

Abstract
The measurement of carbon dioxide production rates as an indication of metabolic activity was applied to study biofilm development and response of Pseudomonas sp. biofilms to an environmental disturbance in the form of a moving air-liquid interface (i.e., shear). A differential response in biofilm cohesiveness was observed after bubble perturbation, and the biofilm layers were operationally defined as either shear-susceptible or non-shear-susceptible. Confocal laser scanning microscopy and image analysis showed a significant reduction in biofilm thickness and biomass after the removal of the shear-susceptible biofilm layer, as well as notable changes in the roughness coefficient and surface-to-biovolume ratio. These changes were accompanied by a 72% reduction of whole-biofilm CO2 production; however, the non-shear-susceptible region of the biofilm responded rapidly after the removal of the overlying cells and extracellular polymeric substances (EPS) along with the associated changes in nutrient and O2 flux, with CO2 production rates returning to preperturbation levels within 24 h. The adaptable nature and the ability of bacteria to respond to environmental conditions were further demonstrated by the outer shear-susceptible region of the biofilm; the average CO2 production rate of cells from this region increased within 0.25 h from 9.45 ± 5.40 fmol of CO2·cell−1·h−1 to 22.6 ± 7.58 fmol of CO2·cell−1·h−1 when cells were removed from the biofilm and maintained in suspension without an additional nutrient supply. These results also demonstrate the need for sufficient monitoring of biofilm recovery at the solid substratum if mechanical methods are used for biofouling control.
Spatial differences in biofilm cohesiveness have been observed after the application of increased shear forces. Coufort et al. (8) subjected both aerobic and anaerobic biofilms, cultivated on ethanol or wastewater, to increased shear stress and found that the biofilm layer at the bulk liquid interface was removed by slight increases in shear rates (0.2 Pa), whereas the middle and base biofilm layers were able to resist removal when exposed to up to 2 Pa and 13 Pa, respectively (8). Total organic carbon (TOC) analyses indicated that the sensitive top layer of the biofilm contained approximately 60% of the total biofilm biomass while the remaining two layers each represented approximately 20%. In a follow-up study, biofilms grown under similar conditions exhibited comparable degrees of heterogeneity in the susceptibility of the various biofilm layers to shear and abrasion (9). It was also indicated that the basal biofilm layer contained active microorganisms, as characterized by oxygen uptake rates, but no details were provided on the methodology or time lapse after the removal of the less-cohesive upper biofilm layers.
Spatial differentiation in metabolic activity in biofilms has also been noted. Most experimental strategies to determine biofilm activity have been centered on microscopy in combination with fluorescent reporter genes or probes that target various indicators of physiological activity in the cell. Several fluorescent stains have been applied previously, such as 5-cyano-2,3-ditolyl tetrazolium chloride (CTC) (15) and acridine orange (27) as well as the commercially available BacLight viability kit (17). Reporter gene expression is another means to evaluate physiological activity in a biofilm. Alkaline phosphatase activity correlated well with oxygen penetration into the upper layers (30 μm) of 117- to 151-μm-thick biofilms (28).
Although all of the above approaches have been shown to be effective, most suffer from inherent disadvantages (26), including incomplete penetration of fluorescent stains and the production of artifacts, and, perhaps most significantly, generally allow only end point analysis due to cellular toxicity. Reporter gene technologies may circumvent this problem but require prior genetic manipulation, and it is unknown what, if any, changes in cell physiology may occur as a result of expression of the reporter gene. The need for genetic manipulation further constrains analysis to pure culture studies.
The basis for spatial heterogeneity in biofilm physiological activity is widely accepted, as previously reviewed (25, 26). Limited diffusion of nutrients and oxygen into the biofilm from the bulk liquid and waste products from a multilayered biofilm are among the simplest explanations since the absence of a complete exchange with the environment, in concert with microbial activity, leads to the formation of chemical gradients in the biofilm. The bacteria in the biofilm respond to the gradients, likely by altering gene expression patterns as determined by global regulators. The remarkable recalcitrance of biofilms toward many antimicrobials may in part be due to the insensitivity of dormant cells in the regions of the biofilm where limited diffusion reduces metabolic activity.
The effect of air bubbles on biofilm stability has mostly been studied in a dental context, where biofilm removal is the goal. Gomez-Suarez et al. (11) utilized a single bubble to investigate the strength of bacterial cell adhesion to various surfaces (11). According to the authors, the probability of cell detachment due to the movement of an air bubble over an attached cell is determined by several factors, namely, collision efficiency, bubble-bacteria attachment efficiency, and the stability of the bubble-bacteria aggregate. For a bubble spanning the entire width of a flow chamber, the collision efficiency is expected to be equal to 1 although the velocity of the bubble may also influence the detachment efficiency since a rapidly moving bubble will result in a thicker liquid film surrounding the bubble, which in turn decreases the collision efficiency. Bacterium-substratum adhesion forces of ∼10−9 N were estimated, which is significantly smaller than the detachment force of a bubble moving over an attached cell (up to 10−7 N).
Liquid flow in most environments—in nature, industry, and clinical or dental settings—typically shows much variation. It can be expected that microbial biofilms have evolved to manage this variability and even to utilize the resulting differences in flow to optimize activity (e.g., the prevention of excessive biomass accumulation for the maintenance of optimum gradients of nutrients and gases) or to relocate to more favorable environments.
Furthermore, increased shear is a recognized strategy to remove unwanted microbial growth from surfaces; therefore, methods to measure the effect of shear on biofilms, including biofilm recovery after partial shear-induced removal, should contribute to our overall understanding of this important form of microbial existence. We developed an approach that measures CO2 production as an indication of biofilm activity in real time and combined this method with confocal laser scanning microscopy (CLSM) and cell yield measurements to study activity-structure relationships in biofilms. This approach is an extension of the one we described in 2009 (18) and enables us to comment on differences in metabolic activity of the whole biofilm versus that of the shear-susceptible biofilm region and to compare biofilm-derived planktonic cells with those growing in batch culture.
MATERIALS AND METHODS
Strain and culture conditions.
Pure culture biofilms of Pseudomonas sp. CT07 (2), labeled with the green fluorescent protein as previously described (1), were cultivated in flow cells under continuous flow of a sterile, defined growth medium [final concentrations of 1.51 mM (NH4)2SO4, 3.37 mM Na2HPO4, 2.20 mM KH2PO4, 179 mM NaCl, 0.1 mM MgCl2·6H2O, 0.01 mM CaCl2·2H2O, and 0.001 mM FeCl3] (7) with 5 mM sodium citrate as the sole carbon source.
