

Abstract
High-density functional gene arrays have become a powerful tool for environmental microbial detection and characterization. However, microarray data normalization and comparison for this type of microarray remain a challenge in environmental microbiology studies because some commonly used normalization methods (e.g., genomic DNA) for the study of pure cultures are not applicable. In this study, we developed a common oligonucleotide reference standard (CORS) method to address this problem. A unique 50-mer reference oligonucleotide probe was selected to co-spot with gene probes for each array feature. The complementary sequence was synthesized and labeled for use as the reference target, which was then spiked and cohybridized with each sample. The signal intensity of this reference target was used for microarray data normalization and comparison. The optimal amount or concentration were determined to be ca. 0.5 to 2.5% of a gene probe for the reference probe and ca. 0.25 to 1.25 fmol/μl for the reference target based on our evaluation with a pilot array. The CORS method was then compared to dye swap and genomic DNA normalization methods using the Desulfovibrio vulgaris whole-genome microarray, and significant linear correlations were observed. This method was then applied to a functional gene array to analyze soil microbial communities, and the results demonstrated that the variation of signal intensities among replicates based on the CORS method was significantly lower than the total intensity normalization method. The developed CORS provides a useful approach for microarray data normalization and comparison for studies of complex microbial communities.
Microarray-based technology has become a robust genomic tool to detect, track, and profile hundreds to thousands of different microbial populations simultaneously in complex environments such as soils and sediments. For example, GeoChip, a comprehensive functional gene array, has been developed for investigating biogeochemical, ecological, and environmental processes (12, 18, 23, 27, 29, 32). Although a massive amount of microarray data can be generated rapidly, one of the bottlenecks in using microarrays for environmental microbial community studies is the lack of an appropriate standard for data comparison and normalization (6). Currently, it is difficult to compare microarray data across different sites, experiments, laboratories, and/or time periods (10). This limits the power of the technology to address ecological and environmental questions.
In pure culture-based functional genomics studies, genomic DNAs (gDNAs) have been used as a common reference for hybridizations in which the same amount of gDNAs are used to cohybridize with each target cDNA sample and then to normalize different target cDNAs based on the gDNA standard (4, 5, 8, 9, 19, 21, 23). Several normalization methods such as scale normalization, quantile normalization, and Lowess normalization have been used for gene expression studies (2). Using the gDNA standard method can minimize or eliminate differences in target cDNA quantity, spot morphology, uneven hybridization, labeling, and sequence-specific hybridization behaviors (5), and this allows the comparison of microarray data across different sites, laboratories, experiments, and/or times. The main rationale for gDNA as a common reference is that it provides complete coverage for all genes represented on the array because the DNA composition from a particular organism should be identical across different treatment samples even though RNA expression is different (8). However, this approach is not applicable to microbial community studies because not all communities have identical DNA compositions. Pooling of equal amounts of gDNA or RNA from every target sample to make a common sample could be used as an alternative reference for cohybridization (1, 22). However, the disadvantage of the sample pooling approach is that samples do not provide large amounts of DNA or RNA in a reliable and reproducible way. For example, groundwater samples usually have a very low biomass and thus would not provide enough DNA for pooling. In addition, the sample pool itself is uncharacterized, and gene abundance may be diluted out so that insufficient DNA is present to result in a positive signal some array features, especially for those genes in low abundance. Moreover, a new sample pool would be required for every new experiment, making comparison across experiments difficult. Thus, other approaches need to be developed for microbial community studies.
Dudley et al. (7) used a 25-mer oligonucleotide that matched a small portion of the parental EST clone vector contained in every PCR product printed on the array for normalization of pure culture RNA expression. Although the oligonucleotide generated a stable hybridization signal on every array feature, this method requires a universal sequence tag as a “capture” sequence, limiting its general use in microbial community studies. Thus, in the present study, we developed a common oligonucleotide reference standard (CORS) approach by co-spotting a common oligonucleotide with each array feature to improve the accuracy and comparability of microarray data for microbial community studies. This method was evaluated by using a pilot array, a whole-genome array, and a functional gene array, and all results demonstrate that the developed CORS is a reliable and reproducible method for microarray data normalization and comparison for microbial community studies.
MATERIALS AND METHODS
Bacterial strains and environmental samples.
Shewanella oneidensis MR-1 was from our laboratory culture collection and Rhodopseudomonas palustris CGA009 was provided by Caroline Harwood, Department of Microbiology, University of Washington (Seattle, WA). Desulfovibrio vulgaris Hildenborough (ATCC 29579) was obtained from the American Type Culture Collection (Manassas, VA). The D. vulgaris Δfur mutant was constructed with a marker-exchange method for gene deletion as described previously (3). S. oneidensis was grown in Luria-Bertani broth, and R. palustris was grown in nutrient broth. Wild-type D. vulgaris and the Δfur mutant were grown in LS4D medium containing 5 μM FeCl2 instead of 60 μM FeCl2 (3). Cells were harvested at the exponential phase and frozen at −80°C.
