

Abstract
Interferon (IFN) signaling is initiated by the recognition of viral components by host pattern recognition receptors. Dengue virus (DEN) triggers IFN-β induction through a molecular mechanism involving the cellular RIG-I/MAVS signaling pathway. Here we report that the MAVS protein level is reduced in DEN-infected cells and that caspase-1 and caspase-3 cleave MAVS at residue D429. In addition to its well-known function in IFN induction, MAVS is also a proapoptotic molecule that triggers disruption of the mitochondrial membrane potential and activation of caspases. Although different domains are required for the induction of cytotoxicity and IFN, caspase cleavage at residue 429 abolished both functions of MAVS. The apoptotic role of MAVS in viral infection and double-stranded RNA (dsRNA) stimulation was demonstrated in cells with reduced endogenous MAVS expression induced by RNA interference. Even though IFN-β promoter activation was largely suppressed, DEN production was not affected greatly in MAVS knockdown cells. Instead, DEN- and dsRNA-induced cell death and caspase activation were delayed and attenuated in the cells with reduced levels of MAVS. These results reveal a new role of MAVS in the regulation of cell death beyond its well-known function of IFN induction in antiviral innate immunity.
In the battle of hosts and microbes, the innate immune system uses pathogen recognition receptors (PRRs) to sense pathogen-associated molecular patterns (23). There are several functionally distinct classes of PRRs, such as the transmembrane (TM) Toll-like receptors (TLRs) and the intracellular retinoic acid-inducible gene I (RIG-I)-like helicase (RLH) receptors (15, 23, 25, 38). RLHs, including RIG-I and melanoma differentiation-associated gene 5 (MDA5), comprise an N-terminal caspase recruitment domain (CARD), a middle DEXD/H box RNA helicase domain, and a C-terminal domain. RLHs sense intracellular viral RNA and initiate an antiviral interferon (IFN) response (1, 43). RIG-I binding to viral RNA triggers conformational changes that expose the CARD for subsequent signaling (42). The adaptor molecule providing a link between RIG-I and downstream events was identified independently by four research groups as a mitochondrial CARD-containing protein, which was named mitochondrial antiviral signaling protein (MAVS) (34), IFN-β promoter stimulator 1 (IPS-1) (12), virus-induced signaling adaptor (VISA) (40), and CARD adaptor-inducing IFN-β (Cardif) (24). We refer to this adaptor as MAVS in this paper. MAVS transduces signals from RIG-I through CARD-CARD interactions, which then lead to interferon regulatory factor 3 (IRF-3) and NF-κB activation of IFN-β induction through a signaling cascade involving IKKα/β/γ, IKKɛ, and TBK1 (15). Recently, a protein termed STING (11) or MITA (47) was identified as a mediator that acts downstream of RIG-I and MAVS and upstream of TBK1.
MAVS protein contains an N-terminal CARD required for signaling, a proline-rich domain that interacts with TRAF3, and a C-terminal TM region that targets MAVS to the mitochondrial outer membrane (29). Several cellular and viral proteins target MAVS in the attenuation of the IFN induction pathway. Cleavage of MAVS by hepatitis C virus (HCV) and hepatitis A virus (HAV) proteases, at residues C508 (18, 24) and Q428 (41), respectively, results in the loss of MAVS mitochondrial localization, thereby disrupting its function in IFN induction. Another mitochondrial outer membrane protein, NLRX1, can sequester MAVS from its association with RIG-I and act as a negative regulator of the IFN pathway (28). MAVS was recently found to be cleaved and inactivated by caspases during apoptosis (31, 33).
The caspases are a well-known family of cysteinyl aspartate-specific proteases. The diverse roles of caspases in the cell cycle, proliferation, differentiation, cytokine production, innate immune regulation, and microbial infection suggest various functions of caspases beyond apoptosis (13, 14). The caspases can be separated into two subfamilies, namely, the cell death and inflammation subfamilies. In response to apoptotic stimuli, the initiators caspase-2, -8, -9, and -10 and effectors caspase-3, -6, and -7 mediate cell death events. Caspase-1, -4, -5, and -12 are known as the inflammatory caspases. Caspase-1 is involved in the cleavage and maturation of cytokines (8, 17). Caspase-8 and -10 were discovered as essential components that mediate antiviral signaling (37). Caspase-1 and -3 are activated in innate immune signaling (32). These findings indicate that caspases are involved in the regulation of innate immunity, in addition to their well-known apoptotic role. However, the details of how caspases are activated, the role of caspase activation, and how caspases manipulate the signaling pathways in innate immunity are still obscure.
The family Flaviviridae contains three genera: Hepacivirus, Flavivirus, and Pestivirus. Infections with flaviviruses, such as dengue virus (DEN), Japanese encephalitis virus, and West Nile virus, are emerging worldwide. DEN triggers IFN-β through a molecular mechanism involving the RIG-I/MAVS signaling pathway (5, 20). In this study, we found that MAVS is cleaved during DEN serotype 2 (DEN-2) infection, in a caspase-dependent manner; this contrasts with viral protease-dependent cleavage of MAVS during infection with HCV and HAV. In a cell-free caspase assay system, MAVS was cleaved at residue D429 by caspase-1 and caspase-3. Cleaved MAVS failed to induce IFN production and caspase activation, and overexpression of MAVS also triggered caspase activation, which then negatively regulated its own function. Importantly, the role of MAVS in viral infection was verified by knockdown of MAVS expression. We discuss the possible regulatory mechanisms of MAVS and the biological significance of this cleavage event by caspases in the context of understanding how these apoptosis-related proteases might achieve cross talk with the innate immune pathway during viral infection.
MATERIALS AND METHODS
Viruses, cells, and chemicals.
Virus propagation and titration of the DEN-2 PL046 strain were performed as described previously (45). Lentivirus was prepared following the protocol of the National RNAi Core Facility (Academia Sinica, Taipei, Taiwan). N18, a mouse neuroblastoma cell line, was cultured in RPMI 1640 medium supplemented with 5% fetal bovine serum (FBS; Gibco). 293FT cells, obtained from Invitrogen for high-titer lentivirus production, were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS. A549, a human lung carcinoma cell line, was cultured in F-12 medium supplemented with 10% FBS. GeneJammer transfection reagent (Stratagene) was used according to the manufacturer's instructions. Lipofectamine 2000 (Invitrogen) was used for delivery of poly(I)-poly(C) (GE Healthcare). The caspase inhibitors z-VAD-fmk, z-YVAD-fmk, and z-DEVD-fmk were purchased from Calbiochem.
Plasmids.
