

Abstract
Friend virus induces an erythroleukemia in susceptible mice that is initiated by the interaction of the Friend virus-encoded glycoprotein gp55 with the erythropoietin (Epo) receptor and the product of the host Fv2 gene, a naturally occurring truncated form of the Stk receptor tyrosine kinase (Sf-Stk). We have previously demonstrated that the activation of Sf-Stk, recruitment of a Grb2/Gab2/Stat3 signaling complex, and induction of Pu.1 expression by Stat3 are required for the development of the early stage of Friend disease both in vitro and in vivo. Here we demonstrate that the interaction of gp55 with Sf-Stk is dependent on cysteine residues in the ecotropic domain of gp55 and the extracellular domain of Sf-Stk. Point mutation of these cysteine residues or deletion of these domains inhibits the ability of gp55 to interact with Sf-Stk, resulting in the inability of these proteins to promote the Epo-independent growth of erythroid progenitor cells. We also demonstrate that the interaction of gp55 with Sf-Stk does not promote dimerization of Sf-Stk but results in enhanced phosphorylation of Sf-Stk and the relocalization of Sf-Stk from the cytosol to the plasma membrane. Finally, we demonstrate that a constitutively active form of Sf-Stk (Sf-StkM330T), as well as its human counterpart, Sf-Ron, promotes Epo-independent colony formation in the absence of gp55 and that this response is also dependent on the cysteines in the extracellular domains of Sf-StkM330T and Sf-Ron. These data suggest that the cysteines in the extracellular domains of Sf-Stk and Sf-Ron may also mediate the interaction of these truncated receptors with other cellular factors that regulate their ability to promote cytokine-independent growth.
Since Friend disease was first reported in 1957 (19), the acute erythroleukemia induced by the various strains of Friend virus have provided an excellent model to study multistage carcinogenesis (5). In the first stage, the virus infects erythroid progenitor cells and a viral glycoprotein, gp55, interacts with both the erythropoietin receptor (EpoR) and a naturally occurring truncated form of the stem cell-derived tyrosine kinase (Stk), Sf-Stk, resulting in the Epo-independent (Epoind) expansion of erythroid progenitor cells. The late stage of erythroleukemia in Friend disease is marked by inactivation of the p53 locus (6, 28, 38, 39, 51) and proviral integration into the Spi-1 locus (36, 43, 44), resulting in enhanced expression of Pu.1, which causes a block in erythroid differentiation and promoting the onset of acute erythroleukemia.
Friend virus is a complex of two viruses, the spleen focus-forming virus (SFFV), which is a replication-defective C-type retrovirus, and the ecotropic Friend murine leukemia virus (F-MuLV). SFFV is responsible for the rapid splenomegaly and acute erythroleukemia induced by Friend virus infection (7, 64, 65, 67), while F-MuLV provides helper function and can be substituted for by other murine leukemia viruses (35). Specifically, the glycoprotein gp55, encoded by the SFFV env gene, acts as the transforming viral oncoprotein (2, 65).
Several loci in the mouse genome that control Friend virus susceptibility have been identified. Fv1, Fv3, and Fv4 affect the ability of Friend virus to infect early erythroid progenitor cells. The Fv1 gene product inhibits Friend virus infection by interacting with the viral capsid protein (60). The Fv3 gene encodes cytidine deaminase Apobec3, which broadly inhibits retrovirus infection (42, 53, 57). The Fv4 gene product affects viral binding by competing for receptors on the cell membrane (59). Another set of genes, W, Sl, f, and Fv2, are required for the development or expansion of infected progenitor cells. Our previous work demonstrated that W, Sl, and f, which encode the kit receptor, its ligand SCF, and Smad5, respectively, also play key roles in the BMP4-dependent stress erythropoiesis pathway(46, 47, 55). Analysis of those mutants showed that Friend virus activates this pathway, leading to acute amplification of stress progenitors, which are targets of Friend virus in the spleen, and resulting in rapid onset of disease.
The Friend virus susceptibility gene Fv2 encodes the stem cell-derived tyrosine kinase (Stk) receptor (48). A naturally occurring N-terminally truncated form of Stk, short-form Stk (Sf-Stk), is required for Friend virus susceptibility. Fv2r/r mice, including C57BL/6, lack expression of Sf-Stk and are resistant to Friend virus infection, while full-length Stk expression is unaffected in these mice. An internal promoter within the Stk locus drives Sf-Stk expression, and Fv2r/r mice harbor mutations in the internal promoter. Sf-Stk lacks the N-terminal ligand binding domain of full-length Stk but retains the transmembrane and tyrosine kinase domains. In vitro and in vivo expression of Sf-Stk in C57BL/6 bone marrow cells has been shown to confer Friend virus susceptibility to Fv2r/r mice (18).
Sf-Stk covalently interacts with gp55, resulting in constitutive activation of Sf-Stk (41). However, the mechanism by which this occurs is currently unknown. Here, we identify cysteines in the extracellular domains of Sf-Stk and gp55 that mediate this interaction. Furthermore, we demonstrate that while the association with gp55 is not required for the dimerization of Sf-Stk, the interaction of gp55 with Sf-Stk promotes tyrosine phosphorylation of Sf-Stk. In addition, while the extracellular cysteines in Sf-Stk promote retention of Sf-Stk in the cytoplasm in the absence of gp55, the interaction of Sf-Stk with gp55 through these cysteines results in enhanced cell surface localization of Sf-Stk. These changes in receptor activation and subcellular localization mediate the ability of Sf-Stk to induce gene expression and promote the Epoind growth of primary erythroblasts.
MATERIALS AND METHODS
Antibodies and cell culture reagents.
HEK 293 cells and CHO cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The Mirus-293 and TransIT-CHO transfection reagents were purchased from Mirus Bio, LLC (Madison, WI). The dual-luciferase reporter assay system was purchased from Promega Corporation (Madison, WI). Antibodies against the myc tag, hemagglutinin (HA) tag, phosphotyrosine, phospho-Erk1/2, Erk1/2, and horseradish peroxidase (HRP)-linked anti-rat IgG were purchased from Cell Signaling (Danvers, MA). Antibody against actin and HRP linked anti-mouse IgG were purchased from Sigma-Aldrich, Inc (St. Louis, Mo). Mouse True Blot Ultra HRP-anti-mouse IgG was purchase from eBiosciences (San Diego, CA). HRP-linked anti-rabbit IgG was purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). Rat antiserum against gp55 was kindly provided by Sandra Ruscetti (National Cancer Institute). Murine interleukin-3 (IL-3) was purchased from Peprotech (Rocky Hill, NJ). Erythropoietin (Epo) was purchased from R&D Systems (Minneapolis, MN). Methocult medium M3234 was purchased from Stem Cell Technologies (Vancouver, British Columbia, Canada). All PCR primers were ordered from OpeRon Biotechnologies, Inc. (Huntsville, AL). Restriction enzymes and protein G magnetic beads were purchased from New England Biolabs (Ipswich, MA). PfuTurbo DNA polymerase was purchased from Stratagene (La Jolla, CA). ECL Plus Western blotting detection reagents were purchased from GE Healthcare (Piscataway, NJ). The Pierce cell surface protein isolation kit was purchased from Thermo Fisher Scientific Inc. (Rockford, IL).
Gene construction and mutagenesis.
