

Abstract
Reduced migratory function of circulating angiogenic progenitor cells (CPCs) has been associated with impaired neovascularization in patients with cardiovascular disease (CVD). Previous findings underline the role of the kallikrein-kinin system in angiogenesis. We now demonstrate the involvement of the kinin B2 receptor (B2R) in the recruitment of CPCs to sites of ischemia and in their proangiogenic action. In healthy subjects, B2R was abundantly present on CD133+ and CD34+ CPCs as well as cultured endothelial progenitor cells (EPCs) derived from blood mononuclear cells (MNCs), whereas kinin B1 receptor expression was barely detectable. In transwell migration assays, bradykinin (BK) exerts a potent chemoattractant activity on CD133+ and CD34+ CPCs and EPCs via a B2R/phosphoinositide 3-kinase/eNOS-mediated mechanism. Migration toward BK was able to attract an MNC subpopulation enriched in CPCs with in vitro proangiogenic activity, as assessed by Matrigel assay. CPCs from cardiovascular disease patients showed low B2R levels and decreased migratory capacity toward BK. When injected systemically into wild-type mice with unilateral limb ischemia, bone marrow MNCs from syngenic B2R-deficient mice resulted in reduced homing of sca-1+ and cKit+flk1+ progenitors to ischemic muscles, impaired reparative neovascularization, and delayed perfusion recovery as compared with wild-type MNCs. Similarly, blockade of the B2R by systemic administration of icatibant prevented the beneficial effect of bone marrow MNC transplantation. BK-induced migration represents a novel mechanism mediating homing of circulating angiogenic progenitors. Reduction of BK sensitivity in progenitor cells from cardiovascular disease patients might contribute to impaired neovascularization after ischemic complications.
Keywords: bradykinin, migration, progenitor recruitment, angiogenesis, therapeutic neovascularization
Accumulating evidence corroborates the relevance of a heterogeneous population of bone marrow (BM)-derived circulating cells, widely referred to as endothelial progenitor cells (EPCs), for the maintenance of endothelial function and vascular integrity and for postnatal vessel growth.1 Efficient recruitment of EPCs to the injured tissue is fundamental for their proangiogenic function. Consistently, in ischemic pathologies reduced vascularization is associated with an impairment of progenitor cell migration.2,3
Various cytokines and growth factors, including vascular endothelial growth factor (VEGF), stromal cell-derived factor-1 (SDF-1), and interleukin-8 (IL-8) participate in EPC recruitment.4 The most extensively characterized pathway mediating EPC migration centers on the kinase Akt, which phosphorylates among other substrates glycogen synthase kinase (GSK)3β and endothelial nitric oxide synthase (eNOS), promoting the activation of β-catenin and the production of nitric oxide (NO), respectively.5,6 Akt itself is activated through phosphoinositide-3-kinase (PI3K) in response to various stimuli, including ligand-mediated receptor activation.7,8
Apart from “classic” growth factors, the kallikrein–kinin system (KKS) contributes to the revascularization of ischemic tissues.9 Kinins, generated through kininogen cleavage by kallikreins, are present in various tissues. Subject to enzymatic degradation, ie, by kininase II (angiotensin-converting enzyme [ACE]), kinins have a short half-life after release into the circulation but can persist longer in tissues.10 Kinin gradients are generated in ischemic tissues or because of local tissue kallikrein (KLK1) gene transfer.11,12 Kinin-mediated effects are cell type-specific, which is mainly accomplished by the differential expression, activation, and downstream signaling of its 2 G protein-coupled receptors (GPCRs), the inducible B1 (B1R), and the constitutively expressed B2 (B2R). Both receptors mediate proangiogenic effects of kinins, although in different pathological circumstances, through, for example, promoting vascular cell proliferation and survival, as well as kinin chemoattraction of endothelial cells and leukocytes.13-18
We now report the relevance of the B2R for the recruitment of distinct populations of proangiogenic progenitors to sites of ischemia and the impairment of B2R signaling in progenitor cells from patients with cardiovascular disease (CVD). Furthermore, we demonstrate that proangiogenic cells are enriched in the fraction of circulating mononuclear cells (MNCs), which are able to migrate toward bradykinin (BK), a result that may have important implications in a clinical and therapeutic perspective.
Materials and Methods
Cell Isolation
MNCs were obtained from anticoagulated peripheral blood (PB) of healthy subjects (n=37) or from patients with stable class I angina (SA) (n=37) or acute myocardial infarction (aMI) (n=31). Late-outgrowth EPCs were enriched by an adhesion-based method. Likewise, MNCs or EPCs were enriched from bone marrow (BM) of wild-type (WT) or B1R- or B2R-deficient (B1R−/−, B2R−/−) C57Bl/6 mice.
