasticity. Many years ago, we proposed that calpain activation, by triggering spectrin degradation and cytoskeletal reorganization, was critically involved in the formation of long-term potentiation at glutamatergic synapses (Lynch and Baudry, 1984). More recently, theta-burst stimulation, an electrophysiological protocol commonly used to induce LTP in neuronal preparations, was found to be optimal for BDNF release from neuronal terminals (Thoenen, 2000; Yamada et al., 2002; Lu, 2003; Pang and Lu, 2004; Rex et al., 2006; Lynch et al., 2007). Theta-burst stimulation also activates calpain and increases calpain-mediated spectrin cleavage within dendritic spines (Vanderklish et al., 1995, 2000). BDNF plays several important roles in LTP-associated synaptic plasticity, such as inducing AMPA receptor translocation from the cytoplasm to the cell surface, increasing actin polymerization in dendritic spines, and enhancing local protein synthesis (Narisawa-Saito et al., 2002; Rex et al., 2006, 2007; Bramham, 2007; Chen et al., 2007; Jourdi et al., 2009). All these events have been linked to the induction and maintenance of long-term structural changes at synapses (Lynch and Baudry, 1984; Yamamoto et al., 2005; Taniguchi et al., 2006; Rex et al., 2007; Tominaga-Yoshino et al., 2008).
More recently, the MAPK pathway has also been shown to play a central role in LTP formation, although the exact targets of this pathway have generally been assumed to be some elements of the transcriptional and/or translational machinery (Selcher et al., 2003; Giovannini, 2006; Shalin et al., 2006). Taking into consideration the role of BDNF in LTP induction and its recently uncovered roles in regulation of the neuronal actin cytoskeleton, our results further implicate BDNF and calpain in facilitating cytoskeletal changes at the submembrane level. More recently, it has become widely acknowledged that calpain, by partially truncating a variety of cytoskeletal proteins, plays a critical role in the regulation of shape and motility in numerous cell types (for review, see Carragher and Frame, 2002; Perrin et al., 2006). A recent study indicates that calpain functions as a molecular switch in Chinese hamster ovary cells to control cell spreading and retraction (Flevaris et al., 2007). In particular, calpain-mediated spectrin truncation as well as calpain regulation of actin filament reorganization could play major roles in regulating the structural reorganization generally proposed to underlie long-term modifications of synaptic efficacy. BDNF-induced spectrin degradation is likely to play a permissive role for LTP induction. For instance, BDNF-induced spectrin degradation may precede and facilitate AMPA receptor translocation to the cell surface. Anchoring of AMPA receptors may follow and require de novo actin polymerization. Recently, it has also been shown that BDNF activates calpain both in vitro and in vivo and, as a result, produces the truncation of SCOP (suprachiasmatic nucleus circadian oscillatory protein), a negative regulator of a Ras protein (Shimizu et al., 2007). In this way, calpain activation would facilitate MAPK activation. Our results therefore suggest the possibility that BDNF, by stimulating the MAPK pathway, produces calpain activation, which would further stimulate MAP kinase activity and participate in the dendritic spine reorganization that underlies stabilization of synaptic modifications involved in LTP consolidation. BDNF has also been shown to produce calpain-mediated truncation of ARMS (ankyrin-rich membrane spanning protein; also known as Kidins220), and this effect has been proposed to participate in BDNF-mediated synaptic reorganization (Wu and Chao, 2007). In good agreement with the hypothesis that calpain participates in cytoskeletal reorganization, we found that BDNF-mediated actin polymerization was dependent on calpain activation.
In conclusion, our results demonstrate that m-calpain is rapidly activated in dendrites and dendritic spines by BDNF as a result of MAP kinase-mediated phosphorylation and participates in cytoskeletal reorganization. These results not only provide a new link between calpain and synaptic plasticity but also shed new light on the paradox that m-calpain, although widely expressed in CNS, requires unphysiological calcium levels for activation. In addition, these findings reveal the existence of a critical link between several elements long proposed to participate in synaptic plasticity, learning and memory, and strengthen the hypothesis that m-calpain plays a critical role in activity-dependent modifications of synaptic efficacy.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant P01NS045260-01 (principle investigator: Dr. C. M. Gall).