Murine stem cell virus (MSCV)-myc-Sf-StkC8A mutagenesis was generated from MSCV-myc-Sf-Stk by using the primers 5′-GTGGGTGGTGAGGTCGCCCAACATGAGCTCCG-3′ and 5′-CGGAGCTCATGTTGGGCGACCTCACCACCCAC-3′. MSCV-myc-Sf-StkC19A and MSCV-myc-Sf-StkC8,19A mutageneses were generated from MSCV-myc-Sf-Stk and MSCV-myc-Sf-StkC8, respectively, by using the primers 5′-GGGGATGTGGTGATCGCCCCCCTGCCCCCTTC-3′ and 5′-GAAGGGGGCAGGGGGGCGATCACCACATCCCC-3′. MSCV-myc-Sf-StkC37A mutagenesis was generated by using the primers 5′-GTCCCATTGCAGGTCGCTGTAGACGGTGGGTG-3′ and 5′-CACCCACCGTCTACAGCGACCTGCAATGGGAC-3′. MSCV-myc-Sf-StkC42A mutagenesis was generated by using the primers 5′-CTGTGTAGACGGTGGGGCTCACATCCTGAGCC-3′ and 5′-GGCTCAGGATGTGAGCCCCACCGTCTACACG-3′. MSCV-myc-Sf-StkC37,42A mutagenesis was generated from Sf-StkC37A by using the primers 5′-CGCTGTAGACGGTGGGGCTCACATCCTGAGCC-3′ and 5′-GGCTCAGGATGTGAGCCCCACCGTCTACAGCG-3′. MSCV-myc-Sf-StkC4A mutagenesis was generated from Sf-StkC37,42A by using the primers for Sf-StkC8A and Sf-StkC19A mutations. MSCV-myc-Sf-StkM330T and MSCV-myc-Sf-StkC4AM330T mutageneses were generated from MSCV-myc-Sf-Stk and MSCV-myc-Sf-StkC4A by using the primers 5′-CCTGCCAGTCAAATGGACGGCACTGGAGAGCCTGC-3′ and 5′-GCAGGCTCTCCAGTGCCGTCCATTTGACTGGCAGG-3′. To construct MSCV-myc-Sf-StkΔ19, the Sf-StkΔ19 fragment was PCR amplified from MSCV-myc-Sf-Stk by using the primers 5′-GGGAATTCCCCCCTGCCCCCTTCCCTG-3′ and 5′-GAGACGTGCTACTTCCATTTGTC-3′. The Sf-StkΔ19 PCR fragment was then digested with EcoRI and cloned into EcoRI-digested MSCV-myc-Sf-Stk vector. To construct MSCV-myc-Sf-StkΔ42, the Sf-StkΔ42 fragment was PCR amplified from MSCV-myc-Sf-Stk by using the primers 5′-GGGAATTCTCACATCCTGAGCCAAGTGC-3′ and 5′-GAGACGTGCTACTTCCATTTGTC-3′. the Sf-StkΔ42 PCR fragment was then digested with EcoRI and cloned into EcoRI-digested MSCV-myc-Sf-Stk vector. To construct MSCV-myc-Sf-StkΔE, the Sf-StkΔE fragment was PCR amplified from MSCV-myc-Sf-Stk by using the primers 5′-GGGAATTCGATACTCCTTATTGCTCTTCTGG-3′ and 5′-GAGACGTGCTACTTCCATTTGTC-3′. The Sf-StkΔE PCR fragment was then digested with EcoRI and cloned into EcoRI-digested MSCV-myc-Sf-Stk vector. To construct MSCV-myc-Sf-StkΔETM, the Sf-StkΔETM fragment was PCR amplified from MSCV-myc-Sf-Stk by using the primers 5′-CGGAATTCTAACTCCCGAAGACGGAAAAAGC-3′ and 5′-GAGACGTGCTACTTCCATTTGTC-3′. The Sf-StkΔETM PCR fragment was then digested with EcoRI and cloned into EcoRI-digested MSCV-myc-Sf-Stk vector. To construct MSCV-Sf-Stk-HA, the Sf-Stk fragment was PCR amplified from MSCV-myc-Sf-Stk by using the primers 5′-GGCAGATCTTGTGACTGTGAACATG-3′ and 5′-AGTGGGCAGGGGTGGCTCTG-3′. The PCR SfStk fragment was then purified and inserted into the EcoRV site of pcDNA3.1-HAc. The Sf-Stk-HA fragment was cut out using BglII/XhoI and ligated into BglII/XhoI-digested MSCV-neo vector. To construct MSCV-myc-Sf-Ron, the Sf-Ron fragment was PCR amplified from pCDNA-Ron by using the primers 5′-CCGGAATTCCATGGTTGTCTGCCCCCTGCCC-3′ and 5′-GAACGAATTCAAGTGGGCCGAGGAGGCTC-3′. The Sf-Ron PCR fragment was then digested with EcoRI and cloned into EcoRI-digested MSCV-myc-Sf-Stk vector. To construct MSCV-myc-Sf-RonC3A, MSCV-myc-Sf-RonC4thA mutagenesis was first generated by using the primers 5′-CTTGAATTCCATGGTTGTCGCCCCCCTGCCCCATCCCTGC-3′ and 5′-GCAGGGATGGGGGCAGGGGGGCGACAACCATGGAATTCAAG-3′. MSCV-myc-Sf-RonC4,27A mutagenesis was then generated from MSCV-myc-Sf-RonC4thA by using the primers 5′-GGTCTGCGTAGATGGTGAAGCTCATATCCTGGGTAGAGTGG-3′ and 5′-CCACTCTACCCAGGATATGAGCTTCACCATCTACGCAGACC-3′. MSCV-myc-Sf-RonC3A mutagenesis was then generated from MSCV-myc-Sf-RonC4,27A by using the primers 5′-GGTGCCCCATTGCAGGTCGCCGTAGATGGTGAAGCTCATATC-3′ and 5′-GATATGAGCTTCACCATCTACGGCGACCTGCAATGGGGCACC-3′. MSCV-gp55C306A mutagenesis was generated by using the primers 5′-CCCTGATAAAATTCAAGAGGCCTGGTTATGCCTAGTGTCTGG 3′ and 5′-CCAGACACTAGGCATAACCAGGCCTCTTGAATTTTATCAGGG-3′. MSCV-gp55C309A mutagenesis was generated by using the primers 5′-CAAGAGTGCTGGTTAGCCCTAGTGTCTGGACCCCCC-3′ and 5′-GGGGGGTCCAGACACTAGGGCTAACCAGCACTCTTG-3′. MSCV-gp55C306,309A mutagenesis was generated by using the primers 5′-CAAGAGGCCTGGTTAGCCCTAGTGTCTGGACCCCCC-3′ and 5′-GGGGGGTCCAGACACTAGGGCTAACCAGGCCTCTTG-3′. MSCV-gp55C337A mutagenesis was generated by using the primers 5′-GCCCTAAAAGAAAAAGCTTGTTTCTATGCTGACCATACAGGCC-3′ and 5′-GGCCTGTATGGTCAGCATAGAAACAAGCTTTTTCTTTTAGGGC-3′. MSCV-gp55C338A mutagenesis was generated by using the primers 5′-GCCCTAAAAGAAAAATGTGCTTTCTATGCTGACCATACAGGCC-3′ and 5′-GGCCTGTATGGTCAGCATAGAAAGCACATTTTTCTTTTAGGGC-3′. MSCV-gp55C337,338A mutagenesis was generated by using the primers 5′-GCCCTAAAAGAAAAAGCTGCTTTCTATGCTGACCATACAGGCC-3′ and 5′-GGCCTGTATGGTCAGCATAGAAAGCAGCTTTTTCTTTTAGGGC-3′. MSCV-gp55C4A mutagenesis was generated from MSCV-gp55 C306,309A by using the same primers for MSCV-gp55 C337, 338A mutagenesis. To construct pCDNA-YFP1-gp55, pCDNA-YFP2-gp55, pCDNA-YFP1-gp55C4A, and pCDNA-YFP2-gp55C4A, the gp55 and gp55C4A fragments were PCR amplified from MSCV-gp55-HA and MSCV-gp55C4A-HA by using the primers 5′-GTTCCGGAATGGAAGGTCCAGCGTCCTC-3′ and 5′-GCTCTAGACTGCCTTGGGAAAAGCGCCTC-3′. The PCR-amplified gp55 and gp55C4A fragments and pCDNA-YFP1 and pCDNA-YFP2 plasmids were double digested with BspEI and XbaI and ligated.
Cell transfection and luciferase assay.
A total of 5 × 104 HEK 293 cells were plated into 24-well plates. Twenty hours later, a designated mixture of 20 ng Sf-Stk or gp55, 20 ng of AP.1 luciferase reporter plasmid, and 0.5 ng Renilla reporter plasmid was used for transient transfection in each well with the Mirus-293 transfection reagent according to the manufacturer's protocol. For a control vector, 20 ng MSCV-neo plasmid was used in transfection of each well. At 48 h after transfection, luciferase assays were performed according to the manufacturer's instructions.
Immunoprecipitation and Western blot analysis.
A total of 3 × 105 HEK 293 cells/well were plated into six-well plates. Twenty hours later, the cells were transiently transfected with a 300-ng mixture of the designated plasmids per well, and two wells were transfected for every set of samples. At 40 to 48 h posttransfection, cells were suspended in 500 μl ice-cold cell lysis buffer containing 150 mM NaCl, 20 mM Tris-HCl (pH 7.5), 5 mM EDTA, 1% NP-40, 1 mM phenylmethylsulfonyl fluoride, 1 mM Na3VO4, and 10 mM NaF. Cell lysates were centrifuged at 1,500 rpm for 20 min. Following centrifugation, the supernatant lysates were transferred to prechilled tubes. Forty microliters of cell lysate was mixed with 4× denaturing SDS loading buffer and heated at 100°C for 5 min. Eight hundred microliters of lysate was used for immunoprecipitation as follows. Cell lysates were incubated with the appropriate amount of primary antibody at 4°C for 30 min, incubated with protein G magnetic beads for 2 h, and washed with ice-cold phosphate-buffered saline (PBS) three times. Beads were resuspended in 40 μl 1× SDS denaturing loading buffer and heated at 100°C for 5 min. Samples for Western blotting and immunoprecipitation were separated by SDS-PAGE and then transferred to polyvinylidene difluoride membranes. Membranes were then blocked with 5% nonfat milk in Tris-buffered saline with Tween 20 (TBST) for 1 h and probed with primary antibody at 4°C overnight. Membranes were washed three times in TBST and incubated with horseradish peroxidase-conjugated IgG for another hour. Membranes were washed three times in TBST before ECL plus Western blotting detection reagents were applied for visualization. For reprobing, membranes were stripped with 62.5 mM Tris-HCl (pH 6.8), 2% SDS, and 0.7% β-mercaptoethanol at 55°C for 30 min.
Retrovirus generation and in vitro infection of primary murine bone marrow cells.