To evaluate the performance of the CORS for microarray data normalization and comparison in microbial community analysis, soil samples obtained from Daqing oilfield in northeast China were used. The oilfield was explored in the 1960s and has the largest production of crude oil among all oilfields in China. Some sites in the Daqing oilfield were contaminated as a result of oil exploration, production, maintenance, transportation, storage, and accidental release. Soil samples were collected to a depth of 10 cm in September 2006 from both contaminated and uncontaminated sites to compare the response of the microbial communities to crude oil contamination. Soil samples were sealed in sterile sampling bags and transported to the lab on ice. The physical and chemical properties were measured immediately as described previously (18).
Nucleic acid extraction and purification.
The gDNAs of the pure cultures were isolated as previously described (31) and treated with RNase A (Sigma, St. Louis, MO). Soil community DNAs were isolated and purified as described previously (30). The molecular weights of all DNA samples were checked by using agarose gels stained with ethidium bromide and quantified by using Quant-It PicoGreen (Invitrogen, Carlsbad, CA).
RNA of wild-type D. vulgaris and the Δfur mutant were isolated by using TRIzol (Invitrogen) and purified with the RNeasy minikit (Qiagen, Valencia, CA) and an RNase-free DNase set (Qiagen). All DNA and RNA samples were stored at −80°C.
Oligonucleotide reference probe design and microarray construction.
To avoid confusion, the following terms were used in the present study. (i) Test probes or gene probes refer to oligonucleotide probes targeting specific genes on an array. (ii) Test samples or targets refer to genomic DNAs or total RNAs from pure cultures or microbial communities (environmental samples) used for array hybridizations. (iii) Reference probe refers to the common oligonucleotide probe that is mixed at a certain proportion with each gene probe. (iv) Reference target refers to the labeled oligonucleotide that is complementary to the reference probe and is spiked into each sample. (v) Finally, for the co-spot the reference probe is mixed with each gene probe at a certain proportion and the mixture of both probes is printed on an array for each array feature.
The reference probe (50-mer) was randomly designed to have ∼50% G+C content and compared to all sequences in current databases to make sure that the sequence would not have any cross-hybridization with other sequences. The reference target (50-mer) was labeled with Cy3 at the 5′ end during synthesis (Table 1).
TABLE 1.
Microarrays and samples used in this studya
| Array | Probe type (oligonucleotide) | Surface coating and specification | Intrareplicate | Reference probe (amt [%] of test probes) | Test targets/samples | Normalization method(s)b |
|---|---|---|---|---|---|---|
| Pilot array | 50-mer | 30 functional genes derived from D. vulgaris, R. palustris, and S. oneidensis | Six replicates on a slide | 10, 5, 2.5, 1, 0.5, 0.25, 0.1, and 0.05 | gDNAs of D. vulgaris, R. palustris, and S. oneidensis | I, IV |
| D. vulgaris whole-genome array | 70-mer | ORFs for the genome of D. vulgaris Hildenborough | Duplicates on a slide | 2.5 | RNAs and gDNA of wild type D. vulgaris and Δfur mutant | II, III, IV |
| GeoChip | 50-mer | 37,000 gene sequences for more than 290 gene families | No intrareplicates | 2.5 | Microbial gDNAs from environmental soil samples of oilfield | I, IV |
For the CORS sequence, the reference probe was 5′-CCGCACCTCGGACCGCACACAATCGTTTGAGGACGTGTAGCTGTGCTGGC-3′. The reference target was complementary to the probe with Cy3 labeled at the 5′ end.
I, mean signal intensity normalization; II, dye swap normalization; III, gDNA normalization; IV, CORS normalization. See Materials and Methods for additional details.
Three types of arrays were used in the present study (Table 1). (i) A pilot array was constructed to determine the optimal amount and/or concentrations of reference probe printed on the array and the reference target spiked into the sample target. The pilot array was constructed with 30 gene probes (50-mer oligonucleotide: dnaK, glmS, murA, murL, nadE, nusG, proS, recA, rpoB, and rpsK of D. vulgaris, R. palustris, and S. oneidensis); a detailed description of test probe design is described elsewhere (13, 17). To determine the optimal amount of reference probe, reference probe was spiked into gene probes at various proportions of 10, 5, 2.5, 1, 0.5, 0.25, 0.1, and 0.05% and coprinted on the pilot array with six replicates. (ii) The D. vulgaris Hildenborough whole-genome microarray was used to evaluate the performance of this method in detecting the difference in gene expression between the D. vulgaris wild type and the Δfur mutant in comparison with other normalization methods. The D. vulgaris microarray (70-mer oligonucleotide) was constructed to cover all open reading frames (ORFs) for this organism as described previously (11, 20), with the addition of the reference probe (2.5% of gene probe) co-spotted on each array feature. (iii) GeoChip 3.0, an updated version of GeoChip 2.0 (12), was used to test the applicability of the developed CORS approach for studying microbial communities in natural settings. GeoChip 3.0 contains 24,676 probes co-spotted with the reference probe (2.5% of a gene probe). It covers ∼37,000 gene sequences from more than 290 gene families.
Preparation of fluorescently labeled DNA and RNA.