Flag-MAVS/pcDNA3 was kindly provided by Zhijian James Chen (34). The Bcl-xL and Bcl-xS genes (35) were cloned into Flag/pcDNA3 to generate Flag-tagged bcl-xL and bcl-xS. The N-terminally deleted Δ133 IRF-3 gene (5) was subcloned into pEGFP-C2 vector (Clontech) to generate dnIRF-3-GFP. For overexpression and purification of Flag- and His-tagged MAVS, the MAVS fragment was cloned into the HindIII site of pET21b (Novagen). MAVS deletion constructs Flag-MAVS(1-508), Flag-MAVS(1-429), Flag-MAVS(87-540), and Flag-MAVS(87-429) were generated by PCR cloning, using primers with inserted stop codons at the expected positions. Single-point mutant MAVS constructs Flag-MAVS(D86A), Flag-MAVS(D429A), and Flag-MAVS(D429E) were generated by single-primer mutagenesis (22), using the primers 5′-CTGTGAGCTAGTTGATCTCGCCGCGGAAGTGGCCTCTGTCTACGAGA-3′, 5′-GTGCTGGCATCCCAGGTCGCGAGCCCGTTCTCGGGCT-3′, and 5′-GTGCTGGCATCCCAGGTAGAAAGCCCGTTCTCGGGCT-3′, respectively. All constructs were verified by DNA sequencing. The RNA interference (RNAi) lentiviral control vector pLKO.1 and those expressing short hairpin RNAs (shRNAs) targeting nucleotides (nt) 972 to 992 of MAVS (targeting sequence, 5′-CAAGTTGCCAACTAGCTCAAA-3′) and LacZ (targeting sequence, 5′-TGTTCGCATTATCCGAACCAT-3′) were obtained from the National RNAi Core Facility, Taiwan.
MAVS in vitro cleavage assay.
The active recombinant human caspase-1, -3, -8, and -9 were purchased from Chemicon and used in a reaction solution containing 50 mM HEPES, 50 mM NaCl, 0.1% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 10 mM EDTA, 5% glycerol, and 10 mM dithiothreitol (DTT) according to the manufacturer's instructions. Expression of recombinant Flag-MAVS-His was induced by isopropyl-β-d-thiogalactopyranoside (IPTG), and the protein was then purified using a HisBind kit (Novagen) according to the manufacturer's instructions. Thirty micrograms of Flag-MAVS-His and 2 U of recombinant caspase-1, -3, -8, or -9 were incubated in reaction buffer at 37°C for 1.5 h, and the reaction mixtures were separated by SDS-PAGE and analyzed by silver staining (Bio-Rad). For the cleavage of isotope-labeled MAVS, Flag-tagged MAVS and its mutants were transcribed and translated in vitro by using TNT rabbit reticulocyte lysate (Promega) supplemented with [35S]methionine (PerkinElmer), and the Flag-tagged proteins were purified by binding to Flag-Sepharose beads (Sigma-Aldrich). Ten microliters of 35S-labeled Flag-tagged protein-binding beads was incubated with 1 U of recombinant active caspase-3 in reaction buffer as described above. After 1 h of incubation at 37°C, the reaction products were eluted in sample buffer, resolved by SDS-PAGE, and analyzed by autoradiography.
Immunofluorescence assay (IFA).
Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) and then permeabilized by 0.5% Triton X-100. After blocking of the cells with skim milk in PBS with 0.1% Tween 20 (PBS-T), primary mouse anti-Flag M2 antibody (1:2,500; Sigma-Aldrich) and goat anti-mouse Alexa Fluor 488-conjugated secondary antibody (1:1,000; Molecular Probes) were added sequentially for 1 h at room temperature. Cells were examined and photographed with an inverted fluorescence microscope.
Western immunoblot analysis.
Cells were lysed with RIPA buffer (10 mM Tris, pH 7.5, 5 mM EDTA, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate) containing a cocktail of protease inhibitors (Roche). Protein samples were separated by SDS-PAGE and transferred to a nitrocellulose membrane (Hybond-C Super; Amersham). The nonspecific antibody binding sites were blocked with skim milk in PBS-T and then incubated with the following primary antibodies: anti-MAVS (1:1,000; Axxora), anti-phospho-IRF-3 (Ser396), anti-IRF-3, anti-caspase-3 (1:1,000; Cell Signaling), anti-actin (1:10,000; Chemicon), anti-His (1:1,000; Novagen), and anti-Flag M2 (1:5,000; Sigma-Aldrich). Blots were treated with horseradish peroxidase-conjugated secondary antibody (1:2,000; Jackson ImmunoResearch), and the signals were detected using enhanced chemiluminescence (ECL; Amersham Biosciences).
Enzyme activity assays.
The firefly (p125-Luc) and Renilla (pRL-TK; internal control) luciferase activities were measured using a dual-luciferase assay system (Promega). The release of the cytoplasmic enzyme lactate dehydrogenase (LDH) was measured using a cytotoxicity detection kit (Roche). To measure caspase activity, 293FT cells were seeded in black clear-bottom 96-well plates, transfected with plasmids as described above, and incubated for 48 h in serum-containing medium. Caspase activity was quantified using the Caspase-Glo 3/7, 8, and 9 assay systems (Promega) according to the manufacturer's instructions.
Flow cytometry analysis.
An annexin V-fluorescein isothiocyanate (annexin V-FITC) apoptosis detection kit (BioVision) and a MitoProbe JC-1 assay kit (Molecular Probes) were used according to the manufacturer's instructions for annexin V-propidium iodide (PI) staining and mitochondrial membrane potential (MMP) analysis, respectively. For MMP and annexin V analyses, cells were detached and resuspended in 1 ml warm medium at 1 × 106 per ml. After 15 to 30 min of labeling with JC-1 (1 μM), cells were stained with annexin V-Cy5 (BioVision) according to the manufacturer's instructions and then analyzed using a FACSCanto flow cytometer and FACSDiva software (Becton Dickinson). For MMP disruption control, CCCP (carbonyl cyanide 3-chlorophenylhydrazone) (50 μM) was added simultaneously with JC-1.
RESULTS
MAVS is cleaved by caspase-1 and caspase-3 at D429.
To investigate the role of MAVS in viral infection, we determined the protein expression pattern of MAVS in mock- and DEN-2-infected N18 cells, a mouse neuroblastoma cell line that is susceptible to DEN-2 infection and has been used for studies of DEN-2 (35, 45). In addition to the band for full-length MAVS, another major protein band of smaller molecular size was noted, and its abundance increased further during DEN-2 infection (Fig. 1A). Addition of z-VAD-fmk, a pan-caspase inhibitor, rescued full-length MAVS protein expression in both mock- and DEN-2-infected cells, suggesting that MAVS is cleaved by caspases but not by viral protease.
FIG. 1.