To generate MSCV retroviral supernatants, 3 × 105 293T cells/well were plated into six-well plates. Twenty hours later, the cells were transiently transfected with 300 ng pEco and 700 ng MSCV plasmid for each well. At 40 to 48 h posttransfection, viral supernatants were collected and filtered through 0.45-μm sterile syringe filters (Pall Life Sciences, Ann Arbor, MI) before use. For Friend polycythemia virus (FVP) supernatant generation, FP63 cells (a kind gift from Alan Bernstein, Mount Sinai Hospital, Toronto, Ontario, Canada) were cultured in DMEM with 10% FBS. The FVP supernatants were collected and filtered through 0.45-μm sterile syringe filters, aliquoted, and stored at −80°C. For in vitro infection of primary bone marrow, cells were harvested from femurs of 8-week-old mice and resuspended in DMEM with 40% FBS. The bone marrow cells were infected by incubation with retroviral supernatants for 20 h along with 10 ng/ml interleukin-3. For Epo-independent erythroid burst-forming unit (BFU-E) colony assays, the bone marrow cells were resuspended with FVP supernatants and incubated on ice for 1 h. The cells were then added to Methocult medium M3234, along with 2.5 ng/ml IL-3, in triplicate with or without 1 U/ml Epo. Cultures were incubated for 5 days with 5% CO2 at 37°C. Erythroid colonies were visualized by acid-benzidine staining.
Cell transfection and cell surface protein isolation.
A total of 1 × 106 to 2 × 106 CHO cells per ml were plated into 10-cm plates. Twenty hours later, a designated mixture of 1 to 2 μg plasmid was used for transient transfection in each plate with the Mirus-CHO transfection reagent according to the manufacturer's protocol. For a control vector, 1 to 2 μg MSCV-neo plasmid was used in transfection of each plate. At 48 h after transfection, cell surface biotinylation and protein isolation were performed according to the manufacturer's instructions. Prepared cell surface and cytosolic proteins were directly subjected to standard Western blot assay.
Flow cytometry.
A total of 3 × 105 293 cells per well were plated into six-well plates. Twenty hours later, the cells were transiently transfected with a 300-ng mixture of the designated plasmids per well. At 40 to 48 h posttransfection, cells were treated with EDTA-trypsin for 20 s and resuspended in 2 ml ice-cold washing buffer containing PBS and 2% FBS. One million cells were transferred to new tubes and centrifuged for 5 min at 1,500 rpm. For 293 cells transfected with yellow fluorescent protein (YFP), cells were resuspended in 1 ml ice-cold washing buffer for analysis by flow cytometry. Cells requiring staining were resuspended in 100 μl washing buffer and incubated with 10 μl Alexa Fluor 647-conjugated myc tag mouse antibody (Cell Signaling) for 30 min on ice. Cells were then washed and centrifuged twice before being resuspended in 1 ml washing buffer. YFP and Alexa Fluor 647 were analyzed with excitation at 647 nm, while enhanced green fluorescent protein (EGFP) was analyzed at 488 nm.
RESULTS
gp55 oligomerization is regulated by cysteines in the ecotropic domain.
Homo-oligomerization is believed to be an intrinsic feature of the gp55 protein (68). Detailed sequence studies have revealed 12 cysteine residues in the gp55 peptide: eight in the dualtropic N-terminal domain and four in the ecotropic C-terminal domain (3, 10, 66). The eight cysteine residues within the gp55 dualtropic domain are homologous to those in the envelope glycoprotein gp70 of Friend mink cell focus-forming virus (F-MCFV) and gp71 of F-MuLV, which share the same evolutionary origin (1). The cysteine residues in gp70 and gp71 are responsible for intrachain disulfide bond formation (32, 33). After replacement of the 3′ region of gp70 with that of gp55, the recombined F-MCFV exhibited pathogenic activity similar to that of F-SFFV (62). This finding suggests that cysteine residues in the gp55 dualtropic domain similarly form intrachain disulfide bonds and are not available for interchain disulfide bond formation. This leaves the possibility that gp55 dimerization is mediated by interchain disulfide bond formation between cysteines at positions 306, 309, 337, and 338 in the ecotropic C-terminal domain of gp55.
In order to address this hypothesis, we generated cysteine-to-alanine mutations in the ecotropic domain of gp55. These mutants were transiently transfected either alone or in the presence of Sf-Stk into 293 cells, and their expression was examined by Western blot analysis under both reducing and nonreducing conditions (Fig. 1A). Wild-type gp55 and gp55 harboring cysteine-to-alanine substitutions at two of the four cysteines (gp55C306,309A and gp55C337,338A) were detected as 55-kDa monomers, 95-kDa dimers, and 170-kDa multimers under nonreducing Western blotting conditions in both the presence and absence of Sf-Stk. However, gp55 harboring cysteine-to-alanine substitutions at all four cysteines in the ecotropic domain (gp55C4A) was detected only as a monomeric band. Our results indicate that the level of the gp55 dimer is very low compared to that of the monomeric form. This is consistent with previous studies which demonstrated that correct folding and dimerization of gp55 is inefficient and that only 3 to 5% of the total gp55 is correctly processed and localized on the cell surface(20, 21, 68), where it is believed to be essential for pathogenesis.
FIG. 1.

Cysteines in the ecotropic domain mediate gp55 homo-oligomerization. (A) 293 cells were transfected with wild-type gp55 or gp55 in which two or four cysteine residues in the ecotropic domain were mutated to alanine in the absence or presence of Sf-Stk, and 48 h later, cell lysates were separated by SDS-PAGE electrophoresis under nondenaturing and denaturing conditions. Membranes were stained with anti-gp55 antiserum. (B) 293 cells were cotransfected with plasmids expressing the fusion proteins YFP1-gp55 and YFP2-gp55 or YFP1-gp55C4A and YFP2-gp55C4A. Plasmids expressing YFP1-gp55 and YFP2-gp55 were also transfected individually. Forty-eight hours later, the cells were analyzed by flow cytometry for YFP fluorescence. *, P < 0.05. Error bars indicate standard deviations.
In order to further address the role of these cysteines in gp55 homodimerization, a yellow fluorescent protein (YFP) fragment complementation assay was performed (24). Plasmids expressing the fusion proteins YFP1-gp55, YFP2-gp55, YFP1-gp55C4A, and YFP2-gp55C4A were constructed and transfected into 293 cells, and 48 h later, cells were collected and sorted by flow cytometry for YFP fluorescence (Fig. 1B). Using this approach, we demonstrated the presence of dimers of wild-type gp55 but not of gp55C4A. Taken together, these results support the hypothesis that the four cysteines in the gp55 ecotropic domain are essential for g55 homodimerization.
gp55 promotes tyrosine phosphorylation, but not dimerization, of Sf-Stk.
Previous work from our laboratory demonstrated that Sf-Stk kinase activity is required for Friend virus-induced Epoind erythroid colony formation (18). A widely accepted model of receptor tyrosine kinase activation is one in which ligand binding to the receptor promotes the dimerization of the receptor, resulting in phosphorylation of the activation loop in the tyrosine kinase domain of the receptor in trans (54). In order to determine whether gp55 promotes Sf-Stk oligomerization, plasmids encoding myc- or HA-tagged Sf-Stk (myc-Sf-Stk and Sf-Stk-HA), were constructed and cotransfected into 293 cells with or without gp55; 48 h later, cells were lysed and oligomerization was assessed by coimmunoprecipitation of the myc and HA tags (Fig. 2B). To our surprise, myc-tagged Sf-Stk coimmunoprecipitated with HA-tagged Sf-Stk in the absence of gp55, and this interaction was not significantly altered in the presence of gp55, suggesting that Sf-Stk oligomerization is independent of gp55. In order to map the region of Sf-Stk responsible for promoting oligomerization, we generated N-terminally truncated forms of Sf-Stk lacking the extracellular domain sequences (Sf-StkΔE) or the extracellular and transmembrane domains (Sf-StkΔETM) (Fig. 2A). As shown in Fig. 2B, myc- and HA-tagged versions of both Sf-StkΔE and Sf-StkΔETM were also found to coimmunoprecipitate. These data demonstrate that the Sf-Stk extracellular and transmembrane domains are not required for Sf-Stk homo-oligomerization, which suggests that the intracellular juxtamembrane and kinase domains are responsible for homo-oligomerization.
FIG. 2.

Sf-Stk oligomerization is independent of gp55 and mediated by the cytoplasmic domain of Sf-Stk. (A) Schematic representation of myc- or HA-tagged Sf-Stk, Sf-StkΔE, and Sf-StkΔETM used in this experiment. TM, transmembrane domain; TK, tyrosine kinase domain. (B) 293 cells were transfected with the indicated wild-type or mutant forms of Sf-Stk in the presence or absence of gp55. Cell lysates were immunoprecipitated with anti-myc antibody and blotted with anti-HA antibody.
The fact that Sf-Stk oligomerization is independent of gp55 prompted us to further investigate whether gp55-independent Sf-Stk oligomerization is sufficient to promote Sf-Stk tyrosine phosphorylation. myc-Sf-Stk and Sf-Stk-HA were transfected into 293 cells in the presence of increasing concentrations of gp55. Sf-Stk tyrosine phosphorylation and oligomerization were assessed by immunoprecipitation and Western blot analysis (Fig. 3). Consistent with our previous results, gp55 did not enhance the coimmunoprecipitation of myc-Sf-Stk and Sf-Stk-HA. However, gp55 expression strongly induced the phosphorylation of Sf-Stk in a dose-dependent manner, suggesting that gp55 is required for efficient Sf-Stk tyrosine phosphorylation. These results suggest that rather than promoting dimerization of Sf-Stk, gp55 likely induces a conformational change in Sf-Stk oligomers resulting in enhanced tyrosine kinase activity and receptor autophosphorylation.
FIG. 3.