The gDNAs of pure cultures were fluorescently labeled with Cy5 or Cy3 using a BioPrime DNA labeling kit (Invitrogen). The gDNA was mixed with 15 μg of random primer, denatured by boiling for 5 min at 98°C, and then fluorescently labeled at 37°C for 3 h in a reaction solution containing 50 μM dATP, dCTP, and dGTP and 20 μM dTTP (USB Corp., Cleveland, OH); 1 mM Cy5 or Cy3 dUTP (Amersham Pharmacia Biotech, Piscataway, NJ); and 40 U of Klenow fragment (Invitrogen).
For environmental soil DNA samples, an aliquot of 100 ng of DNA from each sample was amplified in triplicate using the TempliPhi kit (Amersham Biosciences, Piscataway, NJ) in a modified buffer containing single-stranded binding protein (200 ng/μl) and spermidine (0.04 mM) to increase the sensitivity of amplification and incubated at 30°C for 3 h (28). All amplified DNAs were labeled with Cy5 as detailed above.
RNA of wild-type D. vulgaris and the Δfur mutant was labeled with Cy3 or Cy5 as described previously (11). RNA (10 μg) was mixed with 10 μg of random primers and incubated at 70°C for 10 min and then fluorescently labeled in a reaction solution containing 10 mM dATP, dCTP, and dGTP and 0.5 mM dTTP (USB Corp.); 1 mM Cy5 or Cy3 dUTP (Amersham Pharmacia Biotech); 40 U of RNase inhibitor (Gibco-BRL/Invitrogen); and 200 U of Superscript RNase H-reverse transcriptase in 1× first-strand buffer, followed by incubation at 42°C for 2 h. All of the labeled products were purified with a QIAquick PCR purification kit (Qiagen) and dried.
Microarray hybridization, scanning, and image processing.
Labeled products were resuspended in hybridization solution containing 50% formamide, 15× SSC (vol/vol; 20×), 3% sodium dodecyl sulfate (vol/vol; 10%), 7% herring sperm DNA (10 mg/ml), and 0.8% dithiothreitol (vol/vol; 0.1 M) and brought to a final volume of 40 μl with manual hybridization and 130 μl with automatic hybridization.
The hybridizations with the pilot array were carried out manually at 45°C overnight as detailed described previously (23). The hybridizations with the D. vulgaris whole-genome array and GeoChip were carried out on a HS4800 Hybridization Station (Tecan US, Durham, NC) at 45 and 42°C, respectively, for 10 h. All hybridizations were performed in triplicate.
Microarrays were scanned on a ScanArray 5000 microarray analysis system (Perkin-Elmer, Wellesley, MA) with 95% laser power and 68% photomultiplier tube gain for Cy5 and 74% for Cy3. Signal intensities were measured with ImaGene 6.0 (Biodiscovery, Inc., El Segundo, CA) by averaging the intensities of every pixel inside the target region (segmentation method). The mean signal intensity was determined for each spot, and the local background signals were subtracted automatically from the hybridization signal of each spot. Any spots with a signal-to-noise ratio (SNR) of <2.0 were defined as empty spots (14), while poor spots were defined as spots whose signals could not be accurately quantified due to their irregular shapes and/or contaminations. All empty, poor, and outlier spots were removed from subsequent analysis. Any gene with more than one of three of positive probe spots was considered positive.
Microarray data analysis.
To evaluate the performance and effectiveness of the developed CORS method for data normalization and comparison, the following four normalization methods were used in the present study: mean signal intensity, dye-swap normalization, gDNA reference, and CORS.
(i) Mean signal intensity normalization.
The hybridization signal was normalized by the mean signal intensity across all genes on the array. The across-array mean was calculated based on all intensities on the arrays after the removal of empty and poor spots and outliers. Then, a ratio was calculated for each positive spot by dividing the signal intensity of the spot by the mean signal intensity to obtain the normalized ratio.
The mean signal intensity xij of the jth replicate in the ith sample was calculated as follows:
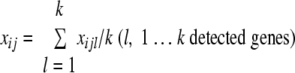 |
The maximum mean signal intensity xmax of all replicates in all samples was calculated as follows:
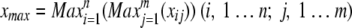 |
The normalize factor Nij was calculated as follows:
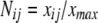 |
This method was used for the pilot array and GeoChip. The average signal intensities across all of the genes were expected to be approximately equal if the same amount of DNA was used for amplification (if needed), labeling, and hybridization (27).
(ii) Dye swap normalization.
Ratios were calculated for the sample pair. Any two of the total replicates were normalized and final ratio values were obtained by taking the median value of all normalized ratios as described previously (22). This method was used for the D. vulgaris whole-genome array only.
(iii) Genomic DNA normalization.
The ratio rij of each gene pair (i, j) among replicates was calculated as follows:
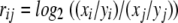 |
where xi and xj are the signal intensities for the ith and jth (i ≠ j) genes from the target DNAs, and yi and yj are their corresponding signals from the hybridization with gDNAs. Genes with two of three or more positive spots of the total number of spots among replicates were considered positive. This method was used only for the D. vulgaris whole-genome array only.
(iv) CORS normalization.
For this method, target DNA was labeled with Cy5 and the reference target was labeled with Cy3. First, normalization among technique replicates was performed by the greatest mean intensity of the Cy5 target signal as described in section i above. Then normalization was performed among samples using the CORS as follows.