Continuous-flow culture of biofilms. (i) Experimental setup.
Multichannel flow cells were milled from Plexiglas, as previously described (29), with individual chamber dimensions of 7.22 mm by 3.00 mm by 60.0 mm (width, depth, and length, respectively). A glass coverslip was attached to the flow cell with silicone adhesive and served as the primary attachment surface for microscopic studies. The flow chambers were connected to growth medium reservoirs with silicone tubing (inner diameter, 1.58 mm), and Watson-Marlow 205S peristaltic pumps supplied the growth medium to the biofilm at a constant flow rate (Fl) of 37.5 ml·h−1 or a linear velocity of 0.47 mm·s−1 (Reynolds number of 2.00; shear rate of 3.58 s−1).
(ii) Disinfection and inoculation.
The system was disinfected after assembly with a 1 in 10 dilution of a commercial bleach solution in distilled H2O (dH2O) for 2 h, followed by continuous irrigation with sterile dH2O for a minimum of 15 h. The dH2O was displaced with sterile growth medium for 20 min before the flow was stopped, and each chamber was inoculated with 0.2 ml of a Pseudomonas sp. CT07::gfp-2 preculture using a sterile needle and syringe. The flow of medium was resumed after 30 min. Since the medium dilution rate in the flow chambers (28.47 h−1) significantly exceeded the maximum specific planktonic growth rate (μmax planktonic) of Pseudomonas sp. CT07::gfp-2 in the 5 mM citrate medium (μmax planktonic, 0.35 ± 0.05 h−1 for six replicate batch cultures), it was concluded that planktonic cell replication did not contribute to the effluent cell numbers; i.e., microbial cells in the flow cell effluent originated from the attached biofilm.
(iii) Experimental conditions and perturbation.
Biofilms were cultured in multiple flow chambers (n = 9) for 8 days. Various parameters were investigated at 24-h intervals, including biofilm CO2 production and total and viable biofilm-derived cell numbers in the effluent (n = 6). The extent of biofilm formation on the glass coverslip was investigated in three additional flow chambers with CLSM.
After 5 days of incubation, a single air bubble was introduced into all of the flow chambers to remove the shear-susceptible fraction of the biofilm. Each bubble was generated by disconnecting the silicone tube from the growth medium reservoir upstream of the peristaltic pump. An air bubble, large enough to fill the entire flow chamber volume, was introduced at a linear flow rate of 0.47 mm·s−1, as controlled by the pump head, and allowed to move through the chamber. The flow cell was tilted vertically with the inlet at the top to ensure that the air bubble spanned the entire chamber cross-section and contacted all sides as it traveled through the chamber. The displaced biofilm material (containing the shear-susceptible biofilm region as well as the bulk liquid) was clearly visible as a slimy, milky-white layer ahead of the bubble, which increased in size as the bubble sheared off the attached biomass as the bubble moved through the chamber before the biomass was collected at the outlet for subsequent analyses. After collection, the up- and downstream tubing were reconnected, and six of the flow chambers were refilled with medium to measure recovery over 2 to 3 days (three flow chambers for CO2 production and effluent cell number enumeration and three flow chambers for CLSM). Cells from the non-shear-susceptible biofilm (i.e., the biomass that resisted shear removal) in the three remaining flow chambers were removed by sonication, and the cell numbers were determined using direct microscopy.
Viable and total cell counts.
Effluent from two of the six chambers was collected directly from each chamber outlet, after the downstream tubing was disconnected, for serial dilution in sterile saline (0.9% NaCl) and spread-plating onto agar plates containing the 5 mM citrate defined medium to confirm bacterial viability in the effluent.
Approximately 1.5 ml of effluent was collected directly from each chamber outlet (n = 6) and preserved for subsequent microscopic enumeration of total cell numbers by the addition of formaldehyde to a final concentration of 2.0% (vol/vol), followed by vortexing and storage at 4°C (14). Each sample was diluted in sterile saline to achieve a minimum of 20 cells per microscope field after incubation with the fluorescent nucleic acid stain 4′,6-diamidino-2-phenylindole (DAPI; final concentration of 5 μg·ml−1) for a minimum of 20 min in the dark. Five milliliters of the stained, diluted sample was vacuum filtered onto a 0.2-μm-pore-size black, polycarbonate filter (25-mm diameter; Nuclepore, Whatman) before each filter was mounted onto a glass slide; a coverslip was fixed into place with a drop of Citifluor antifade mounting medium AF2 (catalogue number 17971-25; Electron Microscopy Sciences). Each filter was examined with a 63× oil immersion lens (Plan Apochromat 63 ×/1.4) with a Zeiss Axiovert 200 M inverted epifluorescent microscope (Zeiss), and 40 to 50 images of microscope fields were captured at random from each filter and used for digital image analysis.
The number of cells in each microscope field was determined with image analysis after a threshold was determined manually with Scion Image for Windows (Scion Corporation and the public-domain NIH Image program [http://rsb.info.nih.gov/nih-image/]), using digital imaging filtering steps as previously described (21).
Dispersion of shear-susceptible and base biofilm layers.
The bubble-displaced content of each flow chamber was collected as described, and the volume was determined (approximately 1.3 ml). This fraction contained liquid medium, single (i.e., nonaggregated) biofilm-derived planktonic cells, and the shear-susceptible portion of the biofilm biomass. In order to enumerate the number of cells in these samples, it was necessary to disperse the cells from the biofilm matrix. Methanol was added to each sample to a final concentration of 1% (vol/vol), followed by sonication in an ultrasonic cleaner (Elma Ultrasonic LC20/H) at 35°C at 35 kHz for 15 min (19). The samples were preserved by the addition of formaldehyde and stored prior to enumeration by direct fluorescent counting as described previously.
The number of cells in the non-shear-susceptible biofilm layer that remained in the flow chambers after the bubble perturbation was also enumerated by direct counting. Three of the emptied flow chambers were filled with 1% methanol, sealed, and sonicated as described previously. After sonication, the content of each chamber was collected in a microcentrifuge tube with repeated up and down pipetting to aid in the removal of biomass from the surfaces. Each chamber was filled with methanol for a second time and sonicated, and the contents were collected to determine the removal efficiency of the first sonication step; analysis of the results indicated that the second sonication step removed 2 orders of magnitude fewer cells than the first, which showed that the first removal step was effective. These samples were preserved in the same manner as the shear-susceptible samples prior to direct fluorescent counting.