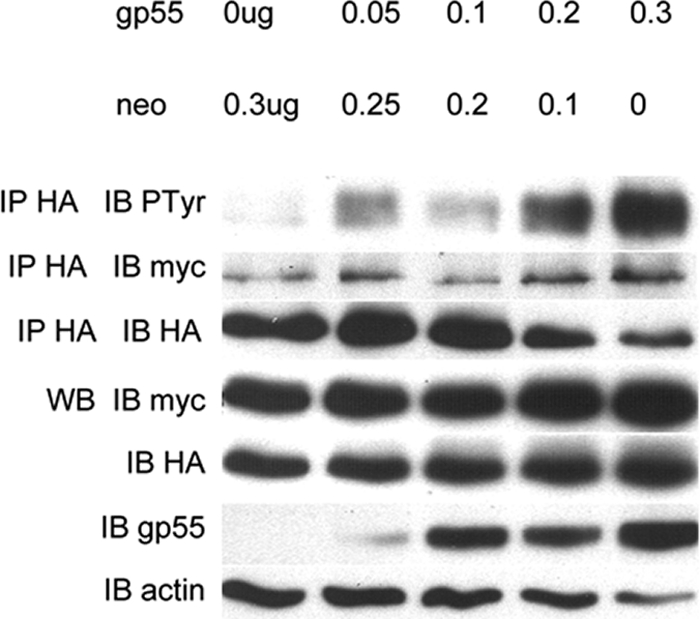
gp55 promotes Sf-Stk tyrosine phosphorylation in a dose-dependent manner. 293 cells were cotransfected with myc-Sf-Stk and Sf-Stk-HA in the presence of increasing concentrations of gp55. Cell lysates were immunoprecipitated with anti-HA and blotted with antiphosphotyrosine and anti-myc antibodies.
The interaction of gp55 with Sf-Stk is regulated by cysteines in the extracellular domain of Sf-Stk.
Previous studies demonstrated that gp55 is capable of transforming rodent fibroblasts in the presence of Sf-Stk and that this transformation is dependent upon the extracellular domain of Sf-Stk (40). In order to map the region of Sf-Stk responsible for the interaction of Sf-Stk with gp55, myc-tagged Sf-Stk, Sf-StkΔ19 (in which the first 19 amino acid residues of the extracellular domain are deleted), and Sf-StkΔ42 (in which the first 42 amino acid residues of the extracellular domain are deleted) were transfected into 293 cells with gp55. The interaction between gp55 and the Sf-Stk deletions was investigated by coimmunoprecipitation and Western blot analysis (Fig. 4B). The results demonstrate that gp55 coimmunoprecipitates with Sf-Stk. While Sf-StkΔ19 retains partial ability to coimmunoprecipitate with gp55, this interaction is completely abrogated with Sf-StkΔ42. This indicates that the first 42 amino acid residues of Sf-Stk extracellular domain are essential for its interaction with gp55. There are four cysteine residues located at amino acids 8, 19, 37, and 42 of the Sf-Stk extracellular domain. In order to determine whether these cysteines mediate the interaction of Sf-Stk with gp55, we introduced cysteine-to-alanine mutations into Sf-Stk individually or in combination. These Sf-Stk mutants were transfected into 293 cells with gp55, and the interaction between Sf-Stk mutants and gp55 was assessed by coimmunoprecipitation and Western blot analysis (Fig. 5B). The results from these studies demonstrate that while the double cysteine mutants, Sf-StkC8,19A and Sf-StkC37,42A, retain partial ability to coimmunoprecipitate with gp55, the interaction with gp55 of Sf-Stk in which all four cysteines are mutated to alanine, Sf-StkC4A, is diminished.
FIG. 4.

The first 42 amino acid residues in the Sf-Stk extracellular domain are required for the interaction of Sf-Stk with gp55. (A) Schematic representation of myc-tagged Sf-Stk, Sf-StkΔ19, and Sf-StkΔ42 used in this experiment. (B) 293 cells were transfected with wild-type or deletion forms of myc-Sf-Stk and gp55. Cell lysates were immunoprecipitated with anti-myc and blotted with anti-gp55 antiserum.
FIG. 5.

Sf-Stk interacts with gp55 through cysteine residues in the extracellular domain. (A) Schematic representation of gp55 and Sf-Stk. The locations of the cysteine residues are indicated. (B) 293 cells were transfected with wild-type gp55 and myc-tagged wild-type or mutated Sf-Stk. Cell lysates were immunoprecipitated with anti-myc antibody and blotted with anti-gp55 antiserum. (C) 293 cells were transfected with myc-tagged wild-type or mutated Sf-Stk and wild-type or mutated gp55. Cell lysates were immunoprecipitated with anti-myc antibody and blotted with anti-gp55 antiserum. (D) 293 cells were transfected with myc-tagged EpoR and wild-type gp55 or gp55C4A. Cell lysates were immunoprecipitated with anti-myc antibody and blotted with anti-gp55 antiserum.
Cysteines in the ecotropic domain of gp55 mediate its interaction with Sf-Stk but not with the EpoR.
To determine whether the cysteines in the ecotropic domain of gp55 promote the interaction of gp55 with Sf-Stk, we coexpressed Sf-Stk or Sf-StkC4A with gp55 or gp55C4A in 293 cells and assessed their ability to coimmunoprecipitate (Fig. 5B and C). Our data clearly indicate that both the cysteines in the extracellular domain of Sf-Stk and the cysteines in the ecotropic domain of gp55 are required for the interaction between gp55 and Sf-Stk. gp55 mutants in which two of the four cysteines are mutated to alanine, gp55C306,309A and gp55C337,338A, retain the ability to coimmunoprecipitate with wild-type Sf-Stk (Fig. 5C). The interaction of these double cysteine-to-alanine mutants with Sf-StkC8,19A and Sf-StkC37,42A are significantly reduced, but not abrogated, indicating that the interchain disulfide bonds formed between gp55 and Sf-Stk are not limited to specific cysteines but that the Sf-Stk/gp55 interaction can be promoted through the formation of different cysteine pairs between gp55 and Sf-Stk.
Two forms of Friend virus exist: FVP, which promotes erytholeukemia and polycythemia, and FVA, which also promotes erythroleukemia but with which the mice are anemic. The transmembrane domain of gp55P mediates the interaction of gp55 with the EpoR (11, 13) and is essential for promoting Epoind signaling downstream of the EpoR. In contrast, gp55A cannot effectively activate the EpoR due to sequence differences in the transmembrane domain (16). We introduced these changes into gp55P (gp55M390IdLL) and demonstrated that gp55M330IdLL can still efficiently bind and promote signaling through Sf-Stk (22). These studies support the conclusion that sequences in the extracellular domain of gp55, rather than in the transmembrane domain, mediate the interaction of gp55 with Sf-Stk. Conversely, we also tested whether the cysteines in gp55 regulate its interaction with the EpoR (Fig. 5D). Wild-type gp55 and gp55C4A were cotransfected into 293 cells with a myc-tagged EpoR. Cell lysates were immunoprecipitated with anti-myc and blotted for gp55. The results from these studies demonstrate coimmunoprecipitation of both wild-type gp55 and gp55C4A with the EpoR, indicating that the cysteines in gp55 are not required for its ability to interact with the EpoR.
Cysteines in the extracellular domain of Sf-Stk and the ecotropic domain of gp55 are required to promote downstream signaling and Epoind BFU-E colony formation.
Our data clearly demonstrate that the cysteines in the extracellular domain of Sf-Stk and the ecotropic domain of gp55 mediate the interaction of Sf-Stk with gp55. In order to determine whether these cysteines are required for the enhanced phosphorylation of Sf-Stk observed in the presence of gp55, 293 cells were cotransfected with wild-type or mutant forms of gp55 and wild-type or mutant forms of Sf-Stk. Cell lysates were immunoprecipitated with antiphosphotyrosine and immunoblotted with anti-myc (Fig. 6A). Consistent with the coimmunoprecipitation results, these results indicate that cysteines in both the extracellular domain of Sf-Stk and the ecotropic domain of gp55 are required for the enhanced phosphorylation of Sf-Stk by gp55. While double cysteine-to-alanine mutants of both gp55 and Sf-Stk partially retained this activity, mutation of all four cysteines on gp55 or Sf-Stk reduced Sf-Stk tyrosine phosphorylation to near-baseline levels. Erk1/2 phosphorylation and AP-1 activity were also examined in 293 cells transiently transfected with gp55 and Sf-Stk. While the levels of Erk phosphorylation in the presence of Sf-Stk alone were low, the presence of gp55 dramatically enhanced Erk phosphorylation. This enhanced Erk phosphorylation was dependent on the cysteines in both Sf-Stk and gp55 (Fig. 6B). Similarly, AP-1-driven luciferase activity was induced in these cells only in the presence of both gp55 and Sf-Stk, and this activity was dependent on the cysteines in both gp55 and Sf-Stk (Fig. 6C).
FIG. 6.

The cysteine residues in Sf-Stk and gp55 are required for Sf-Stk activation and mitogen-activated protein (MAP) kinase signaling. (A) 293 cells were transfected with myc-tagged wild-type or mutant forms of Sf-Stk with wild-type or mutant gp55. Cell lysates were immunoprecipitated with antiphosphotyrosine antibody and blotted with anti-myc antibody. (B) 293 cells were transfected with myc-tagged wild-type Sf-Stk or Sf-StkC4A with wild-type gp55 or gp55C4A. Cell lysates were blotted with anti-phosho-Erk1/2 antibody, stripped, and reprobed with anti-Erk1/2 antibody. (C) 293 cells were transfected with myc-tagged wild-type Sf-Stk or Sf-StkC4A and wild-type gp55 or gp55C4A in the presence of Ap-1 luciferase and Renilla reporter plasmids. At 48 h after transfection, luciferase assays were performed according to the manufacturer's instructions. *, P < 0.01; **, P < 0.01. Error bars indicate standard deviations.