Flow Cytometry
Using monoclonal, directly labeled antibodies, surface expression of antigens CD133, CD34, VEGF receptor 2 (KDR), CXCR4, CD14, CD11b, and CD45 was recognized on human PB-MNCs and migrating (BKmig) and nonmigrating (BKnon) cells. B1R and B2R were detected by unconjugated antibodies followed by anti-rabbit fluorescein-isothiocyanate (FITC) or anti-rabbit phycoerythrin (PE)–AlexaFluor680 antibodies. In digests of mouse adductors, the expression of sca-1, flk-1, and cKit was studied on donor-derived (DiI-labeled) cells. Secondary antibody, isotype and single-staining controls were performed. Fluorescence was analyzed in a fluorescence-activated cell sorter (FACS) Canto II flow cytometer using the FACS Diva software.
Migration
Migration of cultured EPCs was studied as described before,19 in the presence or absence of icatibant, LY294002 or N5-(1-iminoethyl)-ornithine. The lower chamber contained 3×10−8 mol/L BK or its vehicle (unstimulated control). For enrichment, total PB-MNCs were left to migrate toward BK for 5 hour and subsequently harvested from the lower chamber.
In Vivo Homing of Progenitor Cells and Promotion of Blood Flow Recovery
WT mice, submitted to operative unilateral limb ischemia 1 day in advance, were injected systemically with BM-MNCs obtained from WT or B2R−/− mice or with the cell vehicle. Another recipient subgroup received WT BM-MNCs plus icatibant 1 hour before and every 12 hours after cell injection. Homing of injected cells was analyzed 2 days later and limb blood flow recovery was followed for 3 weeks after cell injection.
An expanded Materials and Methods section is available in the online data supplement at http://circres.ahajournals.org.
Results
Expression of KKS Components
Expression of B2R in total PB-MNCs, CD133+ and CD34+ progenitors and cultured EPCs was documented by flow cytometry and quantitative PCR, whereas B1R was low expressed. B2R was more abundant in the progenitor cell fractions as compared with total PB-MNCs (Figure 1 and the online data supplement, Figure I, A, and Figure II). Kininogen mRNA was present, whereas ACE expression was low in CD133+ and CD34+ cells and EPCs (supplemental Figure I, A and B).
Figure 1.
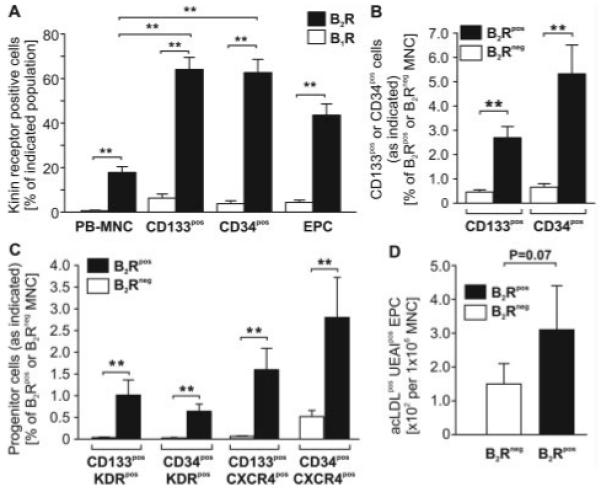
Flow cytometry shows the expression of kinin receptors on freshly isolated PB-MNCs, CD133+, and CD34+ progenitors, as well as cultured EPCs of healthy human subjects. A, B2R is highly abundant in progenitor CD133+ and CD34+ cell fractions as compared with total MNCs, whereas B1R expression is low in the studied cell populations. B and C, Reverse gating shows that in the B2R+ MNC fraction, CD133+ and CD34+ progenitors (B) coexpressing KDR and CXCR4 (C) are more abundant. D, Consistently, outgrowth of acLDL+UEAI+ EPCs is more frequent from FACS sorted B2R+ PB-MNCs. Values are means±SEM; n=6 to 15. **P<0.01.
Enrichment of Putative EPCs Among Human B2R+ PB-MNCs
We next gated for B2R+ MNCs to determine whether this fraction contains more progenitor cells than the B2R− population. Flow cytometry verified a strikingly higher abundance of CD133+KDR+, CD34+KDR+, CD133+CXCR4+, and CD34+CXCR4+ progenitors within B2R+ MNCs as compared with B2R− MNCs (Figure 1B and 1C and supplemental Figure III). Furthermore, fluorescence-activated cell-sorted B2R+ cells tended to give rise to more acLDL+UEAI+ EPCs in culture as compared with B2R− MNCs (Figure 1D).