CLSM and COMSTAT.
The extent of biofilm formation in three independent flow chambers was examined daily using a Zeiss CLSM microscope with a 20× Plan Apochromat objective and excitation from a 488-nm argon laser. The fluorescence emitted by the gfp protein was detected with a long-pass 505-nm filter. Ten microscope fields (area of 0.002 cm2) were chosen at random along a central transect ranging from the chamber inlet to outlet, and a stack of images was captured in the Z direction with the Zeiss LSM 510 software (version 3.2 SP2) for subsequent image analysis.
Each Z image stack was exported from the Zeiss LSM Image Browser (version 4.2.0.121) as a raw series of single images and converted to gray-scale tagged-image file format (TIF) files with the freeware IrfanView (version 4.10) for analysis with the COMSTAT program (http://www.im.dtu.dk/comstat), which runs as a script in MatLab, equipped with the Image Processing Toolbox (13). Only a selected number of the COMSTAT functions were used for the analysis of the biofilm: the biovolume of each image stack, expressed as the volume of biomass per substratum area (μm3·μm−2), the mean thickness of the biofilm (μm), the maximum thickness of the biofilm (μm), the biofilm surface-to-volume ratio (μm2·μm−3), and the dimensionless roughness coefficient, which is a product of biofilm thickness variability and provides a measure of structural heterogeneity.
CMR setup.
The CO2 production measurement reactor (CMR) consisted of a 20-ml serum vial equipped with a butyl rubber stopper containing four ports (one port each for inflowing liquid, outflowing liquid, inflowing gas, and outflowing gas), as well as a magnetic stirrer bar. All of the tubing connecting the flow cell, CMR, and the CO2 gas analyzer consisted of Tygon, which has very low gas permeability. Sterile growth medium with or without dissolved CO2 (for kla determination) or flow cell effluent (to measure the steady-state biofilm CO2 production rate) was delivered to the CMR at a constant flow rate (Fl = 37.5 ml·h−1) provided by a Watson-Marlow peristaltic pump. Air without CO2 (grade TOC of <0.5 ppm of CO2; Linde Canada) was bubbled through the liquid in the CMR at a constant gas flow rate (Fg = 1,530 ml·h−1) provided by a Watson-Marlow peristaltic pump. The gas flow rate was determined by volumetric displacement. Off-gas CO2 was measured with an absolute, nondispersive, infrared LI-820 CO2 gas analyzer (Li-Cor Biosciences, NE).
The CMR was situated downstream of the flow chambers (Fig. 1), which allowed the real-time, nondestructive measurement of the CO2 produced by the whole biofilm as well as of the contributions of the shear-susceptible biofilm layer, the remaining base biofilm layer after the removal of the shear-susceptible layer, and the biofilm-derived planktonic cells exiting the flow cell in the effluent. The CO2 production and contributions of the different biofilm fractions were measured in the gas phase using two different measuring regimes, namely, open- and closed-loop configurations.
FIG. 1.

A schematic diagram of the experimental setup showing the flow cell and the CMR.
Theory for open loop.
Measurement of the total amount of CO2 produced by an intact (in situ) biofilm growing in a flow cell was accomplished by using the CMR in an open-loop configuration (clamp 3 was closed, while clamps 1, 2, and 4 remained open, as shown in Fig. 1). A CO2 mole balance over the flow cell at steady state (with no accumulation) can be written as follows:
 |
(1) |
where Fl,FC,in = Fl,FC,out is the liquid flow rate into and out of the flow cell, respectively (liter·h−1), cFC,in is the CO2 concentration in the sterile liquid medium entering the flow cell (μmol of CO2·liter−1 of liquid medium), cFC,out is the CO2 concentration exiting the flow cell and serves as the feed to the CMR (this assumes that all of the CO2 produced in the flow cell reached the CMR since the connecting Tygon tubing is highly impermeable to CO2, thus recognizing that cFC,out = cA,CMR,in), and CERFC is the CO2 production rate (μmol of CO2·h−1) of all microbial cells within the flow cell (sum of CO2 contributions from all three in situ biofilm fractions, namely, the biofilm-derived planktonic cells and the shear-susceptible and base biofilm layers).
For a gas that does not interact with the liquid phase, the gas mole balance across the CMR can be written as:
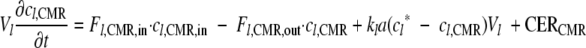 |
(2) |
where Vl is the liquid volume in the CMR (liter); cl,CMR, is the dissolved gas concentration in the liquid phase in the CMR (μmol of gas·liter−1 of liquid medium); Fl,CMR,in is the liquid flow rate into the CMR (liters of liquid medium·h−1); Fl,CMR,out is the liquid flow rate out of the CMR (liters of liquid medium·h−1) where Fl,CMR,in = Fl,CMR,out; kla is the volumetric transfer coefficient (h−1); c*l is the concentration of gas in the liquid phase that is in equilibrium with the gas phase (μmol of gas·liter−1 liquid medium); and CERCMR is the gas produced in the system (μmol of CO2·h−1).