The authors declare no competing financial interests.
References
- Bánóczi Z, Alexa A, Farkas A, Friedrich P, Hudecz F. Novel cell-penetrating calpain substrate. Bioconjug Chem. 2008;19:1375–1381. doi: 10.1021/bc800021y. [DOI] [PubMed] [Google Scholar]
- Baudry M, Lynch G. Remembrance of arguments past: how well is the glutamate receptor hypothesis of LTP holding up after 20 years? Neurobiol Learn Mem. 2001;76:284–297. doi: 10.1006/nlme.2001.4023. [DOI] [PubMed] [Google Scholar]
- Bramham CR. Control of synaptic consolidation in the dentate gyrus: mechanisms, functions, and therapeutic implications. Prog Brain Res. 2007;163:453–471. doi: 10.1016/S0079-6123(07)63025-8. [DOI] [PubMed] [Google Scholar]
- Carragher NO, Frame MC. Calpain: a role in cell transformation and migration. Int J Biochem Cell Biol. 2002;12:1539–1543. doi: 10.1016/s1357-2725(02)00069-9. [DOI] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Casale MS, Gall CM, Lynch G. Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci. 2007;27:5363–5372. doi: 10.1523/JNEUROSCI.0164-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny JB, Polan-Curtain J, Ghuman A, Wayner MJ, Armstrong DL. Calpain inhibitors block long-term potentiation. Brain Res. 1990;534:317–320. doi: 10.1016/0006-8993(90)90148-5. [DOI] [PubMed] [Google Scholar]
- Flevaris P, Stojanovic A, Gong H, Chishti A, Welch E, Du X. A molecular switch that controls cell spreading and retraction. J Cell Biol. 2007;179:553–565. doi: 10.1083/jcb.200703185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco SJ, Huttenlocher A. Regulating cell migration: calpains make the cut. J Cell Sci. 2005;118:3829–3838. doi: 10.1242/jcs.02562. [DOI] [PubMed] [Google Scholar]
- Friedrich P. The intriguing Ca2+ requirement of calpain activation. Biochem Biophys Res Commun. 2004;323:1131–1133. doi: 10.1016/j.bbrc.2004.08.194. [DOI] [PubMed] [Google Scholar]
- Giovannini MG. The role of the extracellular signal-regulated kinase pathway in memory encoding. Rev Neurosci. 2006;17:619–634. doi: 10.1515/revneuro.2006.17.6.619. [DOI] [PubMed] [Google Scholar]
- Glading A, Uberall F, Keyse SM, Lauffenburger DA, Wells A. Membrane proximal ERK signaling is required for m-calpain activation downstream of epidermal growth factor receptor signaling. J Biol Chem. 2001;276:23341–23348. doi: 10.1074/jbc.M008847200. [DOI] [PubMed] [Google Scholar]
- Glading A, Bodnar RJ, Reynolds IJ, Shiraha H, Satish L, Potter DA, Blair HC, Wells A. Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol Cell Biol. 2004;24:2499–2512. doi: 10.1128/MCB.24.6.2499-2512.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdi H, Hsu YT, Zhou M, Qin Q, Bi X, Baudry M. Positive AMPA receptor modulation rapidly stimulates BDNF release and increases dendritic mRNA translation. J Neurosci. 2009;27:8688–8697. doi: 10.1523/JNEUROSCI.6078-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katanaev VL, Wymann MP. Microquantification of cellular and in vitro F-actin by rhodamine phalloidin fluorescence enhancement. Anal Biochem. 1998;264:185–190. doi: 10.1006/abio.1998.2837. [DOI] [PubMed] [Google Scholar]
- Koponen E, Lakso M, Castrén E. Overexpression of the full-length neurotrophin receptor trkB regulates the expression of plasticity-related genes in mouse brain. Mol Brain Res. 2004;130:81–94. doi: 10.1016/j.molbrainres.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Lin B, Kramár EA, Bi X, Brucher FA, Gall CM, Lynch G. Theta stimulation polymerizes actin in dendritic spines of hippocampus. J Neurosci. 2005;25:2062–2069. doi: 10.1523/JNEUROSCI.4283-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Liu MC, Wang KK. Calpain in the CNS: from synaptic function to neurotoxicity. Sci Signal. 2005;1:re.1. doi: 10.1126/stke.114re1. [DOI] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G, Baudry M. The biochemistry of memory: a new and specific hypothesis. Science. 1984;224:1057–1063. doi: 10.1126/science.6144182. [DOI] [PubMed] [Google Scholar]