FVP infection of primary erythroblasts promotes the Epoind formation of BFU-E colonies, due to the ability of gp55 to activate both Sf-Stk and the EpoR. To determine whether the cysteines in Sf-Stk that mediate the interaction of Sf-Stk with gp55 are required for the ability of FVP to induce Epoind colony formation, BFU-E colony assays were performed using bone marrow from C57BL/6 Fv2r/r mice, which lack Sf-Stk expression, transduced with wild-type or mutant forms of Sf-Stk followed by infection with FVP (Fig. 7A). Consistent with our previous studies, FVP induced Epoind colony formation by cells expressing wild-type Sf-Stk at levels comparable to those induced by Epo. While single cysteine mutants retained partial ability to promote Epoind colony formation induced by FVP, Sf-StkC4A failed to support Epoind colony formation.
FIG. 7.

The cysteine residues in Sf-Stk and gp55 are required for Epoind BFU-E colony formation. (A) 293 cells were transfected with MSCV expressing myc-tagged wild-type or mutant forms of Sf-Stk and pEco. At 48 h posttransfection, viral supernatants were collected and incubated overnight with bone marrow cells harvested from C57BL/6 mice. MSCV-infected bone marrow cells were then incubated with FVP on ice for 1 h and plated in methylcellulose medium containing IL-3 (2.5 ng/ml). On day 5, BFU-E colonies were stained with acid-benzidine and counted. *, P < 0.01. Error bars indicate standard deviations. (B) 293 cells were transfected with MSCV expressing wild-type or mutant forms of gp55 and pEco. At 48 h posttransfection, viral supernatants were collected and incubated overnight with bone marrow cells harvested from BALB/c mice. Virus-infected bone marrow cells were then plated in methylcellulose medium containing IL-3 (2.5 ng/ml). On day 5, BFU-E colonies were stained with acid-benzidine and counted. *, P < 0.01.
Expression of gp55 alone in primary erythroblasts of Fv2s/s mice is sufficient to induce Epoind colony formation. In order to determine whether the cysteines in gp55 are required for its ability to promote Epoind colony formation, we transduced BALB/c Fv2s/s bone marrow cells, which express endogenous Sf-Stk, with wild-type and mutant forms of gp55 and assessed BFU-E colony growth (Fig. 7B). While expression of wild-type gp55 was capable of promoting Epoind colony growth in Fv2s/s bone marrow, gp55C4A failed to support Epoind colony formation. As we observed with Sf-Stk, single cysteine mutants of gp55 retained partial activity in this assay. Taken together, these data verify a crucial role for the cysteines in the ecotropic domain of gp55 and the extracellular domain of Sf-Stk in the phenotypic response of primary erythroblasts to infection with Friend virus.
gp55 promotes the localization of Sf-Stk to the plasma membrane.
Although the bulk of gp55 and EpoR is found to coimmunoprecipitate in the rough endoplasmic reticulum (ER) (69), a productive interaction between EpoR and gp55 requires localization on the cell membrane (17, 31). To investigate the effect of gp55 on the subcellular location of Sf-Stk, myc-tagged Sf-Stk was cotransfected with gp55 or empty vector into CHO cells, and 48 h later, the cells were biotinylated and cell surface proteins were isolated. Western blotting of isolated cell surface proteins confirmed that Sf-Stk cell surface localization was increased upon cotransfection with gp55 (Fig. 8A).
FIG. 8.

gp55 and cysteines regulate the cell surface localization of Sf-Stk (A) CHO cells were transfected with myc-Sf-Stk and gp55 or empty vector. Forty-eight hours later, cell surface proteins were biotinylated and isolated. The cell surface proteins were separated by SDS-PAGE. Membranes were blotted with anti-myc antibody. (B) 293 cells were transfected with EGFP, myc-Sf-Stk, and gp55, gp55C4A, or empty vector. Forty-eight hours later, cells were resuspended and stained with Alexa Fluor 647-conjugated myc tag antibody. Cells were washed and analyzed by flow cytometry. EGFP expression populations were gated as transfected cells.
Using wild-type and mutant forms of Sf-Stk tagged at the N terminus with a myc tag, we employed flow cytometry to further evaluate the surface expression of Sf-Stk in the presence and absence of gp55. Plasmids expressing an N-terminal myc-tagged Sf-Stk were cotransfected into 293 cells with EGFP and gp55, gp55C4A, or empty vector; 48 h later, cells were collected and stained with Alexa Fluor 647-conjugated myc tag antibody without permeabilizing the cells in order to visualize myc-Sf-Stk located on the plasma membrane. EGFP-positive cells were gated, and myc expression was detected on the surface of the EGFP-positive cells (Fig. 8B). In cells cotransfected with empty vector (panel 1) or with gp55C4A (panel 9), basal levels of myc-Sf-Stk cell surface localization were detected. However, in the presence of gp55 (panel 5), the myc-Sf-Stk distribution at the plasma membrane was significantly enhanced. Surprisingly, Sf-StkC4A was able to localize to the plasma membrane, even in the absence of gp55 (panel 4). These data indicate that the cysteines in the extracellular domain of Sf-Stk that mediate the interaction of Sf-Stk with gp55 also play a role in regulating the subcellular localization of Sf-Stk in the absence of gp55. However, since our data indicate that Sf-StkC4A is not tyrosine phosphorylated or capable of supporting Epoind BFU-E formation in response to FVP, it appears that cell surface localization alone is not sufficient to activate Sf-Stk signaling.
Cysteines in the extracellular domain of Sf-Stk are required for Epoind growth of primary erythroblasts induced by a constitutively active form of Sf-Stk.
We have shown that the cysteines in the extracellular domain of Sf-Stk promote the phosphorylation of the receptor by mediating its interaction with gp55. In order to assess other potential roles for these cysteines, we utilized a constitutively active form of Sf-Stk containing an M330T mutation in the kinase domain which was reported to induce Epoind colony formation in the absence of FVP (27). We introduced the M330T mutation into both Sf-Stk and Sf-StkC4A (Sf-StkM330T and Sf-StkC4AM330T) and transfected these constructs into 293 cells in the presence or absence of gp55. We then tested the ability of gp55 to promote tyrosine phosphorylation of the Sf-Stk mutants. Consistent with previous studies, Sf-StkM330T exhibits a higher level of tyrosine phosphorylation than wild-type Sf-Stk when gp55 is not present However, this tyrosine phosphorylation is significantly increased when gp55 is coexpressed with Sf-StkM330T. Alternatively, introduction of the M330T mutation in the context of Sf-StkC4A, which localizes to the plasma membrane, resulted in full phosphorylation of the receptor (Fig. 9A). In order to determine whether the cysteines in the extracellular domain of Sf-Stk are required for the Epoind growth of infected erythroblasts in the context of a constitutively active form of Sf-Stk, BFU-E colony assays were performed with bone marrow from C57BL/6 Fv2r/r mice (which lack Sf-Stk expression) transduced with the indicated Sf-Stk mutants, followed by infection with FVP (Fig. 9B). Interestingly, Sf-StkM330T was fully capable of promoting Epoind BFU-E colony formation even in the absence of gp55, while Sf-StkC4AM330T failed to induce Epoind BFU-E colony formation in the absence of gp55, suggesting that the cysteines in the extracellular domain of Sf-Stk may interact with other cellular proteins in the absence of gp55 to elicit this response.
FIG. 9.

The cysteine residues in the extracellular domain of a constitutively active Sf-Stk are required for Epoind BFU-E colony formation. (A) 293 cells were transfected with gp55 and myc-tagged wild-type or mutated Sf-Stk. Cell lysates were immunoprecipitated with anti-tyrosine phosphorylation antibody and blotted with anti-myc antibody. (B) 293 cells were transfected with wild-type or mutated MSCV-myc-Sf-Stk and pEco. At 48 h posttransfection, viral supernatants were collected and incubated overnight with bone marrow cells of C57BL/6 mice. The infected bone marrow cells were plated in methylcellulose medium containing IL-3 (2.5 ng/ml) or incubated with FVP on ice for 1 h prior to plating in methylcellulose medium containing IL-3 (2.5 ng/ml). On day 5, BFU-E colonies were stained with acid-benzidine and counted. *, P < 0.01. Error bars indicate standard deviations.
Cysteines in the extracellular domain of Sf-RON mediate its interaction with gp55 and promote Epoind colony formation in the absence of gp55.