Caspase-1 and caspase-3 cleave MAVS at D429. (A) N18 cells were mock treated or adsorbed with DEN-2 (multiplicity of infection [MOI] of 5) and transfected with 2 μg of wild-type (WT) or D429A- or D429E-mutated Flag-MAVS. The cells were then incubated in the presence (+) or absence (−) of z-VAD-fmk (150 μM) for 30 h. Cell lysates were analyzed by immunoblotting, using the antibodies indicated at the left. (B and C) Recombinant Flag-MAVS-His protein (30 μg) was incubated with 2 U of active caspase-1, -3, -8, or -9 as described in Materials and Methods. The reaction mixtures were resolved by SDS-PAGE and analyzed by silver staining (B) and immunoblotting, using antibodies against Flag (lanes 1 to 5) and His (lanes 6 to 10), as indicated at the top (C). The background reactivity of these caspases with anti-His antibody was also demonstrated by immunoblotting in the absence of Flag-MAVS-His protein (lanes 11 to 14 in panel C). (D) The cleavage assay of in vitro-transcribed and -translated 35S-labeled MAVS and its mutants (D86A and D429A) was performed by coincubation with (+) or without (−) active caspase-3. The proteins were then separated by SDS-PAGE, and the signals were developed by autoradiography. (E) In vitro cleavage of Flag-MAVS-His protein by caspase-1 and caspase-3 was performed in the presence (+) or absence (−) of z-VAD-fmk (1 mM) and analyzed by immunoblotting using anti-Flag antibody. (F) N18 cells were transfected with Flag-tagged MAVS and treated with various caspase inhibitors, as indicated at the top, for 24 h. Cell lysates were harvested and analyzed by immunoblotting, using anti-Flag and anti-actin antibodies, as indicated at the left. (G) A549 cells were infected with DEN-2 (MOI of 5) and then treated with z-VAD-fmk (100 μM). Cell lysates were harvested 48 h after infection and analyzed by immunoblotting, using antibodies against endogenous MAVS, DEN-2 NS3, and actin, as indicated at the left. The open arrows indicate full-length MAVS, and the black arrows indicate cleaved MAVS. DMSO, dimethyl sulfoxide.
An in vitro cleavage assay was used to verify whether MAVS is cleaved by caspases and to determine which caspases are responsible for MAVS cleavage. Using a recombinant expressed MAVS protein with an N-terminal Flag tag and a C-terminal His tag purified from E. coli, we found that the full-length MAVS disappeared almost completely after incubation with caspase-3 and, to a lesser extent, with caspase-1 (Fig. 1B). The predicted caspase cleavage sites (2) indicate that DLA86D and SQV429D are the most likely cleavage motifs for caspase-3 in the MAVS protein. Western blot analysis using antibodies against the tags added at the ends of the MAVS protein showed that the N- and C-terminal fragments of MAVS produced by caspase-1 and caspase-3 cleavage are ∼56 kDa and ∼16 kDa, respectively (Fig. 1C). In agreement with a previous study (31), this suggests that D429 is the cleavage site. This notion was supported by the observation that the D429A mutant, but not the D86A mutant, became resistant to caspase-3 cleavage in vitro (Fig. 1D). In addition, the in vitro MAVS cleavage by caspase-1 and caspase-3 was blocked by z-VAD-fmk (Fig. 1E). In the transfected cells, MAVS cleavage was blocked greatly by z-VAD-fmk (pan-caspase inhibitor) and to a lesser extent by z-YVAD-fmk (caspase-1 inhibitor) and z-DEVD-fmk (caspase-3 inhibitor) (Fig. 1F), suggesting that both caspase-1 and caspase-3 cleave MAVS. These data may explain why MAVS remains cleavable in cells deficient of caspase-3, -8, or -9 (31).
To test whether D429 is the cleavage site in cells, we determined the protein expression pattern of D429A and D429E mutants of MAVS. The 55-kDa cleaved protein bands of MAVS did not appear in the MAVS-D429A/E-transfected cells (Fig. 1A), indicating that residue D429 of MAVS is responsible for MAVS cleavage in transfected cells. Although the cleaved form of MAVS was blocked by D429A/E mutation, the pan-caspase inhibitor z-VAD-fmk restored full-length MAVS protein expression to a higher level, suggesting that caspases may cleave MAVS at other positions besides D429. Alternatively, other proteases whose activities depend on caspases might also be involved in MAVS cleavage. The endogenous MAVS protein was also cleaved in DEN-2-infected A549 cells (Fig. 1G). However, to our surprise, the cleavage of endogenous MAVS was not blocked by z-VAD-fmk (Fig. 1G), suggesting that MAVS might also be cleaved by other, still unidentified proteases besides caspases.
Cleavage of MAVS at D429 abolishes its ability to activate the IFN promoter.
To determine the biological significance of MAVS cleavage by caspases, several deletion mutants of MAVS were generated (Fig. 2), and their expression was verified by Western blotting with anti-Flag antibody (Fig. 3). Consistent with the results of previous studies (12, 24, 34, 40), both the CARD and the TM domain of MAVS were required for IFN induction. That is, only the full-length MAVS constructs (wild type [WT] and D429A and D429E mutants), not those lacking the CARD (87-540 and 87-429) or the TM domain (1-508, 1-429, and 87-429), retained the ability to activate IFN-β signaling, as measured by IRF-3 phosphorylation and by p125-Luc expression (44), which comprises the IFN-β promoter and the luciferase gene reporter (Fig. 3).
FIG. 2.

Schematic diagram and summarized properties of MAVS constructs. The constructs were N-terminally Flag tagged and are numbered according to amino acid residues. The regions corresponding to the three domains of MAVS are also indicated, as follows: CARD, caspase recruitment domain; PR, proline-rich domain; and TM, transmembrane domain. The arrows indicate the HCV protease (C508) and caspase (D429) cleavage sites of MAVS. The abilities of IFN promoter activation, mitochondrial localization, and cytotoxicity induction of these MAVS constructs are also summarized.
FIG. 3.
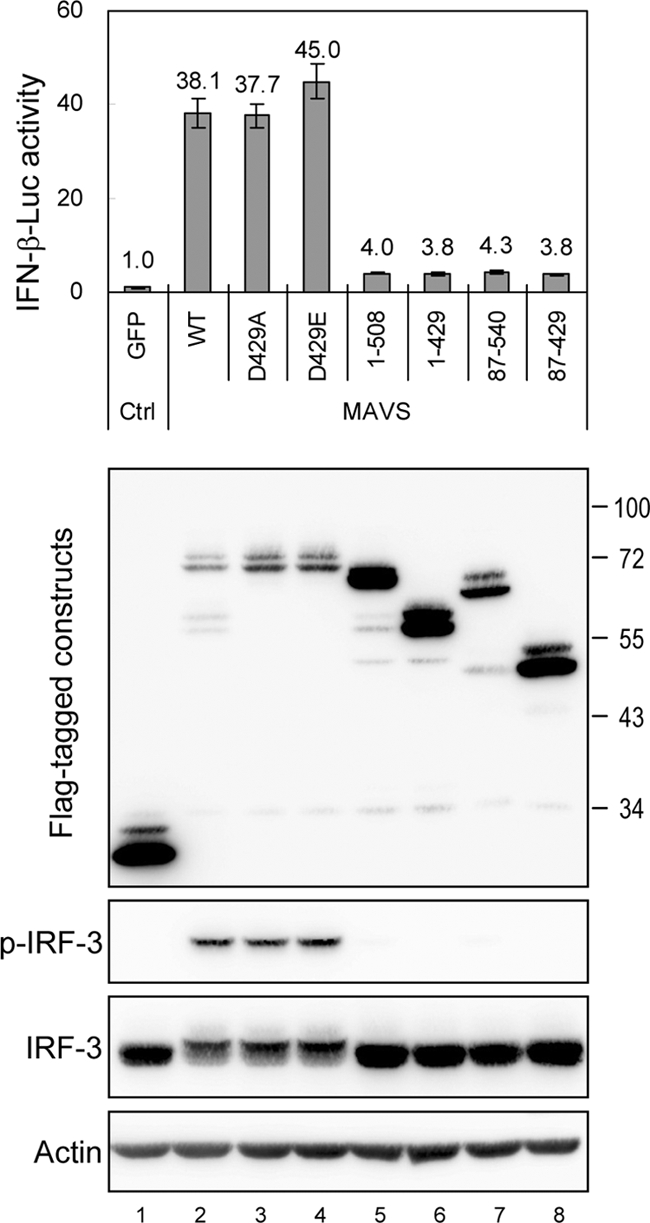
Caspase-cleaved MAVS loses the ability to activate the IFN promoter. N18 cells were cotransfected with p125-Luc (0.3 μg), IRF-3/pCR3.1 (0.3 μg), pRL-TK (0.1 μg), and the indicated MAVS constructs (0.6 μg) for 24 h, and the cell lysates were harvested and analyzed by dual-luciferase assay and by Western blotting, using antibodies against Flag, Ser396-phosphorylated IRF-3, IRF-3, and actin, as indicated at the left. Firefly luciferase activity was normalized to that of Renilla luciferase, and the change in induction relative to that of the green fluorescent protein (GFP) control was determined. The results are expressed as averages and standard deviations (n = 3 per group).