BK-Induced Migration of Human and Murine EPCs
Stimulation of human EPCs with BK resulted in increased formation of filopodia (Figure 2A) and recruitment of rac1 to the cell membrane (Figure 2B), which coincided with an activation of directed migration in transwell (Figure 2C) and motility in scratch assays (Figure 2D). BK-induced EPC polarization, formation of filopodia, and migration were inhibited by the B2R antagonist icatibant (Figure 2A through 2D). Implication of the B2R, but not the B1R, for BK-mediated migration was furthermore confirmed using BM-derived EPCs from gene-deficient C57Bl/6 mice (Figure 2E). In addition, migration toward BK was blocked by PI3K inhibitor LY294002 or eNOS inhibitor l-iminoethyl-l-ornithine (L-NIO) (Figure 2C), as well as rac1silencing (Figure 2F). Efficient silencing of rac1 is supported by reduction of mRNA and protein expression (supplemental Figure IV, A and B).
Figure 2.

A and B, In human EPCs, BK induces B2R-dependent filopodia formation (n=6) (arrows) (A) and polarization of rac1 (n=9) (arrow) (B) as compared with unstimulated control (con). B, Right, Isotype FITC staining. DiI was used as a nonpolarizing label in B. Scale bars, 5 μm. C and F, In transwell assays, directed migration of EPCs toward BK (n=16) was abolished in the presence of the B2R antagonist icatibant (ic) (n=10), PI3K inhibitor LY294002 (LY) (n=6), and eNOS inhibitor L-NIO (n=4) (C) and after silencing of rac1 (n=4) (F). D, Scratch assay demonstrated an increased B2R-dependent motility of BK-stimulated human EPCs (n=10). E, Migration of murine BM-EPCs toward BK was B2R-dependent, being inhibitable by icatibant (WT EPCs) and abrogated by B2R gene deletion (B2R−/− EPCs) but not by B1R gene deletion (B2R−/− EPCs) (n=6 for all). Values are means±SEM. *P<0.05, **P<0.01.
PI3Kγ plays a crucial role in chemokine-dependent EPC migration and reparative angiogenesis in the mouse.19,20 Here, we report for the first time that PI3Kγ is expressed in human EPCs and recruited to the cell membrane in a polarized fashion on BK stimulation (supplemental Figure V, A through E). Whereas total PI3K inhibition by LY294002 reduced EPC migration to baseline levels, PI3Kγ-silenced human EPCs, as well as PI3Kγ-deficient murine BM-EPCs, exhibited a very low basal migratory activity, with induced migration after BK addition (supplemental Figure V, F and G). Efficient silencing of PI3Kγ is supported by reduction of mRNA and protein expression (supplemental Figure V, H and I).
We furthermore found an increased phosphorylation of eNOS at Ser1177 after stimulation of EPCs with BK and a concordant but a less pronounced trend with regard to phosphorylation of Akt at Ser473 and GSK3β at Ser9 (supplemental Figure VI, A through E). β-Catenin was translocated to nuclear/perinuclear regions in BK-treated EPCs (supplemental Figure VI, F). All of the above effects were suspended in the presence of icatibant. BK stimulation NOS-dependently increased NO generation by EPCs (control: 126±20 arbitrary units; BK: 171±40 arbitrary units; P<0.05; n=6).
Enrichment of Putative EPCs in Human BKmig MNCs
We then verified whether EPCs can be enriched from total PB-MNCs based on migratory response to BK. After performing a transwell migration assay toward BK using total PB-MNCs, we harvested migrating (BKmig) and nonmigrating cells (BKnon) from the lower and upper chambers, respectively. Flow cytometry confirmed that B2R expression is predominantly found on BKmig MNCs (65.0±7.9% versus 9.4±0.6% in BKnon; P<0.05; n=6). Importantly, cells carrying progenitor cell markers were enriched up to 10-fold among BKmig as compared with BKnon cells (Figure 3A). In contrast, exposure to BK without migration did not result in higher incidence of CD133+, CD34 +, KDR +, or CXCR4+ MNCs during the time period used for migration (supplemental Figure VII). In culture, BKmig MNCs gave rise to a larger number of acLDL+UEAI+ EPCs compared with BKnon, suggesting that migration may allow for the selection of angio-competent progenitor cells (Figure 3B). To verify this possibility and determine whether BKmig- and BKnon-derived EPCs possess different angio-supporting capacities, we cocultured them with human umbilical vein endothelial cells (HUVECs) in a Matrigel assay. In both groups, a fraction of EPCs was found as elongated elements integrating into HUVEC networks, whereas another subpopulation appeared as rounded cells that stayed separately from HUVEC structures (Figure 3C). Interestingly, however, BKmig-derived EPCs supported HUVEC network formation more efficiently than BKnon-derived EPCs (Figure 3D through 3G). Consistently, BKmig-derived EPCs secreted higher amounts of IL-8 and MCP-1, for which proangiogenic effects have been described, but lower amounts of IL-1β and RANTES (Figure 3H).21,22
Figure 3.