The Ron tyrosine kinase is the human homolog of murine Stk. Overexpression of Ron and its splice variants has been implicated in the progression of multiple cancers (8, 15, 61). Like Sf-Stk, a 55-kDa N-terminally truncated form of Ron (Sf-Ron) is generated from an internal promoter within the Ron locus. Sf-Ron lacks most of the extracellular domain, while the transmembrane and cytoplasmic domains remain intact. Expression of Sf-Ron has been observed in both normal human tissues and malignant human tissues such as ovarian and breast cancer tumors. Overexpression of Sf-Ron demonstrated intrinsic kinase activity, and a T47D breast cancer cell line expressing Sf-Ron exhibited faster growth and motility (4). Sf-Ron shares structural similarities with Sf-Stk, including the presence of three cysteines (Cys4, Cys22, and Cys27) located in the extracellular domain of Sf-Ron. To determine whether Sf-Ron can interact with gp55 in a manner dependent on the cysteines in the extracellular domain, all three cysteines were mutated to alanine in Sf-Ron (Sf-RonC3A). Sf-Ron or Sf-RonC3A was cotransfected with gp55 in 293 cells, and the interaction of gp55 and Sf-Ron was assessed by coimmunoprecipitation (Fig. 10A). As expected, Sf-Ron, but not Sf-RonC3A, coimmunoprecipitated with gp55. The tyrosine phosphorylation of Sf-Ron and Sf-RonC3A was also tested by using the anti-phospho-Ron Tyr1238/1239 antibody (Fig. 10B). Coexpression of Sf-Ron with gp55 significantly enhanced Sf-Ron tyrosine phosphorylation; however, Sf-RonC3A exhibited elevated levels of tyrosine phosphorylation, and gp55 did not enhance the tyrosine phosphorylation of Sf-RonC3A. We further tested whether Sf-Ron can induce Epoind erythroblast growth in bone marrow cells from C57BL/6 Fv2r/r mice (Fig. 10C). As we observed with Sf-StkM330T, Sf-Ron promotes Epoind colony formation even in the absence of gp55, and this response requires the cysteines in the extracellular domain of Sf-Ron. This conservation in function between the murine and human receptors suggests that the cysteines in the extracellular domain of Sf-Stk and Sf-Ron likely play important roles in regulating their normal physiologic functions.
FIG. 10.

The cysteine residues in the extracellular domain of Sf-RON are required for gp55 interaction and Epoind BFU-E colony formation. (A) 293 cells were transfected with wild-type gp55 and myc-tagged wild-type or mutated Sf-RON. Cell lysates were immunoprecipitated with anti-myc antibody and blotted with anti-gp55 antiserum. (B) 293 cells were transfected with gp55 and myc-tagged wild-type or mutated Sf-RON. Cell lysates were blotted with anti-phospho-RON tyrosine 1238/1239 antibody. The membrane was stripped and then blotted with anti-myc antibody. (C) 293 cells were transfected with wild-type or mutated MSCV-myc-Sf-RON and pEco. At 48 h posttransfection, viral supernatants were collected and incubated overnight with bone marrow cells of C57BL/6 mice. The infected bone marrow cells were plated in methylcellulose medium containing IL-3 (2.5 ng/ml) or incubated with FVP on ice for 1 h prior to plating in methylcellulose medium containing IL-3 (2.5 ng/ml). On day 5, BFU-E colonies were stained with acid-benzidine and counted. *, P < 0.01. Error bars indicate standard deviations.
DISCUSSION
The predominance of a transcript encoding an N-terminally truncated form of the Stk receptor tyrosine kinase, compared with the full-length transcript, in cells of hematopoietic origin has long been observed (25). However, the function of Sf-Stk remained unexplored until its identification as the product of the Fv2 gene, which is responsible for restriction of Friend virus susceptibility in C57BL/6 mice (48). It has since become clear that Friend virus-induced erythroleukemia is initiated by the ability of the SFFV-encoded viral glycoprotein gp55 to interact with the Epo receptor and Sf-Stk, expressed by host hematopoietic cells. However, it has remained unclear how the interaction between gp55 and Sf-Stk results in the activation of Sf-Stk signaling. The studies described here shed light on the mechanism by which gp55 promotes the activation of Sf-Stk and may further provide clues to the mechanism by which Sf-Stk activity is regulated in uninfected cells.
Here we demonstrate that the covalent interaction between gp55 and Sf-Stk is regulated through four cysteines located in the ecotropic domain of gp55 and four cysteines located in the extracellular domain of Sf-Stk. Our data further demonstrate that the cysteines in the ecotropic domain also mediate gp55 oligomerization, confirming previous studies showing that gp55 oligomerization is mediated by intermolecular disulfide bond formation (68). However, not all four cysteines are required for gp55 oligomerization, as demonstrated by the ability of the double cysteine-to-alanine mutants, gp55C306,309A and gp55C337,338A, to form dimers. This flexibility in the ability of alternate cysteines to promote gp55 oligomerization likely provides a means by which cysteines in gp55 are also free to form hetero-oligomers in the presence of Sf-Stk via disulfide bond formation. However, while gp55C306,309A and gp55C337,338A interact with Sf-Stk to a similar extent as wild-type gp55, we observed a much lower level of Sf-Stk tyrosine phosphorylation in these complexes than in wild-type gp55. These experiments suggest that through intermolecular disulfide bond formation, the cysteines in the ecotropic domain of gp55 could form molecular clusters of gp55 and Sf-Stk, which further enhance Sf-Stk phosphorylation. While gp55 has also been shown to interact with the EpoR through the dualtropic and transmembrane domains (11, 13, 14), our data showed that the gp55C4A mutant still effectively interacts with the EpoR, supporting a model in which gp55 could simultaneously form complexes with both EpoR and Sf-Stk molecules.
There is increasing evidence that some receptor tyrosine kinases can form homo- or heterodimers in the absence of ligand and that ligand binding stabilizes the dimer and drives a conformational change in the receptor which promotes receptor activation. Using bimolecular fluorescence complementation (BiFC) assays, Tao and Maruyama demonstrated that all epidermal growth factor receptor (EGFR) family receptors form inactive ligand-independent dimers in the ER which then translocate to plasma membrane (58), and a two-hybrid assay suggests that intracellular domains are responsible for this spontaneous dimerization. Further, EpoR is found to form an inactive dimer mediated by the transmembrane domain in the absence of Epo (12). Scanning cysteine mutagenesis in EGFR and EpoR juxtamembrane and transmembrane domains demonstrated that the conformation of the receptor dimers is a critical determinant of receptor activation (34, 37). These data are consistent with our results demonstrating that Sf-Stk spontaneously dimerizes in the absence of gp55 and supporting the hypothesis that gp55 may induce a conformational change in the Sf-Stk dimers that promotes kinase activation.
Previous studies have shown that gp55 homodimerization and glycosylation are required for gp55 to be efficiently translocated to the cell surface (29, 49, 50) and that this translocation is required for pathogenesis. Our data demonstrate that Sf-Stk localizes primarily in the cytosol and that plasma membrane localization is enhanced through its association with gp55. It is reported that an N-terminally truncated EGFR cannot be expressed on the cell surface but that its cell surface localization can be rescued by coexpression of full-length EGFR (30). However, we failed to detect enhanced cell surface localization of Sf-Stk in the presence of full-length Stk (data not shown). We were surprised to find that mutation of all four cysteines in the extracellular domain of Sf-Stk resulted in enhanced cell surface localization of Sf-Stk in the absence of gp55, suggesting that these cysteines may play a role in subcellular trafficking of Sf-Stk, either by promoting the interaction of Sf-Stk with other cellular proteins or by promoting inhibitory intramolecular interactions. Binding of gp55 to these sites could displace these inhibitory interactions, thus promoting cell surface localization of Sf-Stk.
A constitutively activating point mutation in the tyrosine kinase domain of Ret (RetM918T) was first observed in patients with multiple endocrine neoplasia type 2B (MEN 2B) (9, 23). The analogous mutation was later shown to result in the constitutive activation of Met and Ron (MetM1268T, RonM1254T) (26, 63). Generation and expression of the M1231T mutation in Stk (StkM1231T) resulted in enhanced receptor phosphorylation and increased metastasis compared with wild-type Stk (45), and an Sf-StkM330T mutant was reported to promote Epoind colony formation in the absence of gp55 (52). Our data support this report and further demonstrate that the cysteines in the extracellular domain of Sf-Stk are critical for the ability of Sf-StkM330T to promote Epoind colony growth in the absence of gp55. Further, we demonstrate that Sf-Ron can also induce Epoind colony formation in Fv2r/r mice in the absence of gp55 and that this response also requires the cysteines in the extracellular domain of Sf-Ron. This is consistent with previous research demonstrating that Sf-Ron exhibits constitutive tyrosine kinase activity. The conservation in the functions of the murine and human receptors highlights the possibility that Sf-Stk and Sf-Ron may play important roles under normal physiologic conditions.
It is of interest to note that while Sf-RonC3A and Sf-StkM330TC4A are highly phosphorylated, they fail to induce Epoind colony formation in the absence of gp55. These results underscore the possibility that other cellular signals interact with Sf-Stk and Sf-Ron through these cysteines, resulting in the ability of Sf-Stk and Sf-Ron to promote rapid erythroblast expansion. We have shown previously that Friend virus hijacks the BMP4-dependent stress erythropoiesis pathway(55, 56). It is therefore plausible that Sf-Stk or Sf-Ron could promote expansive erythropoiesis during times of acute erythropoietic need, and this response could be mediated, in part, by the cysteines in the extracellular domain of Sf-Stk or Sf-Ron with host proteins. These studies highlight the importance of understanding how Sf-Stk activity is regulated, not only by gp55 but also by other cellular factors. The studies described here will lay the foundation for addressing this important biological question.
Acknowledgments
This work was funded by National Institutes of Health grant R01 HL066471.
Footnotes
Published ahead of print on 16 December 2009.