Ectopic overexpression of MAVS triggers apoptosis in a TM domain-dependent manner.
The cellular distributions of the MAVS constructs were determined using an immunofluorescence assay (Fig. 4A). In contrast to the mitochondrial patterns observed for the WT and the D429E and 87-540 mutants of MAVS, the MAVS constructs lacking the TM domain (1-508, 1-429, and 87-429) showed homogeneous staining in the cytosol, as expected. Moreover, longer incubation periods caused various levels of cytotoxicity among these transfectants (Fig. 4A). Cytotoxicity, measured by annexin V staining (Fig. 4B) and the release of the cytoplasmic enzyme LDH (Fig. 4C), also became obvious after MAVS protein expression. MAVS-induced cytotoxicity was diminished in MAVS constructs expressing residues 1 to 508, 1 to 429, and 87 to 429, which all lack the TM domain (Fig. 4A to C), suggesting that the TM domain but not the CARD of MAVS is essential for induction of cell death.
FIG. 4.

Caspase-cleaved MAVS loses the ability to trigger cell death. (A) (Top) The expression pattern of each MAVS construct in N18 cells was analyzed using an immunofluorescence assay 16 h after transfection. Green, staining with anti-Flag antibody plus Alexa Fluor 488-labeled secondary antibody; blue, nuclear 4′,6-diamidino-2-phenylindole [DAPI] staining. (Bottom) Cell morphology was also photographed by phase-contrast microscopy 30 h after transfection. (B) Twenty-four hours after transfection, the transfected cells were collected and analyzed for annexin V positivity by flow cytometry. (C) The culture supernatants were collected 30 h after transfection, and cytotoxicity was estimated by measuring LDH release. The results are expressed as averages and standard deviations (n = 2 per group). (D) MAVS protein expression and caspase-3 activation were determined in N18 transfectants by immunoblotting with antibodies against the Flag tag and caspase-3, as indicated at the left. Flag-tagged GFP was used as a protein expression control. (E) The levels of caspase-3/7 activation triggered by various MAVS constructs were determined in 293FT cells transfected with the GFP control or the indicated MAVS constructs for 48 h, using Caspase-Glo 3/7 assays. The change in induction relative to that of the GFP control was determined, and the results are expressed as averages and standard deviations (n = 3 per group).
We noted that procaspase-3 cleavage occurred in cells overexpressing MAVS (Fig. 1A). To investigate further the death-signaling trigger by MAVS, immunoblotting was used to study procaspase-3 cleavage in cells transfected with each MAVS-truncated construct. The MAVS-induced procaspase-3 cleavage was diminished in MAVS constructs expressing residues 1 to 508, 1 to 429, and 87 to 429, which all lack the TM domain (Fig. 4D). Quantification of death effector caspase-3/7 activity induced by each of these MAVS constructs in 293FT cells also showed that MAVS requires its TM domain to trigger caspase-3/7 activation (Fig. 4E). These data suggest that the TM domain but not the CARD of MAVS is essential for induction of cell death.
Overexpression of MAVS disrupts the mitochondrial membrane potential and activates caspases.
To evaluate the death signal cascade triggered by MAVS, the activities of the upstream initiation caspases were measured. Both caspase-8 and -9 were activated by MAVS overexpression (Fig. 5A). Because the mitochondrial localization of MAVS is required for caspase activation, we checked whether the integrity of the MMP was affected by MAVS. Mitochondrial depolarization was indicated by a decrease in the red-to-green fluorescence intensity ratio of JC-1 staining, as shown in the protonophore CCCP-treated but not in the mock-treated cells (Fig. 5B). Overexpression of MAVS lowered the JC-1 red/green fluorescence ratio even more than did overexpression of Bcl-xS, a proapoptotic molecule that triggers MMP disruption (7) (Fig. 5B). Treatment with z-VAD-fmk reduced the percentage of annexin V-positivity apoptotic cells from 21.6% to 12.32% in the MAVS-transfected cells (Fig. 5B). z-VAD-fmk treatment did not prevent the MAVS-induced MMP disruption (28.3% versus 43.7%), suggesting that MMP disruption occurs before caspase activation during MAVS-induced cell death.
FIG. 5.

Overexpression of MAVS triggers disruption of the mitochondrial membrane potential and caspase activation. (A) The activation of caspase-3/7, -8, and -9 by MAVS overexpression was determined in transfected 293FT cells by using Caspase-Glo 3/7, 8, and 9 assays as described in the legend to Fig. 4E (n = 3 per group). (B) N18 cells were transfected with MAVS, Bcl-xS, or vector control in the presence or absence of z-VAD-fmk (100 μM) for 24 h and then analyzed by flow cytometry, using JC-1 and annexin V double staining. A decreased red/green fluorescence ratio of JC-1 represents MMP disruption, as shown in the cells treated with CCCP (50 μM), an MMP disrupter.
We then asked whether the MAVS-triggered caspase-3 activation depends on IRF-3 activation. Overexpression of a dominant-negative mutant of IRF-3 that lacks the N-terminal DNA-binding domain (5, 19) had no effect on the MAVS-induced caspase-3 activation (Fig. 6). In contrast, Bcl-xL, an antiapoptotic mitochondrial protein, completely blocked caspase-3 activation triggered by MAVS (Fig. 6). Thus, the ability of MAVS to induce cell death correlates with its mitochondrial targeting, MMP disruption, and caspase cascade activation but not with IRF-3 activation.
FIG. 6.