The mean signal intensity xij of the CORS of the jth replicate in the ith sample was calculated as follows:
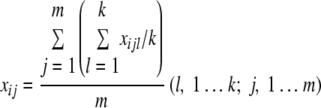 |
The greatest average intensity xmax across all samples of the CORS was calculated as follows:
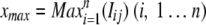 |
The normalization factors Nij was calculated as follows:
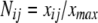 |
Ratios from two dyes were calculated for each spot with the reference target signal as the denominator. The method was used for all three arrays and not limited to a set of experiments as long as same amount of reference probe and reference target were used for all experiments.
All correlation analyses and the paired Student t test were performed with SPSS 13.0 (SPSS, Inc., Chicago, IL). A Mantel test was performed to infer the correlation between soil hydrocarbon concentrations and the functional genes involved in organic contaminant degradation based on Euclidean distance with PC-ORD (MjM Software, Gleneden Beach, Oregon). The P value of the standardized Mantel statistic (r) was calculated from 999 Monte Carlo randomizations.
RESULTS
Determination of the optimal amounts or concentrations of the reference probe and target.
The ideal reference amounts should give detectable and uniform signals on every array feature without affecting the signal intensities of the real sample. To determine the optimal amounts or concentrations of reference probe and target, different concentrations of reference target (12.5, 1.25, 0.625, 0.25, 0.125, 0.0625, 0.025, and 0.0125 fmol/μl) were spiked into samples (500 ng of gDNA equally mixed from D. vulgaris, R. palustris, and S. oneidensis) and cohybridized with the pilot array. The pilot array contained the reference probes with proportions of 10, 5, 2.5, 1, 0.5, 0.25, 0.1, and 0.05% relative to the gene probe. The signal intensities of the CORS increased linearly with the reference target concentrations (r2 > 0.9) (Fig. 1). The average SNR and percentage of positive spots (SNR > 2.0 without saturation) at each reference probe and target amount or concentration were calculated (Table 2). In the middle-range amount or concentrations of reference probe and target, the signal on each array feature was effectively detected without saturation, and the signal intensity of samples appeared not to be affected by the reference probe and reference target tested (Fig. 1). Therefore, the optimal amount of the reference probe was found to be in a range of 0.5 to 2.5% of the gene probe and the reference target concentration to be 0.25 to 1.25 fmol/μl (Table 2). The amounts or concentrations of the reference probe and target used in individual experiments can be adjustable for the array type and size based on the above ranges.
FIG. 1.

Signal intensities of the reference target (ca. 0.0125 to 12.5 fmol/μl) cohybridized with 500 ng of gDNA target of an equal mixture of D. vulgaris, R. palustris, and S. oneidensis. The amounts of the reference probe ranged from ca. 0.05 to 10% of a gene probe. Error bars showing the standard deviations are presented.
TABLE 2.
Average SNR of reference probe and targeta
| Reference probe amt (%) | Reference target concn (fmol/μl) |
|||||||
|---|---|---|---|---|---|---|---|---|
| 12.5 | 1.25 | 0.625 | 0.25 | 0.125 | 0.0625 | 0.025 | 0.0125 | |
| 10.00 | 115.5 (80.1) | 67.2 (99.1) | 52.3 (98.9) | 19.5 (100) | 12.2 (100) | 4.67 (96.4) | 2.16 (39.1) | 0.9 (6.6) |
| 5.00 | 97.0 (91.4) | 52.3 (100) | 36.5 (100) | 15.5 (100) | 5.9 (97.8) | 4.7 (91.8) | 1.1 (9.5) | 0.8 (7.3) |
| 2.50 | 64.8 (97.8) | 36.1 (100) | 28.8 (100) | 12.1 (100) | 6.9 (98.1) | 3.1 (71.0) | 1.5 (21.5) | 0.7 (18.9) |
| 1.00 | 35.6 (100) | 18.1 (100) | 15.4 (100) | 6.8 (94.4) | 2.6 (39.0) | 2.0 (35.8) | 0.7 (4.7) | 0.5 (2.0) |
| 0.50 | 23.9 (100) | 13.8 (100) | 8.7 (99.4) | 4.9 (83.0) | 2.1 (6.2) | 1.3 (14.5) | 0.6 (3.1) | 0.5 (0.4) |
| 0.25 | 13.8 (100) | 7.6 (95.0) | 4.9 (89.7) | 2.7 (44.0) | 1.1 (9.7) | 0.9 (6.8) | 0.4 (0) | 0.4 (0.2) |
| 0.10 | 6.2 (91.2) | 3.4 (54.7) | 2.3 (40.3) | 1.3 (15.4) | 0.7 (3.8) | 0.6 (3.1) | 0.4 (0.2) | 0.4 (0.4) |
| 0.05 | 3.3 (67.7) | 1.7 (19.7) | 1.2 (16.2) | 0.8 (12.2) | 0.4 (0.4) | 0.4 (1.2) | 0.3 (0) | 0.3 (0) |
The percentages of good spots (SNR ≥ 2 without saturation) are indicated in parentheses. The optimal amounts and concentrations of reference probe and target are indicated in boldface.
Reduction in experimental variations with the CORS method.