Dissolved CO2 interacts with water, however, and can convert to bicarbonate, carbonate, and carbonic acid, depending on environmental conditions such as the pH, temperature, and ionic strength of the solution (24). To accommodate the conversion of dissolved CO2 to the bicarbonate ion under the experimental pH conditions, Bonarius et al. (5) introduced cA as a term to include both of the forms of CO2 in the liquid phase, i.e., the sum of the dissolved CO2 (cCO2) and the bicarbonate ion (cHCO3), or cA = cCO2 + cHCO3 (5). The equilibrium constant, K1, which relates the equilibrium of dissolved CO2 and the bicarbonate ion
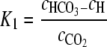 |
can then be used to find a relationship between cA and cCO2:
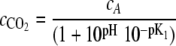 |
(3) |
It should be noted that only the dissolved form of CO2 can move cross the gas-liquid interface and be transferred to the gas phase (i.e., bicarbonate ions cannot). Therefore, at steady state and a CERCMR of 0 (CO2 produced by the biofilm-derived planktonic cells in the CMR from the effluent is negligible; see below), substituting equation 3 into equation 2 (taking the “transfer” of dissolved CO2 to bicarbonate ions due to the pH range of the experiment into account) yields:
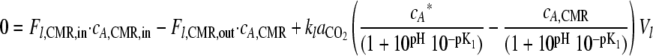 |
(4) |
A gas-phase CO2 balance around the CMR yields:
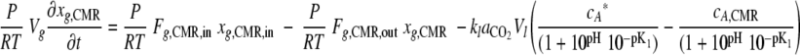 |
(5) |
where Vg is the headspace volume (liter), Fg,CMR,in is the total gas volumetric flow rate into the CMR (liters of gas·h−1), Fg,CMR,out is the total gas volumetric flow rate out of the CMR (Fg,CMR,in = Fg,CMR,out), xg,CMR,in is the CO2 concentration of gas flowing into the CMR (ppm; μl of CO2·liter−1 of total gas), xg,CMR is the CO2 concentration in the gas phase in the CMR (ppm; μl of CO2·liter−1 of total gas), P is the pressure in the CMR (kPa), R is the universal gas constant (liter·kPa·K−1·mol−1), and T is the temperature of the gas in the CMR (in K).
At steady state and with xg,CMR,in = 0 (CO2 free air was used for gas flow), equation 5 becomes:
 |
(6) |
Substituting equation 6 into equation 4 yields:
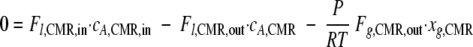 |
(7) |
Considering the Henry's law relationship of CO2 concentration in the gas phase in equilibrium with the liquid phase and equation 3 yields:
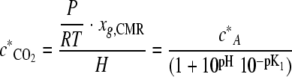 |
(8) |
where H is the dimensionless Henry's law coefficient (liter of liquid·liter−1 of gas). Equation 6 and equation 8 yield:
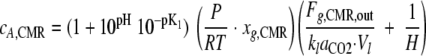 |
(9) |
Substituting equation 9 into equation 7 yields:
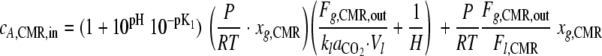 |
(10) |
By substituting equation 10 into equation 1, it is possible to determine the CERFC (CO2 contribution from all of the biomass fractions in the flow cell). The CO2 contribution of the base layer was determined by solving equation 1 after the bubble had removed the shear-susceptible biofilm layer, and the CO2 contribution of the shear-susceptible layer was calculated as the difference between the total biofilm CO2 production and the base layer.
To determine the CO2 contribution of the shear-susceptible biofilm fraction and the planktonic cells (i.e., biofilm-derived planktonic cells as well as planktonic cells harvested during exponential growth in batch culture), the CMR was used in a closed-loop configuration (clamps 1 and 2 were closed after a known volume of effluent was pumped into the CMR, followed by closure of clamp 4; clamp 3 remained open with no flow from the compressed CO2-free air tank) (Fig. 1).
Theory for the closed loop.
In the closed-loop (batch) system, the total amount of CO2 at a specific time was considered a combination of the CO2 in the liquid and gas phases (6):
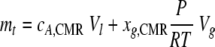 |
(11) |
where mt is the number of the moles of CO2 in the system at time t. In the closed loop it is assumed that the CO2 in the liquid phase is in equilibrium with the gas phase, similar to equation 8 (10):
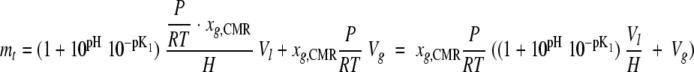 |
(12) |
It can therefore be seen that the total amount of CO2 in the closed vessel can be determined by measuring the CO2 concentration only in the gas phase.
By measuring the total amount of CO2 in the system at different time intervals, it is thus possible to determine a rate of CO2 produced (μmol of CO2·h−1) by microbes in the CMR. During short time intervals (<10 min), the increase in total CO2 of the system can be attributed primarily to respiration and not to a significant increase in cell numbers.
Defining the essential parameters for the CMR. (i) Determining the volumetric transfer coefficient (kla).
The kla (h−1) was determined using the static method, as described previously (3), which involves solving both the gas and liquid mass balances for the CMR. Steady-state off-gas measurements were obtained for known dissolved gas concentrations (cA,CMR,in) in the open-loop system configuration, and the kla was calculated by rearranging equation 10.
Known concentrations of CO2 were generated by agitating solid-state CO2 pellets (dry ice) in the 5 mM citrate growth medium until saturation. This concentrated dissolved CO2 solution was diluted in sterile growth medium to concentrations that resulted in off-gas readings similar to those obtained from the flow cell effluent. The concentration of dissolved CO2 was determined by measuring the inorganic carbon content with a catalytic combustion, nondispersive infrared total carbon analyzer (TOC-VCSH/CSN; Shimadzu, Kyoto, Japan). Growth medium, without the addition of the dissolved CO2, was used as a blank to determine the off-gas reading for growth medium equilibrated with atmospheric CO2.
(ii) Determining the headspace volume (Vg).
Since the CMR system was assembled in the laboratory, it was necessary to determine the headspace (Vg, in liters) experimentally. A liquid-free CMR was prepared and flushed with CO2-free air until no CO2 could be detected in the off-gas. Precise volumes of growth medium with known dissolved CO2 concentrations, prepared as described above, were injected into the CMR, and the CO2 in the off-gas was measured in the closed-loop system configuration. Known concentrations of dissolved CO2 allowed the determination of Vg according to equation 12.
(iii) Calculation of the dimensionless Henry's coefficient (H).
The dimensionless Henry's coefficient (cgas/cliq; liters of gas/liters of liquid) for CO2 was calculated to be 1.12 at 22°C with data from Schumpe et al. (24) and corrected for the ionic strength of the growth medium (24).
Open-loop calculations to determine steady-state whole-biofilm CO2 production.
Whole-biofilm CO2 production rates were measured at 24-h intervals with the CMR in an open-loop configuration, i.e., continuous, once-through flowthrough of both the liquid and gas phases. Effluent exiting the flow cell entered the CMR at a known flow rate (Fl = 37.5 ml·h−1) while being sparged with CO2-free air as before (Fg = 1,530 ml·h−1). Equation 1 was used in conjunction with equation 10 to determine the CO2 production of the in situ whole biofilm. The values for the various parameters were those applied during the kla determination, except that in this case the cA,CMR,in was calculated from the experimental steady-state gas phase CO2 values captured during measurements by the off-gas analyzer.