- Lynch G, Kramar EA, Rex CS, Jia Y, Chappas D, Gall CM, Simmons DA. Brain-derived neurotrophic factor restores synaptic plasticity in a knock-in mouse model of Huntington's disease. J Neurosci. 2007;27:4424–4434. doi: 10.1523/JNEUROSCI.5113-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melloni E, Michetti M, Salamino F, Minafra R, Pontremoli S. Modulation of the calpain autoproteolysis by calpastatin and phospholipids. Biochem Biophys Res Commun. 1996;229:193–197. doi: 10.1006/bbrc.1996.1779. [DOI] [PubMed] [Google Scholar]
- Narisawa-Saito M, Iwakura Y, Kawamura M, Araki K, Kozaki S, Takei N, Nawa H. Brain-derived neurotrophic factor regulates surface expression of alpha-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid receptors by enhancing the N-ethylmaleimide-sensitive factor/GluR2 interaction in developing neocortical neurons. J Biol Chem. 2002;277:40901–40910. doi: 10.1074/jbc.M202158200. [DOI] [PubMed] [Google Scholar]
- Niapour M, Berger S. Flow cytometric measurement of calpain activity in living cells. Cytometry. 2007;71:475–485. doi: 10.1002/cyto.a.20399. [DOI] [PubMed] [Google Scholar]
- Pang PT, Lu B. Regulation of late-phase LTP and long-term memory in normal and aging hippocampus: role of secreted proteins tPA and BDNF. Ageing Res Rev. 2004;3:407–430. doi: 10.1016/j.arr.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER. Some form of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phosphor-MAP kinase. Neuron. 2001;32:123–140. doi: 10.1016/s0896-6273(01)00443-3. [DOI] [PubMed] [Google Scholar]
- Perlmutter LS, Siman R, Gall C, Seubert P, Baudry M, Lynch G. The ultrastructural localization of calcium-activated protease calpain in rat brain. Synapse. 1988;2:79–88. doi: 10.1002/syn.890020111. [DOI] [PubMed] [Google Scholar]
- Perrin BJ, Amann KJ, Huttenlocher A. Proteolysis of cortactin by calpain regulates membrane protrusion during cell migration. Mol Biol Cell. 2006;17:239–250. doi: 10.1091/mbc.E05-06-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Lauterborn JC, Lin CY, Kramár EA, Rogers GA, Gall CM, Lynch G. Restoration of long-term potentiation in middle-aged hippocampus after induction of brain-derived neurotrophic factor. J Neurophysiol. 2006;96:677–685. doi: 10.1152/jn.00336.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Lin CY, Kramár EA, Chen LY, Gall CM, Lynch G. Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J Neurosci. 2007;27:3017–3029. doi: 10.1523/JNEUROSCI.4037-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saido TC, Sorimachi H, Suzuki K. Calpain: new perspective in molecular diversity and physiological/ pathological involvement. FASEB J. 1994;8:814–822. [PubMed] [Google Scholar]
- Selcher JC, Weeber EJ, Christian J, Nekrasova T, Landreth GE, Sweatt JD. A role for ERK MAP kinase in physiological temporal integration in hippocampal area CA1. Learn Mem. 2003;10:26–39. doi: 10.1101/lm.51103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalin SC, Hernandez CM, Dougherty MK, Morrison DK, Sweatt JD. Kinase suppressor or RAS1 compartmentalizes hippocampal signal transduction and subserves synaptic plasticity and memory formation. Neuron. 2006;50:765–779. doi: 10.1016/j.neuron.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Shimizu K, Phan T, Mansuy IM, Storm DR. Proteolytic degradation of SCOP in the hippocampus contributes to activation of MAP kinase and memory. Cell. 2007;128:1219–1229. doi: 10.1016/j.cell.2006.