Variations across microarray hybridizations are relatively high, and this is due to many factors such as irregular spot size and morphology, uneven hybridization, boundary effects of the slides, high local or global background, and freshness of reagents. Using the CORS is expected to reduce these variations during hybridization. To evaluate the effectiveness of the CORS in reducing these variations, the pilot array containing gene probes from D. vulgaris, S. oneidensis, and R. palustris was used. Various amounts of gDNA (1,500, 300, 75, and 15 ng) from an equal mixture of the three bacteria were labeled with Cy5, spiked with a 1.5-fmol/μl concentration of reference target, and hybridized with the pilot array. The hybridization signals of these 30 functional genes among inter- and intrareplicates were normalized by using the mean signal normalization method and the CORS approach. The variations of the normalized hybridization signals with the CORS method were significantly lower than those with the mean signal normalization method (Fig. 2) with the paired Student t test (P < 0.001 for 1,500, 300, and 75 ng of gDNA targets; P < 0.05 for 15 ng of gDNA target), suggesting that the CORS method is effective in reducing experimental variations.
FIG. 2.

Box plots of variations of hybridization signal intensities among inter- and intrareplicates. A reduction of variations was observed when the CORS method was used for normalization compared to the mean signal intensity (mean) as evidenced by the paired Student t test with P < 0.001 for 1,500, 300, and 75 ng of gDNAs and P < 0.05 for 15 ng of gDNAs of equal mixtures of D. vulgaris, R. palustris, and S. oneidensis.
Quantitative capability with CORS.
The quantitative capability of 50-mer oligonucleotide microarrays for detecting microbial functional genes has been reported (23). The quantitative relationship between the normalized hybridization signal intensity and the original DNA concentration was further evaluated. Two normalization methods were compared: mean signal intensity normalization and the CORS normalization. The signal intensity was linear (r2 = 0.95∼0.99) for all detected genes (dnaK, glmS, murA, murL, nadE, nusG, proS, recA, rpoB, and rpsK from D. vulgaris, R. palustris, and S. oneidensis) over a range of 15 to 1,500 ng of genomic DNA using the mean signal intensity normalization method, and r2 = 0.96 to 0.99 when the CORS normalization method was used. These results demonstrate that the CORS method could accurately estimate the relative abundance of target genes in a wide range.
Analysis of gene expression with D. vulgaris wild type and Δfur mutant.
Although the CORS method was primarily developed to normalize functional gene abundance data (DNA) for microarrays used to study microbial communities, it is important to compare this method with other traditional normalization methods, which are primarily used to normalize expression data (RNA) of pure culture data. The gDNA method has been widely used for microarray data normalization and comparison, and the dye swap method has been considered a traditional approach for two-dye microarray data normalization in pure culture studies. To evaluate the accuracy of the developed CORS normalization, it was compared to the gDNA and dye-swap methods.
The gene expression of the wild type and Δfur mutant of D. vulgaris Hildenborough was analyzed after the raw data was normalized by three (gDNA, dye swap, and CORS) different methods. Scatter plots of gene expression ratios (mutant RNA/wild-type RNA) for the CORS normalization against the gDNA and dye swap methods indicated high correlations between the CORS and the other two methods (Fig. 3). The Pearson correlation was 0.908 for the dye swap method versus the CORS method (Fig. 3A) and 0.976 for the gDNA method versus the CORS method (Fig. 3B). All of these correlations were statistically significant (P < 0.001). Good linear relationships were also observed by using nonparametric Spearman rank correlation (data not shown). The results indicate that the CORS method performed equally well compared to two established methods.
FIG. 3.

Quantitative analysis of relationships for expression ratios of all genes (Δfur mutant RNA/wild-type RNA of D. vulgaris) between different normalization methods. (A) Dye swap versus CORS; (B) gDNA versus CORS.
We also compared the positive spots detected by the gDNA method and the CORS approach using the D. vulgaris whole-genome microarray (9). In total, 3,586 genes were printed on the microarray in duplicate (7,172 spots). Both gDNA and the CORS approach detected almost all of the array features, 98.3% for gDNA and 98.2% for the CORS. Consequently, almost all information from the expressed genes was obtained when gDNA or the CORS was used as a reference with 94.1 or 96.2% spots detected for the mutant, respectively, and 99.2 or 98.5% for the wild type, respectively. Therefore, the CORS method represented a high coverage on the reference channel.
We selected 20 genes known to be affected by a fur mutation for further analysis. All of these genes showed a high correlation of gene expression based on the three normalization methods (Fig. 4). For example, it has been reported that three feoAB genes were highly induced in the Δfur mutant of D. vulgaris (3). In the present study, the log2 (mutant/wild-type ratios) of feoAB genes with the CORS method were 2.12, 1.78, and 1.84 for feoB (ORF03249), feoA (ORF03251), and feoA (ORF03253), respectively (Fig. 4), similar to a previous study (3). All of these analyses clearly show that very similar results were obtained with the three different normalization methods and that gene expression profiles normalized by the CORS method were significantly correlated to those from another two commonly used normalization methods.
FIG. 4.

Ratios (Δfur mutant RNA/wild-type RNA of D. vulgaris) of gene expression obtained from three normalization methods, dye swap, gDNA, and CORS.
Application of the CORS method to analyze functional gene array data from environmental microbial communities.