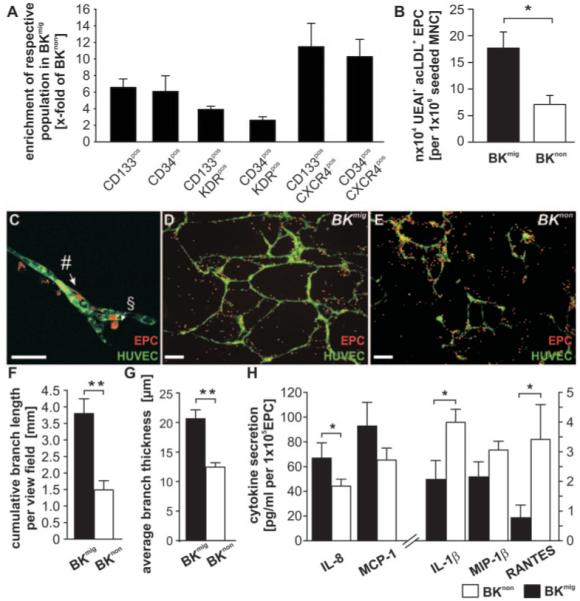
A, CD133+ and CD34+ progenitor cells coexpressing KDR or CXCR4 are enriched in the BKmig population. B, BKmig PB-MNCs give rise to more acLDL+UEAI+ EPCs in vitro as compared with BKnon. EPCs derived from BKmig or BKnon PB-MNCs represent a heterogeneous population harboring elongated cells that integrate into HUVEC networks (#), as well as rounded cells that do not incorporate but stay in proximity to developing networks (§). C, Both populations might modulate network formation. Scale bar, 10 μm). D through H, BKmig-derived EPCs support in vitro network formation of HUVECs more than BKnon-derived EPCs (n=6) (scale bars, 100 μm) (D through G) and secrete more proangiogenic IL-8 and MCP-1 and less IL-1β and RANTES (n=6) (H). Values are means±SEM. *P<0.05, **P<0.01.
B2R Expression and BK Sensitivity in Circulating Progenitor Cells From Patients With Cardiovascular Disease
Consistent with previous reports,2,23 the number of circulating CD133+ and CD34+ progenitor cells was reduced in SA and aMI patients as compared with healthy controls (supplemental Figure VIII). In both SA and aMI patients, coexpression of B2R on circulating CD133+ and CD34+ cells was severely reduced as compared with healthy subjects (Figure 4A and 4B). Similar to CD133+ and CD34+ progenitors, B2R expression on SA EPCs was strikingly reduced (Figure 4C). Because of ethical reasons, we were permitted to obtain blood samples adequate to the study EPC outgrowth from PB-MNCs only in patients with SA but not in those with aMI. We next determined whether the reduced expression of B2R on circulating progenitors translates into impaired migration toward BK. In aMI and SA patients, migration of CD34+ progenitors toward BK was abrogated. A similar migratory dysfunction was observed for CD133+ cells (Figure 4E) and cultured EPCs from SA patients (data not shown). In contrast, in aMI patients, it was still possible to enrich for CD133+ cells, particularly those coexpressing KDR or CXCR4, from PB-MNCs by BK-induced migration (Figure 4E and 4F). Interestingly, the in vitro angio-supporting function of the BKmig cells of aMI patients was comparable to BKmig cells of healthy donors, with BKmig being superior to BKnon. BKmig from SA patients, however, did not support HUVEC network formation and showed no difference to BKnon (Figure 4G).
Figure 4.

A through C, B2R is coexpressed on a lower number of circulating CD34+ (A) and CD133+ progenitor cells (B) of patients with SA (n=23) and aMI (n=27), as well as on a lower number of cultured EPCs from SA patients (n=8) (C), as compared with healthy controls (n=32). The capacity of BK migration assay to enrich for CD34+ (D) and CD133+ (E) progenitor cells from PB-MNCs was evaluated in patients with SA or aMI as compared with healthy subjects (n=8 per group). E and F, Results indicate abrogation of BK responsiveness in all studied populations of SA patients, as documented by a similar enrichment ratio in BK stimulated or unstimulated (con) cells, whereas BK sensitivity was conserved in CD133+ cells coexpressing KDR or CXCR4 of aMI patients. G, Furthermore, BKmig MNCs from aMI patients and healthy donors support network formation by HUVECs more than BKnon, whereas BKmig from SA patients are ineffective and not superior to BKnon. Values are means±SEM or expressed as box and whiskers. *P<0.05, ***P<0.005.