Closed-loop calculations to determine CO2 production of the shear-susceptible biofilm region and planktonic cells.
The closed-loop method was selected as a means to distinguish the CO2 produced by the whole biofilm (originating in the flow cell upstream of the CMR) and that produced by the biofilm-derived planktonic cells swept along by the exiting liquid flow, as well as the shear-susceptible biofilm fraction. The closed-loop method measures the increase in CO2 in the CMR only as a result of respiring suspended cells collected in a known volume of flow cell effluent or bubble-displaced content. In contrast, an open-loop system measures dissolved CO2 in the continuous-flow liquid, thus also measuring CO2 that originated from biofilm cells upstream in the flow cell.
In the closed-loop configuration the liquid flow into and out of the CMR was halted, while continuous stirring of the liquid phase in the CMR was maintained by the magnetic stirrer. The gas was continuously circulated through the system via a peristaltic pump at flow rate of 1,530 ml·h−1. Prior to the closed-loop measurement, the flow cell effluent entered the CMR under open-loop conditions, with the liquid flowing through the CMR at a constant rate and CO2-free air being sparged into the liquid. After a steady-state off-gas CO2 reading was obtained, the liquid flow was halted while the liquid continued to be flushed with the CO2-free air for 3 to 5 min to drive off the residual dissolved CO2 that originated from the flow cell biofilm. The liquid in the CMR was subsequently sparged with ambient air until steady-state CO2 off-gas readings were reached, after which the gas flow was closed, resulting in a continuous recirculation of gas in the headspace through the system. A linear increase in CO2 measurements was recorded for at least 10 min or until the measurement exceeded the upper limit of the CO2 analyzer (as in the case of the shear-susceptible layer responses, where the detection limit was exceeded after approximately 4.5 min).
The CO2 produced in the CMR was calculated by using equation 12 with parameters and variables as before and with Vg as the headspace volume (in liters). The calculation involved the arbitrary choice of two CO2 measurements (xg0 and xg1) at specific time points (t0 and t1).
It was shown that CO2 production by the biofilm-derived effluent cells, when not concentrated, was below the closed-loop detection limit. This confirmed that the effluent cells produced a negligible amount of CO2 relative to the biofilm, so there was no need to take this fraction into account for steady-state open-loop measurements.
To measure the CO2 contribution of planktonic cells exiting the flow cell, effluent from three 48- and 72-h-old biofilms was collected on ice for 1.5 h (approximately 160 ml), and the biofilm-derived effluent cells were concentrated by centrifugation at a relative centrifugal force (RCF) of 3,000 (5,000 rpm) for 20 min at 4°C. After the removal of the supernatant, the remaining cell pellets were resuspended in 3.3 ml of sterile growth medium, of which 3 ml was used to determine CO2 production in the closed-loop configuration, and the remainder was used to determine the viable cell numbers after spread plating.
Planktonic batch cultures were incubated until the exponential phase of growth (as monitored by optical density measurements at 600 nm), prior to the harvesting of cells by centrifugation and resuspension as described for the biofilm-derived effluent cells. The viable cell numbers and CO2 production rate for a fixed volume of this suspension were determined in addition to values for an original batch sample and a 1 in 10 dilution of the latter. The centrifugation and resuspension steps taken in preparation of the batch culture samples were found not to influence the CO2 production rate per cell number for the various batch culture samples (correlation of R2 = 0.998 between the amount of CO2 produced per cell number for the various dilutions of exponential phase cells).
Statistical analysis.
Statistical analysis of replicate measurements (for CO2 production rates, direct cell counts, and biofilm parameters) was performed with analysis of variance (ANOVA) and Tukey's test for comparison of means (P = 0.05).
RESULTS
Validation of the experimental system.
The steady-state CO2 (xg; μl of CO2·liter−1 of air) measured in the off-gas for the different dissolved CO2 concentrations was highly linear (R2 = 0.997) and was within the range of the off-gas measurements taken during the subsequent experimentation. A kla value of 21 h−1 was calculated for the experimental setup described here. The influence of the gas flow rate on kla measurements was also investigated, and lower gas flow rates were found to decrease the kla value, which is in agreement with Blanch and Clark (3).
For known dissolved CO2 concentrations that had off-gas readings in a similar range to the level of the whole biofilm, the open- and closed-loop configurations yielded results that were within 5% of each other.
Both pH and kla may have noticeable influences on the determination of dissolved CO2 concentration from off-gas measurements using equation 10. In the current experimental system, the effluent pH (6.92) did not differ significantly from that of the sterile growth medium (pH 6.97). A common way to measure the CO2 volumetric transfer coefficient is to use a linear relationship with the oxygen transfer coefficient, the latter being simpler to measure experimentally (4). Our results indicate (data not shown) that 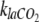 was not constant with varying dissolved CO2 concentrations and that therefore care has to be taken when determining
was not constant with varying dissolved CO2 concentrations and that therefore care has to be taken when determining 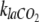 for the particular experimental conditions.
for the particular experimental conditions.
Steady-state whole-biofilm CO2 production.
Whole-biofilm CO2 production rates were measured every 24 h for the first 120 h, at 2 and 4 h after the bubble perturbation, and thereafter again at 24-h intervals for the remainder of the experiment (Fig. 2) using the open-loop system configuration.
FIG. 2.

The average whole-biofilm CO2 production rate (μmol of CO2 produced·h−1) as measured for six biofilms (24 to 120 h) and three biofilms (122 h and 124 to 192 h) using the open-loop system configuration. Up until 120 h, the average CO2 production for six biofilms was determined. After the steady-state measurement of the respiration rate was taken at 120 h, an air bubble was introduced into the each flow cell to remove the shear-susceptible biofilm region. Three of the biofilms were sacrificed at this stage, and open-loop measurements continued on the three remaining flow cells for a total of 192 h.
Removal of the shear-susceptible region led to a significant reduction (P < 0.05) in the rate of CO2 production in the flow cell; only 28% ± 29% of the predisturbance CO2 production rate was maintained by the remaining non-shear-susceptible base biofilm layer 2 h after the perturbation (122 h of incubation). CO2 production in these base biofilm layers recovered to 51% ± 41% of the previous steady-state production rate within 4 h after the removal of the shear-susceptible biofilm fraction and to 104% ± 42% after an additional 24 h. Interestingly, the postdisturbance rate of CO2 evolution stabilized at a higher level than what was observed previously (136% ± 64% at 48 h after the bubble although this increase was not statistically significant at a P value of 0.05).