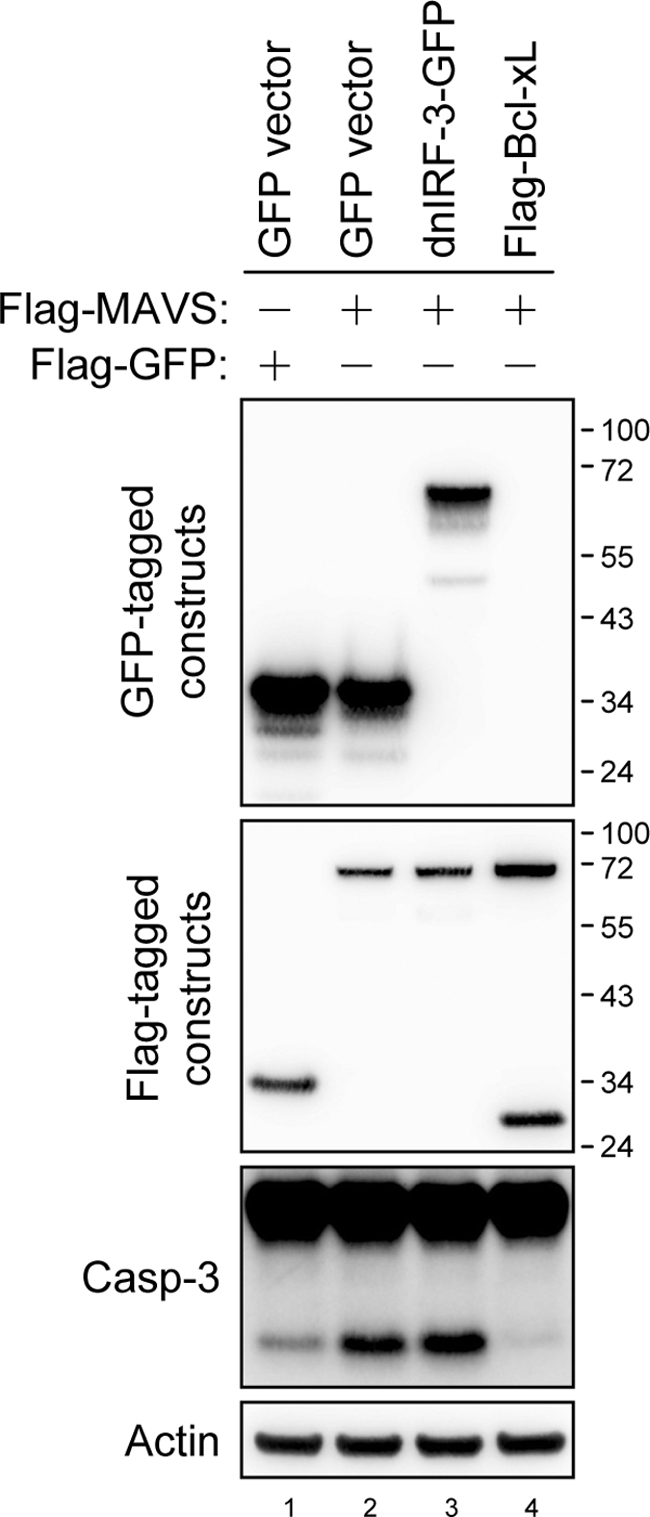
Bcl-xL but not dnIRF-3 blocks MAVS-induced caspase-3 activation. N18 cells were transfected with plasmids encoding dominant-negative IRF-3 fused with GFP, Flag-tagged Bcl-xL, GFP control (1 μg) plus Flag-MAVS, or Flag-GFP control (1 μg), as indicated at the top, for 24 h. Cell lysates were harvested for Western blot analyses with antibodies against GFP, the Flag tag, caspase-3, and actin, as indicated at the left.
Reducing MAVS expression by RNA interference attenuates virus- and dsRNA-induced cell death.
To study the biological function of MAVS in antiviral innate immunity, we used RNAi to reduce endogenous MAVS expression. Human A549 cells were transduced with a lentiviral vector expressing shRNA that targets MAVS (shMAVS), and the stably transduced cells were obtained and verified by Western blotting (Fig. 7B). The MAVS-triggered and DEN-2-induced IFN-β promoter activation, measured by the p125-Luc reporter assay and IRF-3 phosphorylation, decreased markedly in cells expressing shMAVS (Fig. 7). In addition, the levels of DEN-2-induced cell death decreased significantly in cells deprived of MAVS expression (Fig. 8A), whereas DEN-2 progeny production was not affected significantly (Fig. 8C). Attenuation of DEN-2-induced cytopathic effect (CPE) in the MAVS knockdown cells was corroborated by a delayed and less prominent procaspase-3 cleavage detected by immunoblotting (Fig. 8B). We further addressed whether depriving endogenous MAVS expression affects apoptosis induction and MMP integrity of DEN-infected cells. As shown in Fig. 9A, the percentage of annexin V-positive cells dropped from 19.39% in shLacZ control cells to 5.16% in shMAVS cells, and MMP disruption, measured by the JC-1 red/green fluorescence ratio, occurred in 31.1% of shLacZ cells and 14.97% of shMAVS cells, indicating that both apoptosis and MMP disruption triggered by DEN-2 are attenuated in cells with reduced MAVS expression. Treatment with z-VAD-fmk decreased the DEN-2-induced annexin V positivity rate in shLacZ cells, but z-VAD-fmk was impotent to block DEN-2-induced MMP disruption, suggesting that DEN-triggered MMP disruption occurs before caspase activation. Since the levels of IRF-3 phosphorylation triggered by DEN-2 infection were also lower in the cells with reduced MAVS expression (Fig. 8B), we then tested whether DEN-2-triggered apoptosis correlates with IRF-3 activation. Treatment with z-VAD-fmk readily blocked DEN-2-induced caspase-3 activation but had no effect on IRF-3 phosphorylation (Fig. 9B). The DEN-2-induced annexin V-positive rate was decreased in MAVS knockdown cells but not in cells overexpressing a dominant-negative form of IRF-3, dnIRF-3-GFP (Fig. 9C). These data suggest that during DEN-2 infection, MAVS is involved in MMP disruption and apoptosis induction independently of IRF-3 activation. Furthermore, apoptosis triggered by poly(I:C) transfection, measured by procaspase-3 cleavage (Fig. 10A) and annexin V-PI staining (Fig. 10B), was also reduced in the MAVS knockdown cells. Our results suggest that in addition to its well-described role in innate IFN immunity, MAVS is required for full induction of DEN-2- and double-stranded RNA (dsRNA)-triggered caspase activation and that MAVS plays an important role in cell death induction.
FIG. 7.

MAVS- and virus-induced IFN signaling is attenuated in MAVS knockdown cells. (A) Lentivirus-transduced A549 cells with (+) or without (−) MAVS-targeting shRNA were transfected with p125-Luc, IRF-3/pCR3.1, pRL-TK plus GFP, or MAVS as described in the legend to Fig. 3. For DEN-2-induced IFN-β promoter analysis, the cells were adsorbed with DEN-2 (MOI = 5) for 2 h and then transfected with p125-Luc (0.6 μg) and pRL-TK (0.15 μg) for 24 h. The change in IFN-β promoter activation was determined as described in the legend to Fig. 3. The IFN-β induction of the indicated groups was compared by two-tailed Student's t test. (B and C) Cells were prepared as described for panel A, and the cell lysates were analyzed by Western blotting with the antibodies indicated at the left.