REFERENCES
- 1.Adachi, A., K. Sakai, N. Kitamura, S. Nakanishi, O. Niwa, M. Matsuyama, and A. Ishimoto. 1984. Characterization of the env gene and long terminal repeat of molecularly cloned Friend mink cell focus-inducing virus DNA. J. Virol. 50:813-821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aizawa, S., Y. Suda, Y. Furuta, T. Yagi, N. Takeda, N. Watanabe, M. Nagayoshi, and Y. Ikawa. 1990. Env-derived gp55 gene of Friend spleen focus-forming virus specifically induces neoplastic proliferation of erythroid progenitor cells. EMBO J. 9:2107-2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amanuma, H., A. Katori, M. Obata, N. Sagata, and Y. Ikawa. 1983. Complete nucleotide sequence of the gene for the specific glycoprotein (gp55) of Friend spleen focus-forming virus. Proc. Natl. Acad. Sci. U. S. A. 80:3913-3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bardella, C., B. Costa, P. Maggiora, S. Patane, M. Olivero, G. N. Ranzani, M. De Bortoli, P. M. Comoglio, and M. F. Di Renzo. 2004. Truncated RON tyrosine kinase drives tumor cell progression and abrogates cell-cell adhesion through E-cadherin transcriptional repression. Cancer Res. 64:5154-5161. [DOI] [PubMed] [Google Scholar]
- 5.Ben-David, Y., and A. Bernstein. 1991. Friend virus-induced erythroleukemia and the multistage nature of cancer. Cell 66:831-834. [DOI] [PubMed] [Google Scholar]
- 6.Ben-David, Y., V. R. Prideaux, V. Chow, S. Benchimol, and A. Bernstein. 1988. Inactivation of the p53 oncogene by internal deletion or retroviral integration in erythroleukemic cell lines induced by Friend leukemia virus. Oncogene 3:179-185. [PubMed] [Google Scholar]
- 7.Berger, S. A., N. Sanderson, A. Bernstein, and W. D. Hankins. 1985. Induction of the early stages of Friend erythroleukemia with helper-free Friend spleen focus-forming virus. Proc. Natl. Acad. Sci. U. S. A. 82:6913-6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Camp, E. R., W. Liu, F. Fan, A. Yang, R. Somcio, and L. M. Ellis. 2005. RON, a tyrosine kinase receptor involved in tumor progression and metastasis. Ann. Surg. Oncol. 12:273-281. [DOI] [PubMed] [Google Scholar]
- 9.Carlson, K. M., S. Dou, D. Chi, N. Scavarda, K. Toshima, C. E. Jackson, S. A. Wells, Jr., P. J. Goodfellow, and H. Donis-Keller. 1994. Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc. Natl. Acad. Sci. U. S. A. 91:1579-1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark, S. P., and T. W. Mak. 1983. Complete nucleotide sequence of an infectious clone of Friend spleen focus-forming provirus: gp55 is an envelope fusion glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 80:5037-5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Constantinescu, S. N., T. Keren, W. P. Russ, I. Ubarretxena-Belandia, Y. Malka, K. F. Kubatzky, D. M. Engelman, H. F. Lodish, and Y. I. Henis. 2003. The erythropoietin receptor transmembrane domain mediates complex formation with viral anemic and polycythemic gp55 proteins. J. Biol. Chem. 278:43755-43763. [DOI] [PubMed] [Google Scholar]
- 12.Constantinescu, S. N., T. Keren, M. Socolovsky, H. Nam, Y. I. Henis, and H. F. Lodish. 2001. Ligand-independent oligomerization of cell-surface erythropoietin receptor is mediated by the transmembrane domain. Proc. Natl. Acad. Sci. U. S. A. 98:4379-4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Constantinescu, S. N., X. Liu, W. Beyer, A. Fallon, S. Shekar, Y. I. Henis, S. O. Smith, and H. F. Lodish. 1999. Activation of the erythropoietin receptor by the gp55-P viral envelope protein is determined by a single amino acid in its transmembrane domain. EMBO J. 18:3334-3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D'Andrea, A. D. 1992. The interaction of the erythropoietin receptor and gp55. Cancer Surv. 15:19-36. [PubMed] [Google Scholar]
- 15.Eckerich, C., A. Schulte, T. Martens, S. Zapf, M. Westphal, and K. Lamszus. 2009. RON receptor tyrosine kinase in human gliomas: expression, function, and identification of a novel soluble splice variant. J. Neurochem. 109:969-980. [DOI] [PubMed] [Google Scholar]
- 16.Fang, C., E. Choi, L. Nie, and J. P. Li. 1998. Role of the transmembrane sequence of spleen focus-forming virus gp55 in erythroleukemogenesis. Virology 252:46-53. [DOI] [PubMed] [Google Scholar]
- 17.Ferro, F. E., Jr., S. L. Kozak, M. E. Hoatlin, and D. Kabat. 1993. Cell surface site for mitogenic interaction of erythropoietin receptors with the membrane glycoprotein encoded by Friend erythroleukemia virus. J. Biol. Chem. 268:5741-5747. [PubMed] [Google Scholar]
- 18.Finkelstein, L. D., P. A. Ney, Q. P. Liu, R. F. Paulson, and P. H. Correll. 2002. Sf-Stk kinase activity and the Grb2 binding site are required for Epo-independent growth of primary erythroblasts infected with Friend virus. Oncogene 21:3562-3570. [DOI] [PubMed] [Google Scholar]
- 19.Friend, C. 1957. Cell-free transmission in adult Swiss mice of a disease having the character of a leukemia. J. Exp. Med. 105:307-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gliniak, B. C., and D. Kabat. 1989. Leukemogenic membrane glycoprotein encoded by Friend spleen focus-forming virus: transport to cell surfaces and shedding are controlled by disulfide-bonded dimerization and by cleavage of a hydrophobic membrane anchor. J. Virol. 63:3561-3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gliniak, B. C., S. L. Kozak, R. T. Jones, and D. Kabat. 1991. Disulfide bonding controls the processing of retroviral envelope glycoproteins. J. Biol. Chem. 266:22991-22997. [PubMed] [Google Scholar]
- 22.Hegde, S., S. Ni, S. He, D. Yoon, G. S. Feng, S. S. Watowich, R. F. Paulson, and P. A. Hankey. 2009. Stat3 promotes the development of erythroleukemia by inducing Pu.1 expression and inhibiting erythroid differentiation. Oncogene 28:3349-3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hofstra, R. M., R. M. Landsvater, I. Ceccherini, R. P. Stulp, T. Stelwagen, Y. Luo, B. Pasini, J. W. Hoppener, H. K. van Amstel, G. Romeo, et al. 1994. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature 367:375-376. [DOI] [PubMed] [Google Scholar]
- 24.Hu, C. D., Y. Chinenov, and T. K. Kerppola. 2002. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9:789-798. [DOI] [PubMed] [Google Scholar]
- 25.Iwama, A., K. Okano, T. Sudo, Y. Matsuda, and T. Suda. 1994. Molecular cloning of a novel receptor tyrosine kinase gene, STK, derived from enriched hematopoietic stem cells. Blood 83:3160-3169. [PubMed] [Google Scholar]
- 26.Jeffers, M., L. Schmidt, N. Nakaigawa, C. P. Webb, G. Weirich, T. Kishida, B. Zbar, and G. F. Vande Woude. 1997. Activating mutations for the met tyrosine kinase receptor in human cancer. Proc. Natl. Acad. Sci. U. S. A. 94:11445-11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jelacic, T. M., D. Thompson, C. Hanson, J. L. Cmarik, K. Nishigaki, and S. Ruscetti. 2008. The tyrosine kinase sf-Stk and its downstream signals are required for maintenance of Friend spleen focus-forming virus-induced fibroblast transformation. J. Virol. 82:419-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson, P., and S. Benchimol. 1992. Friend virus induced murine erythroleukaemia: the p53 locus. Cancer Surv. 12:137-151. [PubMed] [Google Scholar]
- 29.Kilpatrick, D. R., R. V. Srinivas, and R. W. Compans. 1989. The spleen focus-forming virus envelope glycoprotein is defective in oligomerization. J. Biol. Chem. 264:10732-10737. [PubMed] [Google Scholar]
- 30.Lax, I., F. Bellot, A. M. Honegger, A. Schmidt, A. Ullrich, D. Givol, and J. Schlessinger. 1990. Domain deletion in the extracellular portion of the EGF-receptor reduces ligand binding and impairs cell surface expression. Cell Regul. 1:173-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li, J. P., H. O. Hu, Q. T. Niu, and C. Fang. 1995. Cell surface activation of the erythropoietin receptor by Friend spleen focus-forming virus gp55. J. Virol. 69:1714-1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Linder, M., D. Linder, J. Hahnen, H. H. Schott, and S. Stirm. 1992. Localization of the intrachain disulfide bonds of the envelope glycoprotein 71 from Friend murine leukemia virus. Eur. J. Biochem. 203:65-73. [DOI] [PubMed] [Google Scholar]
- 33.Linder, M., V. Wenzel, D. Linder, and S. Stirm. 1994. Structural elements in glycoprotein 70 from polytropic Friend mink cell focus-inducing virus and glycoprotein 71 from ecotropic Friend murine leukemia virus, as defined by disulfide-bonding pattern and limited proteolysis. J. Virol. 68:5133-5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu, X., A. W. Gross, and H. F. Lodish. 2006. Active conformation of the erythropoietin receptor: random and cysteine-scanning mutagenesis of the extracellular juxtamembrane and transmembrane domains. J. Biol. Chem. 281:7002-7011. [DOI] [PubMed] [Google Scholar]