Biofilm-to-planktonic cell yield.
In addition to respirometry, the number of free-floating cells produced and released into the effluent by the biofilms was determined with direct fluorescent counting and image analysis as well as viable cell counts (Fig. 3). Direct counting allowed the evaluation of the nature of the detached biomass (single cells versus matrix-encased clumps of cells) and facilitated comparison of the cell numbers in the shear-susceptible and base biofilm regions, respectively.
FIG. 3.

The number of cells produced by the biofilms and released into the bulk liquid was enumerated from the flow cell effluent with direct fluorescent counts and plate counts by sampling the effluent at 24-h intervals (n = 6 biofilms from 24 to 120 h, and n = 3 biofilms from 144 to 192 h).
Microscopic examination of the effluent biomass prior to the introduction of the bubble and during biofilm recovery (24, 48, and 72 h subsequent to the removal of the shear-susceptible biofilm layer) indicated that single cells made up the majority of the biomass released from the biofilm as aggregates of matrix-encased cells were rarely present. The yield of cells from the biofilms initially decreased less than a log-fold (a significant decrease for the viable cell counts, but not the direct counts at a P value of <0.05) after the introduction of the bubble despite the fact that a significant fraction of the attached biomass was removed, as suggested by the reduction in the rate of CO2 production after the bubble (Fig. 2) and shown by a series of representative CLSM micrographs of the biofilm taken over the course of the experiment (Fig. 4). Planktonic cell yield from the biofilm recovered to predisturbance levels after 168 to 192 h of incubation (48 and 72 h after the bubble disturbance; statistically there was no significant difference compared to 120 h, with a P value of 0.05).
FIG. 4.

Single CLSM micrographs taken at 24-h intervals at random locations of the gfp-labeled Pseudomonas sp. CT07 biofilms cultivated in conventional flow cells under continuous-flow conditions: 24 h (A), 48 h (B), 72 h (C), 96 h (D), and 120 h (E). (F) The single cells at the glass surface 1 h after the bubble perturbation. The biomass along the edge of the flow cell where the glass coverslip meets the Plexiglas is shown 1 h after the perturbation (G), 24 h after the perturbation (144 h) (H), and 48 h after the perturbation (168 h) (I). Micrographs shown in panels F and G were not included in the image analysis with COMSTAT.
Biofilm architecture.
The bubble perturbation removed a large portion of the biofilm biomass at the glass surface; single cells and some aggregates (Fig. 4F), much smaller than before, were observed attached in isolated regions of the glass along the central transect while more extensive biomass survived the perturbation along the edge of the chamber (Fig. 4G), likely as a result of reduced shear forces in these areas. Image analysis was not performed at this point due to insufficient amounts of biomass at the area normally viewed (i.e., central transect) of the flow chambers. COMSTAT image analysis of the CLSM-acquired images provided information on biofilm architecture in terms of biofilm biomass, average thickness (Fig. 5A), roughness, and the surface area-to-biovolume ratio (Fig. 5B). At the magnification applied, CLSM allowed the observation only of the biofilms growing on the glass coverslips (35% of total surface area available for attachment). However, the observed biomass volume and the average biofilm thickness showed a similar trend as the whole-biofilm CO2 production and did not reach a steady state in the first 120 h of cultivation.
FIG. 5.

COMSTAT image analysis of four biofilm parameters was averaged for three biofilms cultivated under the same conditions, as described previously. The average amount of biofilm biomass at the surface (μm3·μm−2) and average biofilm thickness (μm) are plotted in panel A, and the roughness coefficient and surface area-to-biovolume ratio (μm2·μm−3) are plotted in panel (B). The absence of sufficient biofilm biomass at the glass surface after the bubble perturbation at 121 h (Fig. 4F) did not allow the capture of images suitable for analysis, and hence this data point could not be included.
Biofilm architecture at the glass surface was also significantly altered after the perturbation; the bubble-mediated removal of the shear-susceptible biofilm region resulted in significant changes (P < 0.05) in all of the measured biofilm parameters at 144 h (24 h after the perturbation). Analysis of these parameters indicated that the biofilm was rougher and had a larger surface area-to-volume ratio than the predisturbance biofilm at 120 h and that it consisted of less biomass and exhibited a reduced thickness compared to the predisturbance biofilm at 120 h (Fig. 5A and B). Biofilm biomass and the surface area-to-volume ratio at 144 and 168 h (24 and 48 h after the disturbance) recovered to the levels measured for the 72- and 24-h-old biofilms, respectively, while the mean biofilm thickness recovered to the values observed for a 96-h-old biofilm (P = 0.05). These observations, together with the increased steady-state CO2 production and recovery of biofilm-to-planktonic cell yield, indicated that biofilms remained metabolically active during the first 48 h after the disturbance and that rebuilding of biofilm structure occurred soon after the disturbance.
Comparison of CO2 production rates.
The average rate of CO2 production per cell was compared between planktonic cells in the exponential phase of growth, biofilm-derived effluent cells, the shear-susceptible biofilm layer (both in situ and in the CMR after removal), and the base biofilm layer (Fig. 6). The average cell number of the non-shear-susceptible base biofilm layer at 120 h was 4.84 × 108 ± 1.94 × 108 while that of the shear-susceptible fractions was 6.91 × 108 ± 2.47 × 108. Thus, approximately half of the bacterial cells were removed from the flow cell by the bubble perturbation while only 28% ± 29% of the predisturbance CO2 rate of production was maintained by the remaining base biofilm after the bubble removal. Using the average cell numbers in the respective biofilm layers, it was possible to calculate the steady-state CO2 production rates per cell, as indicated in Fig. 6.
FIG. 6.

The average rate of CO2 production per cell was compared between planktonic cells in the exponential phase of growth, biofilm-derived effluent cells, the shear-susceptible biofilm, and the non-shear-susceptible base biofilm layer. Activity of the shear-susceptible biofilm layer was determined in situ (as part of the biofilm) and as suspended cells after removal from the biofilm.