FIG. 8.

DEN-2-induced cytotoxicity is attenuated by reducing endogenous MAVS protein expression. A549-shMAVS or control (shLacZ) cells were infected with DEN-2 at an MOI of 0.1 or 5. The cells and the culture supernatants were collected at the indicated times postinfection (p.i.). (A) Cells were counted for viability counts by trypan blue exclusion. The results are expressed as averages and standard deviations (n = 3). The cell viabilities of the indicated groups were compared by two-tailed Student's t test. (B) The cells were lysed for immunoblotting using the antibodies indicated at the left. The open arrows indicate full-length MAVS or procaspase-3, and the black arrows indicate cleaved MAVS or caspase-3. (C) The supernatants were used to measure virus titers by plaque-forming assays, and the results are expressed as averages and standard deviations (n = 3).
FIG. 9.

Knockdown of MAVS attenuates DEN-2-triggered MMP disruption. (A) Mock- or DEN-2-infected (MOI of 5) cells were cultured in the presence or absence of z-VAD-fmk (100 μM) for 48 h and then analyzed by flow cytometry for JC-1 and annexin V double staining, as described in the legend to Fig. 5B. (B) The cell lysates described for Fig. 1G were analyzed with the antibodies indicated at the left. (C) Control (shLacZ) and MAVS knockdown (shMAVS) cells were mock infected or infected with DEN-2 (MOI of 5) and transfected with dnIRF-3-GFP or control GFP vector. Cells were harvested at 48 h p.i. and stained with Cy5-conjugated annexin V. GFP-positive cells were gated and analyzed for annexin V positivity by flow cytometry.
FIG. 10.

MAVS facilitates cytosolic poly(I:C)-triggered cell death. (A) The synthetic dsRNA analog poly(I:C) was delivered to A549-shMAVS or A549-shLacZ cells at the doses indicated at the top, and the cells were harvested 24 h after transfection. Endogenous MAVS protein expression and caspase-3 activation were determined by immunoblotting with antibodies against MAVS and caspase-3, respectively. The open arrows indicate full-length MAVS or procaspase-3, and the black arrows indicate cleaved MAVS or caspase-3. Immunoblotting with anti-actin is shown as a protein loading control. (B) The transfected cells were also collected for cell death analysis by flow cytometry of annexin V and PI double staining.
DISCUSSION
Besides its well-described role in IFN induction, MAVS may also be involved in cell death signaling during viral infection. IFN is secreted by virus-infected cells and protects the adjacent cells by binding to cell surface receptors and triggering JAK-STAT signaling and other antiviral processes (21). Destruction of the infected cells after IFN production would be beneficial to the host's ability to eliminate viral infection. Induction of both IFN and apoptosis by MAVS is a double indemnity that conditions neighbor cells to an antiviral state and also kills the virus-producing cells.
An apoptotic role of the RIG-I/MAVS pathway was recently suggested. Overexpression of RIG-I and MAVS activates caspase-3 and induces interleukin-18 (IL-18) processing in HaCaT keratinocytes (32). Transfection of MAVS induces death of HEK293 cells (16), and activation of RIG-I/MDA5 by 5′-triphosphate RNA and poly(I:C) triggers apoptosis in a MAVS-dependent manner in human melanoma cells (3). However, both pro- and antiapoptotic roles of RIG-I/MAVS in viral infection have been reported. For its proapoptotic role, caspase-3/7 activity induced by reovirus is reduced in 293T cells transfected with MAVS small interfering RNA (siRNA) (10), and apoptosis induced by Sendai virus is reduced in mouse embryonic fibroblasts derived from MAVS knockout mice (16). Moreover, MAVS plays an essential role in the protein kinase R (PKR)-mediated IRF-3 activation and cell death signaling pathway in response to vaccinia virus ΔE3L infection (46). However, for its antiapoptotic role, overexpression of MAVS in HEK293 cells has been shown to suppress the cell death induced by vesicular stomatitis virus (VSV), probably through blocking viral replication, and RNAi of MAVS increases VSV viral titers and cytotoxicity (12, 34). Viral replication of VSV and respiratory syncytial virus also increases to higher levels in MAVS-deficient mice (4, 36).
We found that dominant-negative IRF-3 failed to attenuate the MAVS-induced caspase-3 activation (Fig. 6) and that the CARD-lacking MAVS (87-540) mutant could not activate IFN signaling but still triggered cell death (Fig. 4). These results suggest that MAVS by itself is proapoptotic, independent of IRF-3 activation. Our findings are consistent with a recent report that MAVS-induced apoptosis is independent of type I IFN production and still occurs in HEK293 cells deprived of IRF-3 expression by siRNA targeting of IRF-3 (16). The apoptotic role of MAVS is further supported by the finding that ablation of MAVS expression alleviates cytotoxicity and caspase-3 activation during DEN-2 infection and dsRNA stimulation (Fig. 8 to 10). MAVS knockdown has been shown to increase VSV titers (34); our results showing that MAVS knockdown did not increase DEN-2 production may have resulted from the incomplete suppression of MAVS expression in our knockdown cells. Alternatively, this might imply the existence of different interactions of RIG-I/MAVS signaling in response to different viruses. Although the effects of MAVS knockdown on virus-triggered cytotoxicity differ between VSV (34) and DEN-2, poly(I:C) transfection showed a proapoptotic role of MAVS in response to cytosolic foreign RNA. We thus suggest that MAVS plays a proapoptotic role in DEN-2 infection, and possibly in other viral infections.
Several cellular proteins that interact with MAVS and the regulators that modulate RIG-I/MAVS signaling have been reported and reviewed (29). The observation that the function of MAVS in mediating IFN induction requires its localization to the mitochondria establishes an unexpected link between innate immunity and mitochondria (34). MAVS interacts with TRADD, recruits the E3 ubiquitin ligase TRAF3 and the adaptor protein TANK, and forms a complex with FADD and RIP1, leading to the activation of IRF-3 and NF-κB (26). The signaling pathways of proinflammatory tumor necrosis factor (TNF) and antiviral RIG-I are very similar (26). Binding of TNF to TNF receptor I (TNFRI) activates NF-κB and mitogen-activated protein kinases (MAPKs) through a receptor-proximal signaling complex involving TRADD, RIP1, and TRAF2. After TNF binding, TNFRI can also induce cell death by forming an intracellular signaling complex involving FADD, which activates caspase-8 and induces apoptosis (27, 30). The ability of MAVS to trigger cell death depends on its mitochondrial localization; in contrast to its required role in IFN induction, the CARD is dispensable for MAVS-induced cell death (Fig. 4). Because MAVS interacts with FADD and RIP1 through its non-CARD region (12), MAVS may activate caspase-8 by interacting with FADD and TRADD and then turn on the caspase cascade to further activate caspase-3 and caspase-9, eventually causing cell death. Several BH3-only proapoptotic molecules, such as Noxa, Puma, Bim, and Bik, are upregulated upon poly(I:C) transfection, which induces apoptosis through a MAVS-mediated mechanism (3). Our results showing that MAVS triggered MMP disruption (Fig. 5) and that Bcl-xL effectively blocked the MAVS-induced procaspase-3 cleavage (Fig. 6) prompt us to propose that MAVS initiates the death signal by activating these BH3-only proapoptotic proteins, which then disrupt mitochondrial membrane integrity and actuate the caspase cascade.