B2R Relevance for Progenitor Cell Homing to Ischemic Tissue and Promotion of Neovascularization
To verify the importance of B2R in neovascularization supported by progenitor cells, we used BM-MNCs from B2R−/− mice in in vitro angiogenesis assays and in an in vivo ischemia model. Mirroring the human data, acLDL+BSI+ EPC outgrowth from B2R−/− BM-MNCs was lower than from WT BM-MNCs (B2R−/−: 1.2±1.0% versus WT, 5.4±0.1% outgrowing EPCs per seeded BM-MNCs; P<0.05; n=6 per group). Furthermore, B2R−/− BM-EPCs stimulated HUVEC network formation less efficiently than WT BM-EPCs (Figure 5A through 5D). To determine whether deletion of the B2R gene may also compromise the in vivo engraftment and proangiogenic capacity of BM-MNCs, we injected Dillabeled B2R−/− or WT BM-MNCs into the tail vein of syngenic WT recipient mice one day after operative unilateral limb ischemia. An additional WT-injected group received B2R antagonist icatibant in addition. We then assessed the absolute number of incorporated cells using flow cytometry of tissue digests. Furthermore, we computed an index of recruitment by normalizing the number of cells homed to the adductors to the number of cells persisting in the PB. Homing was significantly reduced in ischemic adductors of icatibanttreated recipients or when B2R−/− BM-MNCs were injected as compared with controls given WT BM-MNCs in the absence of icatibant (Figure 6A and 6B). No difference in the recruitment to contralateral adductors (data not shown) or the spleens (supplemental Figure IX, A) was found between groups. Flow cytometric analysis of the incorporated cells confirmed impaired recruitment of donor-derived sca-1+ progenitor cells (representing >95% of homed cells in all groups), as well as the more rare flk-1+cKit+ population under icatibant or B2R deficiency (Figure 6A).
Figure 5.
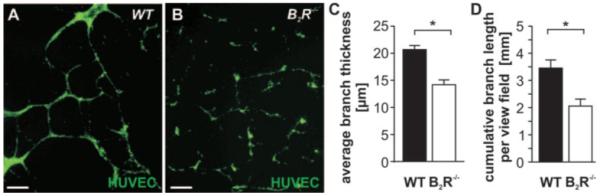
A and B, Network formation by HUVECs is supported by coculture with WT BM-EPCs (A) but not with or B2R−/− BM-EPCs (B). Scale bars, 100 μm. C and D, Bar graphs show average thickness (C) and cumulative length of the branches (D) (n=8 samples per group).
Figure 6.
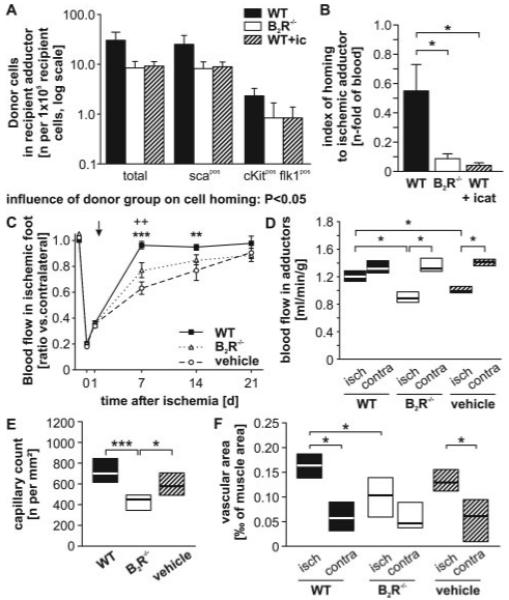
A, Homing of total, sca1+, or cKit+ flk1+ BM-MNCs from the circulation to the ischemic adductor of WT recipients is reduced when B2R−/− BM-MNCs are injected or under systemic blockade of the B2R by icatibant (n=8 per group). B, This remains evident when normalizing to the number of donor cells (total MNCs) persistent in the recipient blood. C and D, Blood flow recovery measured in the paw by Doppler flowmetry (**P<0.01, ++ P<0.01 WT vs vehicle and B2R−/−, respectively) (C) and in the adductor by fluorescent microspheres (*P<0.05) (D) was less efficient in mice receiving vehicle or B2R−/− BM-MNCs as compared with WT BM-MNCs (n=8 per group). E and F, In ischemic adductors of mice receiving B2R−/− BM-MNCs, both capillary density (*P<0.05, ***P<0.005) (E) and vessel area, as normalized per muscle area (*P<0.05) (F), were reduced. Values are means±SEM or expressed as box plot.