DISCUSSION
The goal of this investigation was not to measure the shear forces generated by the moving air bubble or the extent to which the Pseudomonas sp. CT07 biofilms can withstand different shear forces, as has been done previously (8), but, rather, to selectively remove a region of the biofilm to evaluate biofilm metabolic response in terms of CO2 production and biofilm-derived planktonic cell yield, as well as biofilm structural adaptation, by utilizing microscopy and image analysis. Other environmental perturbations, such as increased fluid shear, particle abrasion, or an antimicrobial challenge, could instead be utilized to achieve a similar outcome. The choice of an air bubble proved to simplify execution since it facilitated the quick and efficient removal of the shear-susceptible biofilm region for immediate downstream analysis of changes in CO2 production rates, which was an important consideration in order to capture the expected rapid response in microbial metabolism. This approach yielded reproducible results, as can be seen from the notably similar numbers of cells enumerated from the different shear-susceptible regions after dispersion (no statistically significant difference; P value of 0.05).
The results show that biofilm-derived single cells as well as the cells in the shear-susceptible biofilm layer (in situ) are metabolically less active—as indicated by a lower average CO2 production rate on a per cell basis—than exponentially growing planktonic cells but more active than cells in the base biofilm layer 1 h after exposure to the bulk liquid after the removal of the shear-susceptible layer (Fig. 6). The quick increase in the CO2 production rate by the base biofilm layer provides additional support for numerous suggestions that biofilm cells are less active (i.e., dormant, but not dead) for reasons such as diffusion limitation.
The results indicate that the shear-vulnerable layer, which in this case contained half of the biofilm cells, produced 72% of the CO2 in the system, which is consistent with previous indications of spatial heterogeneity in biofilm metabolic activity. Werner et al. (28) demonstrated stratification of active protein synthesis in Pseudomonas aeruginosa biofilms, where most of the activity was located at the bacterial colony-air interfaces, and at the biofilm-liquid interface for biofilms cultivated under continuous-flow conditions (20, 28). Rani et al. also reported on stratified DNA replication, protein synthesis, and respiratory activity in staphylococcal colonies (23). Moreover, these activities were found to be colocated at the biofilm-air interface (upper 31- to 38-μm layers) and the biofilm-nutritive substratum interface (14- to 16-μm layers) of 153- to 172-μm-thick colonies. The authors identified four cell physiologies, namely, active aerobic growth, active anaerobic growth, nonactive or dormant but viable cells at the interior of colonies, and dead cells (approximately 10% of the total), and suggested that the dormant cells might regain activity if exposed to oxygen and/or nutrients.
The average per cell CO2 production rate of the shear-susceptible layer, after removal from the biofilm, was found to be significantly higher than the in situ rate (P value of 0.05) (Fig. 6). This result demonstrates that biofilms possess a remarkable ability to not only respond rapidly to environmental changes but also retain a powerful metabolic capacity when access to nutrients and oxygen becomes unrestricted. Unlike persister cells that are apparently programmed primarily for survival (12, 16), the primary function of cells from this region is probably to optimize proliferation.
It should be pointed out that the term “persisters” usually refers to cells that can survive high antibiotic concentrations while the disturbance in the present study was shear. In contrast to the small percentage of persisters (less than 1% of the total biofilm population in Escherichia coli according to Harrison et al. [12]), we found that approximately 50% of the biofilm cells could withstand the applied shear in this case. An increase in shear would likely result in the removal of a greater percentage of biofilm biomass, but this was outside the scope of the current investigation. The increase in CO2 production after the base layer was exposed (Fig. 2) indicates that, similar to the cells in the shear-susceptible region that rapidly increase their activity when transferred to a planktonic state, these previously dormant (or less active) cells can speedily respond to reestablish overall biofilm activity. The findings reported here are thus in agreement with previous indications of active microorganisms in the basal biofilm layer (9).
Further applications of this approach may be useful in the study of biofilm form-function relationships. The rapid recovery of metabolic activity after the perturbation suggests that biofilms may, indeed, benefit from shear-related perturbations. The removal of excess biomass may facilitate the maintenance of biofilms with an optimized architecture to ensure maximal utilization of resources, as seen by the increase in biofilm roughness once the biofilm was exposed to the bulk liquid after the removal of the shear-susceptible biofilm layer. The increased roughness measured after the perturbation may facilitate greater rates of nutrient and oxygen transfer to the biofilm, which is confirmed by the higher subsequent metabolic activity. Considering that relatively few real-world environments are bubble and turbulence free, such adaptation can be expected, even if it is not a requirement for survival. It is not known whether the cell distribution or EPS composition in the biofilm was altered in response to the shear-mediated removal of the susceptible layer, as has been observed for biofilms cultivated under higher flow rates (22), and so we cannot speculate on whether the resulting biofilm could withstand subsequent changes in shear to a greater degree.
Finally, the approach described here overcame a number of the limitations associated with the methods available to measure biofilm metabolic activity, as described by Stewart and Franklin (26). A notable improvement is that this approach is not dependent on fluorescent stains that are susceptible to incomplete penetration or to the cellular toxicity characteristic of many of these stains. The approach also overcomes the requirement of working with pure cultures that are amenable to genetic manipulation, typical of reporter gene technologies.
Acknowledgments
Financial support from Bioshield Technologies, AFMNet, Canadian Research Chair Program (G.M.W.), NSERC (G.M.W.), and a commonwealth scholarship (E.B.) is gratefully acknowledged.
Footnotes
Published ahead of print on 18 December 2009.
REFERENCES
- 1.Bester, E., E. Edwards, and G. M. Wolfaardt. 2009. Planktonic cell yield is linked to biofilm development. Can. J. Microbiol. 55:1195-1206. [DOI] [PubMed] [Google Scholar]
- 2.Bester, E., G. Wolfaardt, L. Joubert, K. Garny, and S. Saftic. 2005. Planktonic-cell yield of a pseudomonad biofilm. Appl. Environ. Microbiol. 71:7792-7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blanch, H. W. 1996. Biochemical engineering. M. Dekker, New York, NY.