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraha H, Glading A, Chou J, Jia Z, Wells A. Activation of m-calpain (calpain II) by epidermal growth factor is limited by protein kinase A phosphorylation of m-calpain. Mol Cell Biol. 2002;22:2716–2727. doi: 10.1128/MCB.22.8.2716-2727.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staubli U, Larson J, Thibault O, Baudry M, Lynch G. Chronic administration of a thiol-proteinase inhibitor blocks long-term potentiation of synaptic responses. Brain Res. 1988;444:153–158. doi: 10.1016/0006-8993(88)90922-5. [DOI] [PubMed] [Google Scholar]
- Stockholm D, Bartoli M, Sillon G, Bourg N, Davoust J, Richard I. Imaging calpain protease activity by multiphoton FRET in living mice. J Mol Biol. 2005;346:215–222. doi: 10.1016/j.jmb.2004.11.039. [DOI] [PubMed] [Google Scholar]
- Taniguchi N, Shinoda Y, Takei N, Nawa H, Ogura A, Tominaga-Yoshino K. Possible involvement of BDNF release in long-lasting synapse formation induced by repetitive PKA activation. Neurosci Lett. 2006;406:38–42. doi: 10.1016/j.neulet.2006.06.071. [DOI] [PubMed] [Google Scholar]
- Thoenen H. Neurotrophins and activity-dependent plasticity. Prog Brain Res. 2000;128:183–191. doi: 10.1016/S0079-6123(00)28016-3. [DOI] [PubMed] [Google Scholar]
- Thomas D, Bradshaw RA. Differential utilization of ShcA tyrosine residues and functional domains in the transduction of epidermal growth factor-induced mitogen-activated protein kinase activation in 293T cells and nerve growth factor-induced neurite outgrowth in PC12 cells. Identification of a new Grb2.Sos1 binding site. J Biol Chem. 1997;272:22293–22299. doi: 10.1074/jbc.272.35.22293. [DOI] [PubMed] [Google Scholar]
- Tominaga-Yoshino K, Urakubo T, Okada M, Matsuda H, Ogura A. Repetitive induction of late-phase LTP produces long-lasting synaptic enhancement accompanied by synaptogenesis in cultured hippocampal slices. Hippocampus. 2008;18:281–293. doi: 10.1002/hipo.20391. [DOI] [PubMed] [Google Scholar]
- Vanderklish P, Saido TC, Gall C, Arai A, Lynch G. Proteolysis of spectrin by calpain accompanies theta-burst stimulation in cultured hippocampal slices. Mol Brain Res. 1995;32:25–35. doi: 10.1016/0169-328x(95)00057-y. [DOI] [PubMed] [Google Scholar]
- Vanderklish PW, Krushel LA, Holst BH, Gally JA, Crossin KL, Edelman GM. Marking synaptic activity in dendritic spines with a calpain substrate exhibiting fluorescence resonance energy transfer. Proc Natl Acad Sci U S A. 2000;97:2253–2258. doi: 10.1073/pnas.040565597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Lynch DR. Calpain and synaptic function. Mol Neurobiol. 2006;33:215–236. doi: 10.1385/MN:33:3:215. [DOI] [PubMed] [Google Scholar]
- Wu S, Chao MV. Activity-dependent regulation of ARMS, a Trk receptor substrate. Soc Neurosci Abstr. 2007;33:572–15. [Google Scholar]
- Yamada K, Mizuno M, Nabeshima T. Role for brain-derived neurotrophic factor in learning and memory. Life Sci. 2002;70:735–744. doi: 10.1016/s0024-3205(01)01461-8. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Urakubo T, Tominaga-Yoshino K, Ogura A. Repetition of mGluR dependent LTD causes slowly developing persistent reduction in synaptic strength accompanied by synapse elimination. Brain Res. 2005;1042:6–16. doi: 10.1016/j.brainres.2005.02.028. [DOI] [PubMed] [Google Scholar]
- Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, Bramham CR. Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci. 2002;22:1532–1540. doi: 10.1523/JNEUROSCI.22-05-01532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]