- 35.Mager, D. L., and A. Bernstein. 1985. Induction of clonogenic and erythroleukemic cells by different helper virus pseudotypes of Friend spleen focus-forming virus. Virology 141:337-341. [DOI] [PubMed] [Google Scholar]
- 36.Moreau-Gachelin, F., A. Tavitian, and P. Tambourin. 1988. Spi-1 is a putative oncogene in virally induced murine erythroleukaemias. Nature 331:277-280. [DOI] [PubMed] [Google Scholar]
- 37.Moriki, T., H. Maruyama, and I. N. Maruyama. 2001. Activation of preformed EGF receptor dimers by ligand-induced rotation of the transmembrane domain. J. Mol. Biol. 311:1011-1026. [DOI] [PubMed] [Google Scholar]
- 38.Mowat, M., A. Cheng, N. Kimura, A. Bernstein, and S. Benchimol. 1985. Rearrangements of the cellular p53 gene in erythroleukaemic cells transformed by Friend virus. Nature 314:633-636. [DOI] [PubMed] [Google Scholar]
- 39.Munroe, D. G., B. Rovinski, A. Bernstein, and S. Benchimol. 1988. Loss of a highly conserved domain on p53 as a result of gene deletion during Friend virus-induced erythroleukemia. Oncogene 2:621-624. [PubMed] [Google Scholar]
- 40.Nishigaki, K., C. Hanson, T. Jelacic, D. Thompson, and S. Ruscetti. 2005. Friend spleen focus-forming virus transforms rodent fibroblasts in cooperation with a short form of the receptor tyrosine kinase Stk. Proc. Natl. Acad. Sci. U. S. A. 102:15488-15493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nishigaki, K., D. Thompson, C. Hanson, T. Yugawa, and S. Ruscetti. 2001. The envelope glycoprotein of Friend spleen focus-forming virus covalently interacts with and constitutively activates a truncated form of the receptor tyrosine kinase Stk. J. Virol. 75:7893-7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okeoma, C. M., J. Petersen, and S. R. Ross. 2009. Expression of murine APOBEC3 alleles in different mouse strains and their effect on mouse mammary tumor virus infection. J. Virol. 83:3029-3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paul, R., S. Schuetze, S. L. Kozak, and D. Kabat. 1989. A common site for immortalizing proviral integrations in Friend erythroleukemia: molecular cloning and characterization. J. Virol. 63:4958-4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paul, R., S. Schuetze, S. L. Kozak, C. A. Kozak, and D. Kabat. 1991. The Sfpi-1 proviral integration site of Friend erythroleukemia encodes the ets-related transcription factor Pu.1. J. Virol. 65:464-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peace, B. E., M. J. Hughes, S. J. Degen, and S. E. Waltz. 2001. Point mutations and overexpression of Ron induce transformation, tumor formation, and metastasis. Oncogene 20:6142-6151. [DOI] [PubMed] [Google Scholar]
- 46.Perry, J. M., O. F. Harandi, and R. F. Paulson. 2007. BMP4, SCF, and hypoxia cooperatively regulate the expansion of murine stress erythroid progenitors. Blood 109:4494-4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perry, J. M., O. F. Harandi, P. Porayette, S. Hegde, A. K. Kannan, and R. F. Paulson. 2009. Maintenance of the BMP4-dependent stress erythropoiesis pathway in the murine spleen requires hedgehog signaling. Blood 113:911-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Persons, D. A., R. F. Paulson, M. R. Loyd, M. T. Herley, S. M. Bodner, A. Bernstein, P. H. Correll, and P. A. Ney. 1999. Fv2 encodes a truncated form of the Stk receptor tyrosine kinase. Nat. Genet. 23:159-165. [DOI] [PubMed] [Google Scholar]
- 49.Polonoff, E., C. A. Machida, and D. Kabat. 1982. Glycosylation and intracellular transport of membrane glycoproteins encoded by murine leukemia viruses. Inhibition by amino acid analogues and by tunicamycin. J. Biol. Chem. 257:14023-14028. [PubMed] [Google Scholar]
- 50.Rau, S., R. Geyer, and R. W. Friedrich. 1993. The role of gp55 N-glycosylation in pathogenesis of Friend spleen focus-forming virus. J. Gen. Virol. 74:699-705. [DOI] [PubMed] [Google Scholar]
- 51.Rovinski, B., D. Munroe, J. Peacock, M. Mowat, A. Bernstein, and S. Benchimol. 1987. Deletion of 5′-coding sequences of the cellular p53 gene in mouse erythroleukemia: a novel mechanism of oncogene regulation. Mol. Cell. Biol. 7:847-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rulli, K., T. Yugawa, C. Hanson, D. Thompson, S. Ruscetti, and K. Nishigaki. 2004. Ex vivo and in vivo biological effects of a truncated form of the receptor tyrosine kinase Stk when activated by interaction with the Friend spleen focus-forming virus envelope glycoprotein or by point mutation. J. Virol. 78:4573-4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santiago, M. L., M. Montano, R. Benitez, R. J. Messer, W. Yonemoto, B. Chesebro, K. J. Hasenkrug, and W. C. Greene. 2008. Apobec3 encodes Rfv3, a gene influencing neutralizing antibody control of retrovirus infection. Science 321:1343-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schlessinger, J. 2000. Cell signaling by receptor tyrosine kinases. Cell 103:211-225. [DOI] [PubMed] [Google Scholar]
- 55.Subramanian, A., S. Hegde, P. Porayette, M. Yon, P. Hankey, and R. F. Paulson. 2008. Friend virus utilizes the BMP4-dependent stress erythropoiesis pathway to induce erythroleukemia. J. Virol. 82:382-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Subramanian, A., H. E. Teal, P. H. Correll, and R. F. Paulson. 2005. Resistance to Friend virus-induced erythroleukemia in W/W(v) mice is caused by a spleen-specific defect which results in a severe reduction in target cells and a lack of Sf-Stk expression. J. Virol. 79:14586-14594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takeda, E., S. Tsuji-Kawahara, M. Sakamoto, M. A. Langlois, M. S. Neuberger, C. Rada, and M. Miyazawa. 2008. Mouse APOBEC3 restricts Friend leukemia virus infection and pathogenesis in vivo. J. Virol. 82:10998-11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tao, R. H., and I. N. Maruyama. 2008. All EGF(ErbB) receptors have preformed homo- and heterodimeric structures in living cells. J. Cell Sci. 121:3207-3217. [DOI] [PubMed] [Google Scholar]
- 59.Taylor, G. M., Y. Gao, and D. A. Sanders. 2001. Fv-4: identification of the defect in Env and the mechanism of resistance to ecotropic murine leukemia virus. J. Virol. 75:11244-11248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taylor, W. R., and J. P. Stoye. 2004. Consensus structural models for the amino terminal domain of the retrovirus restriction gene Fv1 and the murine leukaemia virus capsid proteins. BMC Struct. Biol. 4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang, M. H., W. Lee, Y. L. Luo, M. T. Weis, and H. P. Yao. 2007. Altered expression of the RON receptor tyrosine kinase in various epithelial cancers and its contribution to tumourigenic phenotypes in thyroid cancer cells. J. Pathol. 213:402-411. [DOI] [PubMed] [Google Scholar]
- 62.Watanabe, N., M. Nishi, Y. Ikawa, and H. Amanuma. 1991. Conversion of Friend mink cell focus-forming virus to Friend spleen focus-forming virus by modification of the 3′ half of the env gene. J. Virol. 65:132-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Williams, T. A., P. Longati, L. Pugliese, P. Gual, A. Bardelli, and P. Michieli. 1999. MET(PRC) mutations in the Ron receptor result in upregulation of tyrosine kinase activity and acquisition of oncogenic potential. J. Cell. Physiol. 181:507-514. [DOI] [PubMed] [Google Scholar]
- 64.Wolff, L., and S. Ruscetti. 1985. Malignant transformation of erythroid cells in vivo by introduction of a nonreplicating retrovirus vector. Science 228:1549-1552. [DOI] [PubMed] [Google Scholar]
- 65.Wolff, L., and S. Ruscetti. 1988. The spleen focus-forming virus (SFFV) envelope gene, when introduced into mice in the absence of other SFFV genes, induces acute erythroleukemia. J. Virol. 62:2158-2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wolff, L., E. Scolnick, and S. Ruscetti. 1983. Envelope gene of the Friend spleen focus-forming virus: deletion and insertions in 3′ gp70/p15E-encoding region have resulted in unique features in the primary structure of its protein product. Proc. Natl. Acad. Sci. U. S. A. 80:4718-4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wolff, L., P. Tambourin, and S. Ruscetti. 1986. Induction of the autonomous stage of transformation in erythroid cells infected with SFFV: helper virus is not required. Virology 152:272-276. [DOI] [PubMed] [Google Scholar]
- 68.Yang, Y. Y., A. Tojo, N. Watanabe, and H. Amanuma. 1990. Oligomerization of Friend spleen focus-forming virus (SFFV) env glycoproteins. Virology 177:312-316. [DOI] [PubMed] [Google Scholar]
- 69.Yoshimura, A., A. D. D'Andrea, and H. F. Lodish. 1990. Friend spleen focus-forming virus glycoprotein gp55 interacts with the erythropoietin receptor in the endoplasmic reticulum and affects receptor metabolism. Proc. Natl. Acad. Sci. U. S. A. 87:4139-4143. [DOI] [PMC free article] [PubMed] [Google Scholar]