Both of the adaptor proteins of TLR and RLH signaling, Trif and MAVS, were found recently to be cleaved by caspase (31). We demonstrated further that MAVS can be cleaved by virus-induced caspases, suggesting a generalized molecular mechanism that may be adapted by various viruses to blunt the IFN pathway. We also showed that MAVS is cleaved by caspases at residue D429, as reported earlier (31). Furthermore, caspase-1, an inflammatory caspase, and caspase-3, a major apoptotic effector caspase, cleaved MAVS in an in vitro assay, suggesting that the negative regulation of MAVS by caspases plays a role in modulating both cell death and inflammatory cytokine production. The cross talk between cell death and immunity, especially that involving inflammation and cytokine production, is an emerging field of active research (39). The NLR (nucleotide-binding domain, leucine-rich repeat-containing) family of proteins appears to serve as a common platform in host-pathogen interactions by both facilitating the maturation of IL-1β and mediating cell death. A mitochondrion-localized member of the NLR family, NLRX1, was found to inhibit the RIG-I/MAVS-mediated IFN-β promoter activation (28). The detailed interactions and significance of the regulatory loop involving MAVS activation of caspases and caspase cleavage of MAVS warrant further study.
DEN infection is currently the most common arboviral disease, causing dengue fever, dengue hemorrhagic fever, and dengue shock syndrome worldwide (9). Cell death and caspase induction occur mainly in the late stage of DEN infection (6). Using caspases to attenuate IFN-β production in the late stage, when viral replication is complete, might not be an efficient strategy for DEN to escape host immunity. Our result and previous findings (31) showing that the caspase-resistant D429E and D429A mutants of MAVS are not more potent in IFN induction suggest that cleavage of MAVS by caspases plays an important role in alleviating cell death but not in modulating IFN production. We suggest that MAVS participates in host innate immunity by two mechanisms: in the early phase of viral infection, MAVS is involved in IFN production, and in the late phase of infection, MAVS mediates cell death. In innate immunity, both MAVS-mediated events, IFN production and cell death induction, cooperate to eliminate and control viral infection.
Acknowledgments
We thank Zhijian J. Chen for Flag-MAVS/pcDNA3, Takashi Fujita for p125-Luc, and the National RNAi Core Facility, Taiwan (supported by the National Research Program for Genomic Medicine Grants of NSC), for MAVS-targeting shRNA constructs.
This work was supported by grants awarded to Y.-L.L. from the National Science Council (NSC-95-2320-B-001-031-MY3 and NSC 97-3112-B-001-002) and from Academia Sinica, Taiwan.
Footnotes
Published ahead of print on 23 December 2009.
REFERENCES
- 1.Andrejeva, J., K. S. Childs, D. F. Young, T. S. Carlos, N. Stock, S. Goodbourn, and R. E. Randall. 2004. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. USA 101:17264-17269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Backes, C., J. Kuentzer, H. P. Lenhof, N. Comtesse, and E. Meese. 2005. GraBCas: a bioinformatics tool for score-based prediction of caspase- and granzyme B-cleavage sites in protein sequences. Nucleic Acids Res. 33:W208-W213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Besch, R., H. Poeck, T. Hohenauer, D. Senft, G. Hacker, C. Berking, V. Hornung, S. Endres, T. Ruzicka, S. Rothenfusser, and G. Hartmann. 2009. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J. Clin. Invest. 119:2399-2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhoj, V. G., Q. Sun, E. J. Bhoj, C. Somers, X. Chen, J. P. Torres, A. Mejias, A. M. Gomez, H. Jafri, O. Ramilo, and Z. J. Chen. 2008. MAVS and MyD88 are essential for innate immunity but not cytotoxic T lymphocyte response against respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 105:14046-14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang, T. H., C. L. Liao, and Y. L. Lin. 2006. Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-kappaB activation. Microbes Infect. 8:157-171. [DOI] [PubMed] [Google Scholar]
- 6.Courageot, M. P., A. Catteau, and P. Despres. 2003. Mechanisms of dengue virus-induced cell death. Adv. Virus Res. 60:157-186. [DOI] [PubMed] [Google Scholar]
- 7.Fridman, J. S., M. A. Benedict, and J. Maybaum. 1999. bcl-X(S)-induced cell death in 3T3 cells does not require or induce caspase activation. Cancer Res. 59:5999-6004. [PubMed] [Google Scholar]
- 8.Ghayur, T., S. Banerjee, M. Hugunin, D. Butler, L. Herzog, A. Carter, L. Quintal, L. Sekut, R. Talanian, M. Paskind, W. Wong, R. Kamen, D. Tracey, and H. Allen. 1997. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature 386:619-623. [DOI] [PubMed] [Google Scholar]
- 9.Halstead, S. B. 2007. Dengue. Lancet 370:1644-1652. [DOI] [PubMed] [Google Scholar]
- 10.Holm, G. H., J. Zurney, V. Tumilasci, S. Leveille, P. Danthi, J. Hiscott, B. Sherry, and T. S. Dermody. 2007. Retinoic acid-inducible gene-I and interferon-beta promoter stimulator-1 augment proapoptotic responses following mammalian reovirus infection via interferon regulatory factor-3. J. Biol. Chem. 282:21953-21961. [DOI] [PubMed] [Google Scholar]
- 11.Ishikawa, H., and G. N. Barber. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawai, T., K. Takahashi, S. Sato, C. Coban, H. Kumar, H. Kato, K. J. Ishii, O. Takeuchi, and S. Akira. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981-988. [DOI] [PubMed] [Google Scholar]
- 13.Labbe, K., and M. Saleh. 2008. Cell death in the host response to infection. Cell Death Differ. 15:1339-1349. [DOI] [PubMed] [Google Scholar]
- 14.Lamkanfi, M., N. Festjens, W. Declercq, T. Vanden Berghe, and P. Vandenabeele. 2007. Caspases in cell survival, proliferation and differentiation. Cell Death Differ. 14:44-55. [DOI] [PubMed] [Google Scholar]
- 15.Lee, M. S., and Y. J. Kim. 2007. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu. Rev. Biochem. 76:447-480. [DOI] [PubMed] [Google Scholar]