To further determine the applicability of the developed CORS method for microarray-based analysis of complex environmental microbial communities, microbial communities of high-, low-, and no-contamination soil samples were analyzed with the new version of GeoChip (GeoChip 3.0). Variations of three replicates for each gene were significantly lower after normalization by the CORS method (Fig. 5), P < 0.001 for all of the samples based on the paired Student t test. Thus, the developed CORS method significantly reduced the variation among replicates.
FIG. 5.

Box plots of variations of hybridization signals among replicates of high-, low-, and no-contamination soil microbial communities. A reduction in variation was observed when GeoChip data were normalized with the CORS method compared to the conventional mean signal intensity (mean) method as evidenced by the paired Student t test with P < 0.001.
Samples from different sites could be compared in a more accurate way after normalization by the CORS method. Changes in soil microbial functional genes with crude oil contamination were further studied using the CORS normalization method. The functional genes involved in organic contaminant degradation showed significant correlations with hydrocarbon concentrations based on the Mantel test (P < 0.05). A high abundance of alkane degradation genes such as alkB encoding alkane hydroxylase, alkH encoding aldehyde dehydrogenase and alkK encoding acyl-coenzyme A synthetase were detected across all samples. These genes were mainly derived from common genera such as Pseudomonas, Mycobacterium, Rhodopseudomonas, Burkholderia, Bacillus, and Ralstonia. The alk gene abundance was 31.0% higher in the high-contamination soil than in the no- and low-contamination soils. Aromatic hydrocarbons were another major component of crude oil. Functional genes involved in biphenyl degradation detected in the soil samples were bphA, bphC, and bphD derived from Rhodococcus, Pseudomonas, Xanthobacter, and Burkholderia spp. The naphthalene catabolic genes nagG represented the highest abundance in high-contamination soils and were mainly from Roseovarius, Pseudomonas, Bordetella, Silicibacter, Ralstonia, Bradyrhizobium, and Mycobacterium spp., as were another two naphthalene catabolic genes, nagI and nagK, from Polaromonas, Pseudomonas, Sphingomonas, and Burkholderia spp. For organic contaminant degradation, catechol is a key dihydroxylated intermediate in the PAH catabolic pathways. Crude oil also stimulated the abundance of functional genes encoding catechol 1,2-dioxygenase and catechol 2,3-dioxygenase, which were mainly derived from Arthrobacter, Burkholderia, Mycobacterium, Nocardia, Pseudomonas, Ralstonia, and Rhizobium spp. These results showed that the high abundance of genes encoding enzymes degrading various organic chemicals was in good agreement with high organic contamination in the soils, and this also suggests that the CORS method is suitable for the comparison and normalization of functional gene-based microarray (e.g., GeoChip) data for complex environmental microbial community studies.
DISCUSSION
Recently, functional gene-based microarrays have been widely used for the detection, identification, and characterization of microbial communities in natural environments, but data normalization and comparison for these microarrays remain challenging due to the extremely high diversity and complexity of environmental samples. In the present study, we developed a CORS method to address this challenge. This method was evaluated by using different types of microarrays, and the results demonstrated that this CORS method performed well in comparison with other commonly used normalization methods.
The CORS method presented here was developed to allow microbial community microarray data to be normalized and compared across different samples, experiments, time points, and/or laboratories. Although functional gene arrays, such as GeoChip, have been applied for detecting and monitoring microbial communities, data normalization and comparison are difficult mainly due to the lack of a common reference. Currently, microarray data are normalized by a few methods, such as dye swap (16), gDNA (26), and mean/total signal (27). However, most of these methods are only suitable for pure cultures since they have the identical composition of nucleic acids from each organism. For environmental samples, the compositions of microbial communities are generally unknown, so it is extremely difficult, even impossible to use gDNAs as a common reference for a microbial community. The mean/total signal intensity normalization method has been used for analysis of functional gene array data (27), but it is limited to normalize signals among the replicates of a biological sample, or similar biological samples from different experiments or time points, which are analyzed at the same time. For the CORS method developed here, the reference probe and target are synthesized oligonucleotides, and their quality and amounts can be accurately controlled and optimized. Compared to the mean signal intensity normalization method, this method can greatly reduce hybridization variations and normalize the signal intensity of any environmental samples in a more flexible and accurate way. The CORS method is also more convenient and time-saving, and only requires small amounts of the reference probe and the reference target in comparison with the lambda DNA method. For the CORS method, if the same amounts of the reference probe and reference target are used, their signal intensities are expected to be the same across different hybridizations no matter what samples are analyzed, where those samples come from, and which laboratories conduct those experiments. Therefore, the CORS can be used as a common reference for normalization and comparison of functional gene array data in the analysis of microbial communities.
Several other characteristics also make the CORS method attractive for microarray data analysis. First, the reference probe is co-spotted with gene probes on each array feature so that the variations generated from microarray fabrication processes can be minimized (25). Second, since the reference probe is expected to have the same hybridization characteristics as gene probes on the same array, biases from uneven hybridization are expected to be minimized or eliminated if the signal ratio of each gene probe to the reference probe is used. Third, the CORS method can be used for quality control of experiments as the reference probe is expected to give relatively uniform signals for all spots. For example, a failure of depositing probes on the array and artifacts during hybridization (e.g., air bubble) could be easily tracked from the reference channel (15). In addition, since both reference probe and target are used in a very low amounts, this method costs much less in comparison with other common references, such as genomic DNA and sample pooling since those approaches used 50% of the hybridization resources to produce a control or common reference signal (24). Finally, an oligonucleotide reference probe or target can be synthesized in large scale and thus sufficient for a long-term use in different experiments and laboratories.