- 4.Bloemen, H. H. J., L. Wu, W. M. Van Gulik, J. J. Heijnen, and M. H. G. Verhaegen. 2003. Reconstruction of the O2 uptake rate and CO2 evolution rate on a time scale of seconds. AICHE J. 49:1895-1908. [Google Scholar]
- 5.Bonarius, H. P. J., C. D. De Gooijer, J. Tramper, and G. Schmid. 1995. Determination of the respiration quotient in mammalian cell culture in bicarbonate buffered media. Biotechnol. Bioeng. 45:524-535. [DOI] [PubMed] [Google Scholar]
- 6.Chai, X.-S., C. Dong, and Y. Deng. 2008. In situ determination of bacterial growth by multiple headspace extraction gas chromatography. Anal. Chem. 80:7820-7825. [DOI] [PubMed] [Google Scholar]
- 7.Clark, D. J., and O. Maaloe. 1967. DNA replication and the division cycle in Escherichia coli. J. Mol. Microbiol. 23:99-112. [Google Scholar]
- 8.Coufort, C., N. Derlon, J. Ochoa-Chaves, A. Liné, and E. Paul. 2007. Cohesion and detachment in biofilm systems for different electron acceptor and donors. Water Sci. Technol. 55:421-428. [DOI] [PubMed] [Google Scholar]
- 9.Derlon, N., A. Massé, R. Escudié, N. Bernet, and E. Paul. 2008. Stratification in the cohesion of biofilms grown under various environmental conditions. Water Res. 42:2102-2110. [DOI] [PubMed] [Google Scholar]
- 10.Dřímal, P., J. Hrncčiřík, and J. Hoffmann. 2006. Assessing aerobic biodegradability of plastics in aqueous environment by GC-analyzing composition of equilibrium gaseous phase. J. Polym. Environ. 14:309-316. [Google Scholar]
- 11.Gómez-Suárez, C., H. J. Busscher, and H. C. van der Mei. 2001. Analysis of bacterial detachment from substratum surfaces by the passage of air-liquid interfaces. Appl. Environ. Microbiol. 67:2531-2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison, J. J., R. J. Turner, and H. Ceri. 2005. Persister cells, the biofilm matrix and tolerance to metal cations in biofilm and planktonic Pseudomonas aeruginosa. Environ. Microbiol. 7:981-994. [DOI] [PubMed] [Google Scholar]
- 13.Heydorn, A., A. T. Nielsen, M. Hentzer, C. Sternberg, M. Givskov, B. K. Ersboll, and S. Molin. 2000. Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology 146:2395-2407. [DOI] [PubMed] [Google Scholar]
- 14.Hobbie, J. E., R. J. Daley, and S. Jasper. 1977. Use of nucleopore filters for counting bacteria by fluorescence microscopy. Appl. Environ. Microbiol. 33:1225-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang, C.-T., F. P. Yu, G. A. McFeters, and P. S. Stewart. 1995. Nonuniform spatial patterns of respiratory activity within biofilms during disinfection. Appl. Environ. Microbiol. 61:2252-2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keren, I., D. Shah, A. Spoering, N. Kaldalu, and K. Lewis. 2004. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 186:8172-8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korber, D. R., A. Choi, G. M. Wolfaardt, S. C. Ingham, and D. E. Caldwell. 1997. Substratum topography influences susceptibility of Salmonella enteritidis biofilms to trisodium phosphate. Appl. Environ. Microbiol. 63:3352-3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kroukamp, O., and G. M. Wolfaardt. 2009. CO2 production as an indicator of biofilm metabolism. Appl. Environ. Microbiol. 75:4391-4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lunau, M., A. Lemke, K. Walther, W. Martens-Habbena, and M. Simon. 2005. An improved method for counting bacteria from sediments and turbid environments by epifluorescence microscopy. Environ. Microbiol. 7:961-968. [DOI] [PubMed] [Google Scholar]
- 20.Mangalappalli-Illathu, A. K., J. R. Lawrence, and D. R. Korber. 2009. Cells in shearable and nonshearable regions of Salmonella enterica serovar Enteritidis biofilms are morphologically and physiologically distinct. Can. J. Microbiol. 55:955-966. [DOI] [PubMed] [Google Scholar]
- 21.Massana, R., J. M. Gasol, P. K. Bjørnsen, N. Blackburn, A. Hagstrom̈, S. Hietanen, B. H. Hygum, J. Kuparinen, and C. Pedrós-Alió. 1997. Measurement of bacterial size via image analysis of epifluorescence preparations: Description of an inexpensive system and solutions to some of the most common problems. Sci. Mar. 61:397-407. [Google Scholar]
- 22.Pereira, M. O., M. Kuehn, S. Wuertz, T. Neu, and L. F. Melo. 2002. Effect of flow regime on the architecture of a Pseudomonas fluorescens biofilm. Biotechnol. Bioeng. 78:164-171. [DOI] [PubMed] [Google Scholar]
- 23.Rani, S. A., B. Pitts, H. Beyenal, R. A. Veluchamy, Z. Lewandowski, W. M. Davison, K. Buckingham-Meyer, and P. S. Stewart. 2007. Spatial patterns of DNA replication, protein synthesis, and oxygen concentration within bacterial biofilms reveal diverse physiological states. J. Bacteriol. 189:4223-4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schumpe, A., G. Quicker, and W.-D. Deckwer. 1982. Gas solubilities in microbial culture media, p. 1-38. In H. Binder et al. (ed.), Reaction engineering. Springer-Verlag, Berlin, Germany.
- 25.Spormann, A. M. 2008. Physiology of microbes in biofilms, p. 17-36. In T. Romeo (ed.), Bacterial biofilms. Springer, Berlin, Germany. [DOI] [PubMed]
- 26.Stewart, P. S., and M. J. Franklin. 2008. Physiological heterogeneity in biofilms. Nat. Rev. Microbiol. 6:199-210. [DOI] [PubMed] [Google Scholar]
- 27.Wentland, E. J., P. S. Stewart, C.-T. Huang, and G. A. McFeters. 1996. Spatial variations in growth rate within Klebsiella pneumoniae colonies and biofilm. Biotechnol. Prog. 12:316-321. [DOI] [PubMed] [Google Scholar]
- 28.Werner, E., F. Roe, A. Bugnicourt, M. J. Franklin, A. Heydorn, S. Molin, B. Pitts, and P. S. Stewart. 2004. Stratified growth in Pseudomonas aeruginosa biofilms. Appl. Environ. Microbiol. 70:6188-6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolfaardt, G. M., J. R. Lawrence, R. D. Robarts, S. J. Caldwell, and D. E. Caldwell. 1994. Multicellular organization in a degradative biofilm community. Appl. Environ. Microbiol. 60:434-446. [DOI] [PMC free article] [PubMed] [Google Scholar]