- 16.Lei, Y., C. B. Moore, R. M. Liesman, B. P. O'Connor, D. T. Bergstralh, Z. J. Chen, R. J. Pickles, and J. P. Ting. 2009. MAVS-mediated apoptosis and its inhibition by viral proteins. PLoS One 4:e5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li, P., H. Allen, S. Banerjee, S. Franklin, L. Herzog, C. Johnston, J. McDowell, M. Paskind, L. Rodman, J. Salfeld, et al. 1995. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 80:401-411. [DOI] [PubMed] [Google Scholar]
- 18.Li, X. D., L. Sun, R. B. Seth, G. Pineda, and Z. J. Chen. 2005. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 102:17717-17722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin, R., C. Heylbroeck, P. M. Pitha, and J. Hiscott. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18:2986-2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loo, Y. M., J. Fornek, N. Crochet, G. Bajwa, O. Perwitasari, L. Martinez-Sobrido, S. Akira, M. A. Gill, A. Garcia-Sastre, M. G. Katze, and M. Gale, Jr. 2008. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 82:335-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loo, Y. M., and M. Gale, Jr. 2007. Viral regulation and evasion of the host response. Curr. Top. Microbiol. Immunol. 316:295-313. [DOI] [PubMed] [Google Scholar]
- 22.Makarova, O., E. Kamberov, and B. Margolis. 2000. Generation of deletion and point mutations with one primer in a single cloning step. Biotechniques 29:970-972. [DOI] [PubMed] [Google Scholar]
- 23.Medzhitov, R. 2007. Recognition of microorganisms and activation of the immune response. Nature 449:819-826. [DOI] [PubMed] [Google Scholar]
- 24.Meylan, E., J. Curran, K. Hofmann, D. Moradpour, M. Binder, R. Bartenschlager, and J. Tschopp. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167-1172. [DOI] [PubMed] [Google Scholar]
- 25.Meylan, E., and J. Tschopp. 2006. Toll-like receptors and RNA helicases: two parallel ways to trigger antiviral responses. Mol. Cell 22:561-569. [DOI] [PubMed] [Google Scholar]
- 26.Michallet, M. C., E. Meylan, M. A. Ermolaeva, J. Vazquez, M. Rebsamen, J. Curran, H. Poeck, M. Bscheider, G. Hartmann, M. Konig, U. Kalinke, M. Pasparakis, and J. Tschopp. 2008. TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity 28:651-661. [DOI] [PubMed] [Google Scholar]
- 27.Micheau, O., and J. Tschopp. 2003. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114:181-190. [DOI] [PubMed] [Google Scholar]
- 28.Moore, C. B., D. T. Bergstralh, J. A. Duncan, Y. Lei, T. E. Morrison, A. G. Zimmermann, M. A. Accavitti-Loper, V. J. Madden, L. Sun, Z. Ye, J. D. Lich, M. T. Heise, Z. Chen, and J. P. Ting. 2008. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451:573-577. [DOI] [PubMed] [Google Scholar]
- 29.Moore, C. B., and J. P. Ting. 2008. Regulation of mitochondrial antiviral signaling pathways. Immunity 28:735-739. [DOI] [PubMed] [Google Scholar]
- 30.Muppidi, J. R., J. Tschopp, and R. M. Siegel. 2004. Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity 21:461-465. [DOI] [PubMed] [Google Scholar]
- 31.Rebsamen, M., E. Meylan, J. Curran, and J. Tschopp. 2008. The antiviral adaptor proteins Cardif and Trif are processed and inactivated by caspases. Cell Death Differ. 15:1804-1811. [DOI] [PubMed] [Google Scholar]
- 32.Rintahaka, J., D. Wiik, P. E. Kovanen, H. Alenius, and S. Matikainen. 2008. Cytosolic antiviral RNA recognition pathway activates caspases 1 and 3. J. Immunol. 180:1749-1757. [DOI] [PubMed] [Google Scholar]
- 33.Scott, I., and K. L. Norris. 2008. The mitochondrial antiviral signaling protein, MAVS, is cleaved during apoptosis. Biochem. Biophys. Res. Commun. 375:101-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seth, R. B., L. Sun, C. K. Ea, and Z. J. Chen. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669-682. [DOI] [PubMed] [Google Scholar]
- 35.Su, H. L., Y. L. Lin, H. P. Yu, C. H. Tsao, L. K. Chen, Y. T. Liu, and C. L. Liao. 2001. The effect of human bcl-2 and bcl-X genes on dengue virus-induced apoptosis in cultured cells. Virology 282:141-153. [DOI] [PubMed] [Google Scholar]
- 36.Sun, Q., L. Sun, H. H. Liu, X. Chen, R. B. Seth, J. Forman, and Z. J. Chen. 2006. The specific and essential role of MAVS in antiviral innate immune responses. Immunity 24:633-642. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi, K., T. Kawai, H. Kumar, S. Sato, S. Yonehara, and S. Akira. 2006. Roles of caspase-8 and caspase-10 in innate immune responses to double-stranded RNA. J. Immunol. 176:4520-4524. [DOI] [PubMed] [Google Scholar]
- 38.Takeuchi, O., and S. Akira. 2007. Recognition of viruses by innate immunity. Immunol. Rev. 220:214-224. [DOI] [PubMed] [Google Scholar]
- 39.Ting, J. P., S. B. Willingham, and D. T. Bergstralh. 2008. NLRs at the intersection of cell death and immunity. Nat. Rev. Immunol. 8:372-379. [DOI] [PubMed] [Google Scholar]
- 40.Xu, L. G., Y. Y. Wang, K. J. Han, L. Y. Li, Z. Zhai, and H. B. Shu. 2005. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 19:727-740. [DOI] [PubMed] [Google Scholar]
- 41.Yang, Y., Y. Liang, L. Qu, Z. Chen, M. Yi, K. Li, and S. M. Lemon. 2007. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. USA 104:7253-7258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoneyama, M., and T. Fujita. 2008. Structural mechanism of RNA recognition by the RIG-I-like receptors. Immunity 29:178-181. [DOI] [PubMed] [Google Scholar]
- 43.Yoneyama, M., M. Kikuchi, T. Natsukawa, N. Shinobu, T. Imaizumi, M. Miyagishi, K. Taira, S. Akira, and T. Fujita. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730-737. [DOI] [PubMed] [Google Scholar]
- 44.Yoneyama, M., W. Suhara, Y. Fukuhara, M. Sato, K. Ozato, and T. Fujita. 1996. Autocrine amplification of type I interferon gene expression mediated by interferon stimulated gene factor 3 (ISGF3). J. Biochem. 120:160-169. [DOI] [PubMed] [Google Scholar]
- 45.Yu, C. Y., Y. W. Hsu, C. L. Liao, and Y. L. Lin. 2006. Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J. Virol. 80:11868-11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang, P., and C. E. Samuel. 2008. Induction of protein kinase PKR-dependent activation of interferon regulatory factor 3 by vaccinia virus occurs through adapter IPS-1 signaling. J. Biol. Chem. 283:34580-34587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhong, B., Y. Yang, S. Li, Y. Y. Wang, Y. Li, F. Diao, C. Lei, X. He, L. Zhang, P. Tien, and H. B. Shu. 2008. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29:538-550. [DOI] [PubMed] [Google Scholar]