Using Doppler flowmetry, we followed the recovery of superficial blood flow in the ischemic feet of WT mice who had received B2R−/− or WT BM-MNCs or vehicle. All groups showed similarly reduced blood flow at 1 day postischemia, when donor cells were injected. Subsequent recovery was faster in the mice which had received WT BM-MNCs than in the vehicle or B2R−/− group (Figure 6C and supplemental Figure IX, B and C). Analysis of the area under the curve (multiple measurements of ischemic-to-contralateral blood flow ratio over time) confirmed an effect of treatment on the overall recovery, with WT BM-MNC recipients performing better than vehicle or B2R−/− group (supplemental Figure IX, B). To confirm the results of Doppler flowmetry, we measured adductor muscle blood flow by fluorescent microspheres. In pilot experiments, we demonstrated the presence of profound ischemia in adductor muscles at 1 day from surgery, ie, the time of cell injection (supplemental Figure IX, D). At 3 weeks postischemia/post–cell transplantation, blood flow to ischemic adductors was still reduced in the B2R−/− or vehicle groups (P<0.05 versus contralateral), whereas WT BM-MNCs recipients had completely recovered (Figure 6D). Furthermore, the ischemic adductors of mice injected with WT BM-MNCs exhibited a higher number of capillaries and a higher percentage of muscle section area was occupied by vessels, which is compatible with increased neovascularization at late stages of recovery from ischemia (Figure 6E and 6F and supplemental Figure IX, E).
Discussion
After acute ischemia, a chemokine gradient is established, directing the homing of BM-derived circulating EPCs. Recruited EPCs then complement the reparative response effected by local cells in a direct and indirect (paracrine) fashion.24,25 We now provide novel evidence that besides the well-described SDF-1/CXCR4 and VEGF/KDR duos, also BK/B2R signaling contributes to recruitment of cells with neovascularization potential.26,27
The relevance of KKS components, especially tissue kallikrein (hK1), B1R, and B2R, in cardiovascular remodeling, endothelial cell function, and angiogenesis have been investigated before.14,15,27 We now show that circulating human CD34+ and CD133+ progenitors and cultured EPCs express crucial KKS components, in particular, B2R and kininogens, enabling them to sense kinin gradients as well as to produce kinins themselves and so modulate adjacent cells. Interestingly, expression of ACE, a major kinin-degrading enzyme, was very low in CD34+ and CD133+ cells, suggesting a more prolonged effect of kinins on progenitor cells as compared with mature endothelial cells, which abundantly express ACE. The expression of hK1 (G Spinetti, N Kraenkel, P Madeddu, unpublished data and data not shown, 2008) might facilitate cell migration and invasion, as has been suggested previously.28 Those data suggest a role for KKS in circulating angiogenic progenitor cell (CPC) function, as has been supported by recent reports: elevated levels of hK1 and kinins have been reported in ischemic tissues and PB of patients with critical vascular disease.11,28,29 KLK1 overexpression in the ischemic myocardium increases circulating EPC levels, as well as the number of cKit+ progenitors in the infarct border zone, without altering the homing of proinflammatory CD45+ and CD68+ cells.30 Our data document now the recruitment of CPCs to the ischemic hindlimb in a B2R-dependent fashion. In fact, we observed diminished homing of sca-1+ and flk-1+cKit+ progenitors to the ischemic muscle in a mouse model of unilateral hindlimb ischemia if the cells were deficient for B2R or when B2R signaling was blocked systemically in recipient mice with icatibant. Furthermore, the BM-EPC angio-supportive effect was abrogated by B2R deficiency both in vitro and in vivo, in the latter case with deleterious effects for limb muscle reperfusion.