Although the developed CORS is a useful way to compare and normalize microarray data across different environmental samples, some limitations remain. For example, as the oligonucleotide target is artificially prelabeled, it can only minimize the variations during the hybridization process. Variations from sample amplification and labeling do exist. Thus far, there is no satisfactory solution to these problems. Another limitation is that this method requires a mixture of the reference probe and a gene probe to co-spot for each feature, however, this strategy would be difficult to implement in in situ-synthesized oligonucleotide arrays. Synthesis of the reference probe randomly across the array surface may allow for the use of the CORS for these types of arrays, although this has not been tested.
In conclusion, the developed CORS method is a useful approach for the normalization and comparison of functional gene array data from different samples, experiments, time points, and/or laboratories. It performs equally well in comparison with other commonly used normalization methods when tested with pure culture arrays. Thus, this method can be adapted for normalizing and comparing the data of other types of spotted arrays in general, especially for arrays used to study microbial communities since the more commonly used normalization methods will not work with such complex samples.
Acknowledgments
We thank Gene Wickham for design of the random reference probe.
This study was supported by the U.S. Department of Energy under the Genomics GTL program through the Virtual Institute of Microbial Stress and Survival (http://vimss.lbl.gov); by the Environmental Remediation Science Program, Office of Biological and Environmental Research; by the Office of Science, Oklahoma Center for the Advancement of Science and Technology, under the Oklahoma Applied Research Support Program; and by the National Natural Scientific Foundation of China (no. 40730738).
Footnotes
Published ahead of print on 28 December 2009.
REFERENCES
- 1.Andersen, M., and C. Foy. 2005. The development of microarray standards. Anal. Bioanal. Chem. 381:87-89. [DOI] [PubMed] [Google Scholar]
- 2.Barbacioru, C., Y. Wang, R. Canales, Y. Sun, D. Keys, F. Chan, K. Poulter, and R. Samaha. 2006. Effect of various normalization methods on Applied Biosystems expression array system data. BMC Bioinform. 7:533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bender, K., H. Yen, C. Hemme, Z. Yang, Z. He, Q. He, J. Zhou, K. Huang, E. Alm, T. Hazen, A. Arkin, and J. Wall. 2007. Analysis of a ferric uptake regulator (Fur) mutant of Desulfovibrio vulgaris Hildenborough. Appl. Environ. Microbiol. 73:5389-5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bodrossy, L., N. Stralis-Pavese, M. Konrad-Koszler, A. Weilharter, T. Reichenauer, D. Schofer, and A. Sessitsch. 2006. mRNA-based parallel detection of active methanotroph populations by use of a diagnostic microarray. Appl. Environ. Microbiol. 72:1672-1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho, J., and J. Tiedje. 2001. Bacterial species determination from DNA-DNA hybridization by using genome fragments and DNA microarrays. Appl. Environ. Microbiol. 67:3677-3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cho, J., and J. Tiedje. 2002. Quantitative detection of microbial genes by using DNA microarrays. Appl. Environ. Microbiol. 68:1425-1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dudley, A., J. Aach, M. Steffen, and G. Church. 2002. Measuring absolute expression with microarrays with a calibrated reference sample and an extended signal intensity range. Proc. Natl. Acad. Sci. U. S. A. 99:7554-7559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fields, M., C. Bagwell, S. Carroll, T. Yan, X. Liu, D. Watson, P. Jardine, C. Criddle, T. Hazen, and J. Zhou. 2006. Phylogenetic and functional biomakers as indicators of bacterial community responses to mixed-waste contamination. Environ. Sci. Technol. 40:2601-2607. [DOI] [PubMed] [Google Scholar]
- 9.Fierer, N., and R. Jackson. 2006. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. U. S. A. 103:626-631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gentry, T., G. Wickham, C. Schadt, Z. He, and J. Zhou. 2006. Microarray applications in microbial ecology research. Microb. Ecol. 52:159-175. [DOI] [PubMed] [Google Scholar]
- 11.He, Q., K. Huang, Z. He, E. Alm, M. Fields, T. Hazen, A. Arkin, J. Wall, and J. Zhou. 2006. Energetic consequences of nitrite stress in Desulfovibrio vulgaris Hildenborough, inferred from global transcriptional analysis. Appl. Environ. Microbiol. 72:4370-4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He, Z., T. Gentry, C. Schadt, L. Wu, J. Liebich, S. Chong, Z. Huang, W. Wu, B. Gu, P. Jardine, C. Criddle, and J. Zhou. 2007. GeoChip: a comprehensive microarray for investigating biogeochemical, ecological and environmental processes. ISME J. 1:67-77. [DOI] [PubMed] [Google Scholar]