The PI3K/Akt/eNOS pathway is fundamental for EPC liberation from the BM and subsequent homing to ischemic sites.31,32 Here, we newly report directed migration of human EPCs in response to BK stimulation and mediated by B2R/PI3K/eNOS signaling. Stimulation of human EPCs with BK induced eNOS phosphorylation, which was paralleled by augmented NO release. Phosphorylation of Akt and GSK3β in BK-stimulated EPCs was less pronounced, yet B2R-dependent. This supports the possibility that BK activates eNOS through Akt-dependent and independent mechanisms. Previously, stimulation of endothelial cells with BK has been shown to induce eNOS liberation from inhibitory membrane-bound complexes with B2R or caveolin-1, thus allowing eNOS translocation to the cytoplasm and subsequent activation through separate phosphorylation events.33
Among different PI3K isoforms, PI3Kγ is specifically associated with GPCRs and, therefore, would be expected to mediate phosphorylation steps downstream to B2R activation. The interest in PI3Kγ acting as a key regulator of EPC migratory function and vasculogenesis has been vitalized by a series of recent studies in murine models of ischemia.19,20 Here, we report for the first time that PI3Kγ is expressed in human EPCs and polarized at the cell membrane following BK challenge. We also found a severe reduction of spontaneous migration in PI3Kγ-silenced PB-EPCs and PI3Kγ −/− BM-EPCs. Silenced EPCs were still responsive to BK, which was in contrast to total blockade of migration when all PI3K isoforms were inhibited by LY294002. It is, therefore, conceivable that BK-induced EPC migration is only mediated, in part, by PI3Kγ and that other isoforms might associate with the B2R. Interestingly, although PI3Kγ is uniquely coupled to GPCRs, PI3Kα and -β isoforms, which are also expressed in vascular cells and leukocytes, can be promiscuously activated by both GPCRs and the tyrosine kinase receptors.34,35 An earlier report by Miura et al describing transactivation between the B2R and the tyrosine kinase receptor KDR supports the possibility that kinin might exert part of its effects through PI3Kα and -β.36
In recent cell therapy trials, immuno-magnetic sorting has been used to enrich for angio-competent progenitor cells.37,38 Here, we provide novel evidence that selection of B2R+ PB-MNCs results in a higher proportion of EPCs by antigenic and culture definitions. Furthermore, we introduced a migration assay to enrich for functionally responsive progenitor cells. The assay allowed us to document that progenitors which respond to BK have increased angiogenic capacity in vitro as compared with unresponsive cells. These results may have important clinical implications. Several recent studies underline the importance of EPC functional capacity for a successful therapy.4 The newly developed separation procedure, therefore, could be applied on freshly obtained or immunosorted MNCs to further select for functional progenitor cells before administration to the patient. Although cardiovascular patients exhibit a reduced absolute number of “enrichable” progenitor cells, functionally superior cells are accumulating among the population of migrating cells, as is supported by our data: in healthy subjects, angio-competent CD133+ and CD34+ CPCs are several folds more abundant in B2R+, as well as in BKmig, cell populations as compared with B2R− or BKnon PB-MNCs. However, in CPCs from CVD patients, B2R abundance was drastically reduced, together with an impairment of migratory activity toward BK. This has previously been reported for other receptors, such as SDF-1 receptor CXCR4, involved in angiogenesis and EPC function.25 Reduced EPC sensitivity toward growth factors is indeed thought to be a mediator of reduced endothelial function and reparative neovascularization in patients with CVD. Reduced BK sensitivity might be another manifestation of this cellular dysfunction.
Analysis of progenitor cell migratory function indicates a substantial difference between subpopulations of progenitor cells from SA and aMI patients. Whereas migration of CD34+ cells was universally impaired, CD133+ cells of aMI patients, more specifically the CD133+KDR+ and CD133+CXCR4+ subpopulations, could be enriched in the BKmig fraction to an extent comparable to healthy subjects. Recent reports describe an increased mobilization of CD133+ or CXCR4+ cells in aMI as compared with SA, resulting in higher levels per mL of blood.39,40 This is thought to be an autologous response to acute ischemia, associated with mobilization of different MNC populations into the circulation. In fact, we found that the absolute but not the relative abundance of progenitor cells is increased in the circulation of aMI as compared with SA patients. To expand on the above-mentioned results, BKmig or BKnon cells, obtained from freshly isolated PB-MNCs of aMI and SA patients, were incubated together with HUVECs to study support of endothelial cell network formation. Results indicated that BKmig cells from aMI patients support HUVEC network formation in a way comparable to healthy BKmig cells, whereas BKmig from SA patients are ineffective and not superior to BKnon. These data encourage further research exploring the utility of migration as a way to enrich for functionally competent progenitors in the perspective of optimized cell therapy of acute ischemic disease.
Taken together, we demonstrated, for the first time, that B2R/BK signaling is important for the attraction of angio-competent progenitor cells. In patients with CVD, the impairment of B2R signaling might contribute to a reduced recruitment of proangiogenic progenitors to the site of ischemia and the suspended blood flow recovery, similar to the situation observed in our limb ischemia model.