- 13.He, Z., L. Wu, X. Li, M. Fields, and J. Zhou. 2005. Empirical establishment of oligonucleotide probe design criteria. Appl. Environ. Microbiol. 71:3753-3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He, Z., and J. Zhou. 2008. Empirical evaluation of a new method for calculating signal-to-noise ratio for microarray data analysis. Appl. Environ. Microbiol. 74:2957-2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan, R., G. Gonye, G. Gao, and J. Schwaber. 2006. A universal reference sample derived from clone vector for improved detection of different gene expression. BMC Genomics 7:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Konig, R., D. Baldessari, N. Pollet, C. Niehrs, and R. Eils. 2004. Reliability of gene expression ratios for cDNA microarrays in multiconditional experiments with a reference design. Nucleic Acids Res. 32:e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li, X., Z. He, and J. Zhou. 2005. Selection of optimal oligonucleotide probes for microarrays using multiple criteria, global alignment and parameter estimation. Nucleic Acids Res. 33:6114-6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang, Y., X. Zhang, D. Dai, and G. Li. 2009. Porous biocarrier-enhanced biodegradation of crude oil contaminated soil. Int. Biodeter. Biodegr. 63:80-87. [Google Scholar]
- 19.Loy, A., C. Schulz, S. Lucker, A. Schopfer-Wendels, K. Stoecker, C. Baranyi, A. Lehner, and M. Wagner. 2005. 16S rRNA gene-based oligonucleotide microarray for environmental monitoring of the betaproteobacterial order “Rhodocyclales.” Appl. Environ. Microbiol. 71:1373-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukhopadhyay, A., Z. He, E. Alm, A. Arkin, E. Baidoo, S. Borglin, W. Chen, T. Hazen, Q. He, H. Holman, K. Huang, R. Huang, D. Joyner, N. Katz, M. Keller, P. Oeller, A. Redding, J. Sun, J. Wall, J. Wei, Z. Yang, H. Yen, J. Zhou, and J. Keasling. 2006. Salt stress in Desulfovibrio vulgaris Hildenborough: an integrated genomics approach. J. Bacteriol. 188:4068-4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.North, N., S. Dollhopf, L. Petrie, J. Istok, D. Balkwill, and J. Kostka. 2004. Change in bacterial community structure during in situ Biostimulation of subsurface sediment cocontaminated with uranium and nitrate. Appl. Environ. Microbiol. 70:4911-4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peixoto, B., R. Vencio, C. Egidio, L. Mota-Vieira, S. Verjovski-Almeida, and E. Reis. 2006. Evaluation of reference-based two-color methods for measurement of gene expression ratios using spotted cDNA microarrays. BMC Genomics 7:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rhee, S., X. Liu, L. Wu, S. Chong, X. Wan, and J. Zhou. 2004. Detection of genes involved in biodegradation and biotransformation in microbial communities by using 50-mer oligonucleotide microarrays. Appl. Environ. Microbiol. 70:4303-4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vinciotti, V., R. Khanin, D. D'Alimonte, X. Liu, N. Cattini, G. Hotchkiss, G. Bucca, O. de Jesus, J. Rasaiyaah, C. Smith, P. Kellam, and E. Wit. 2005. An experimental evaluation of a loop versus a reference design for two-channel microarrays. Bioinformatics 21:492-501. [DOI] [PubMed] [Google Scholar]
- 25.Wang, X., S. Jia, L. Meyer, M. Yassai, Y. Naumov, J. Gorski, and M. Hessner. 2007. Quantitative measurement of pathogen specific human memory T-cell repertoire diversity using a CDR3 beta-specific microarray. BMC Genomics 8:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weil, M., T. Macatee, and H. Garner. 2002. Toward a universal standard: comparing two methods for standardizing spotted microarray data. Biotechniques 32:1310-1314. [DOI] [PubMed] [Google Scholar]
- 27.Wu, L., L. Kellogg, A. H. Devol, J. M. Tiedje, and J. Zhou. 2008. Microarray-based characterization of microbial community functional structure and heterogeneity in marine sediments from the Gulf of Mexico. Appl. Environ. Microbiol. 74:4516-4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu, L., X. Liu, C. W. Schadt, and J. Zhou. 2006. Microarray-based analysis of subnanogram quantities of microbial community DNAs by using whole-community genome amplification. Appl. Environ. Microbiol. 72:4931-4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yergeau, E., S. Kang, Z. He, J. Zhou, and G. Kowalchuk. 2007. Functional microarray analysis of nitrogen and carbon cycling genes across an Antarctic latitudinal transect. ISME J. 1:163-179. [DOI] [PubMed] [Google Scholar]
- 30.Zhou, J., M. Bruns, and J. Tiedje. 1996. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 62:316-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou, J., M. Fries, J. Cheesanford, and J. Tiedje. 1995. Phylogenetic analyses of a new group of denitrifiers capable of anaerobic growth on toluene and description of Azoarcus tolulyticus sp. nov. Int. J. Syst. Evol. Microbiol. 45:500-506. [DOI] [PubMed] [Google Scholar]
- 32.Zhou, J., S. Kang, C. W. Schadt, and C. T. Garten. 2008. Spatial scaling of functional gene diversity across various microbial taxa. Proc. Natl. Acad. Sci. U. S. A. 105:7768-7773. [DOI] [PMC free article] [PubMed] [Google Scholar]