Supplementary Material
Acknowledgments
Icatibant was kindly provided by Sanofi, Germany; small interfering RNAs against rac1 by Dr Harry Mellor (University of Bristol); and PI3Kγ−/− and B2R−/− mice by Prof Emilio Hirsch (University of Turin, Italy), Prof Michael Bader (Max-Delbrück Center, Berlin, Germany), and Prof Joao B. Pesquero (University of São Paulo, Brazil). We thank Dr Marco Meloni and Dr Ayman Al Haj Zen (University of Bristol) and Dr Marco Vaglimigli (University of Ferrara, Italy) for technical support, valuable comments, and critical revision of the manuscript.
Sources of Funding
The study was supported by a joint research grant from the European Foundation for the Study of Diabetes, the Juvenile Diabetes Research Foundation, and NovoNordisk; and by British Heart Foundation project grants PG 06/035/20641 and PG/06/096/21325.
Footnotes
Disclosures
None.
References
- 1.Kawamoto A, Losordo DW. Endothelial progenitor cells for cardiovascular regeneration. Trends Cardiovasc Med. 2008;18:33–37. doi: 10.1016/j.tcm.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, Zeiher AM, Dimmeler S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001;89:e1–e7. doi: 10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- 3.Segal MS, Shah R, Afzal A, Perrault CM, Chang K, Schuler A, Beem E, Shaw LC, Calzi SL, Harrison JK, Tran-Son-Tay R, Grant MB. Nitric oxide cytoskeletal-induced alterations reverse the endothelial progenitor cell migratory defect associated with diabetes. Diabetes. 2006;55:102–109. [PubMed] [Google Scholar]
- 4.Chavakis E, Urbich C, Dimmeler S. Homing and engraftment of progenitor cells: a prerequisite for cell therapy. J Mol Cell Cardiol. 2008 Jan 18; doi: 10.1016/j.yjmcc.2008.01.004. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 5.Choi J, Hur J, Yoon C, Kim J, Lee C, Youn S, Oh I, Skurk C, Murohara T, Park Y, Walsh K, Kim H. Augmentation of therapeutic angiogenesis using genetically modified human endothelial progenitor cells with altered glycogen synthase kinase-3beta activity. J Biol Chem. 2004;279:49430–49438. doi: 10.1074/jbc.M402088200. [DOI] [PubMed] [Google Scholar]
- 6.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fisslthaler B, Dimmeler S, Hermann C, Busse R, Fleming I. Phosphorylation and activation of the endothelial nitric oxide synthase by fluid shear stress. Acta Physiol Scand. 2000;168:81–88. doi: 10.1046/j.1365-201x.2000.00627.x. [DOI] [PubMed] [Google Scholar]
- 8.Zeng G, Nystrom FH, Ravichandran LV, Cong LN, Kirby M, Mostowski H, Quon MJ. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation. 2000;101:1539–1545. doi: 10.1161/01.cir.101.13.1539. [DOI] [PubMed] [Google Scholar]
- 9.Madeddu P, Emanueli C, El-Dahr S. Mechanisms of disease: the tissue kallikrein-kinin system in hypertension and vascular remodeling. Nat Clin Pract Nephrol. 2007;3:208–221. doi: 10.1038/ncpneph0444. [DOI] [PubMed] [Google Scholar]
- 10.Linz W, Wiemer G, Gohlke P, Unger T, Schölkens BA. Contribution of kinins to the cardiovascular actions of angiotensin-converting enzyme inhibitors. Pharmacol Rev. 1995;47:25–49. [PubMed] [Google Scholar]
- 11.Duncan AM, Burrell LM, Kladis A, Campbell DJ. Angiotensin and bradykinin peptides in rats with myocardial infarction. J Card Fail. 1997;3:41–52. doi: 10.1016/s1071-9164(97)90007-5. [DOI] [PubMed] [Google Scholar]
- 12.Emanueli C, Zacheo A, Minasi A, Chao J, Chao L, Salis MB, Stacca T, Straino S, Capogrossi MC, Madeddu P. Adenovirus-mediated human tissue kallikrein gene delivery induces angiogenesis in normoperfused skeletal muscle. Arterioscler Thromb Vasc Biol. 2000;20:2379–2385. doi: 10.1161/01.atv.20.11.2379. [DOI] [PubMed] [Google Scholar]
- 13.Morbidelli L, Parenti A, Giovannelli L, Granger HJ, Ledda F, Ziche M. B1 receptor involvement in the effect of bradykinin on venular endothelial cell proliferation and potentiation of FGF-2 effects. Br J Pharmacol. 1998;124:1286–1292. doi: 10.1038/sj.bjp.0701943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emanueli C, Salis MB, Stacca T, Pintus G, Kirchmair R, Isner JM, Pinna A, Gaspa L, Regoli D, Cayla C, Pesquero JB, Bader M, Madeddu P. Targeting kinin B(1) receptor for therapeutic neovascularization. Circulation. 2002;105:360–366. doi: 10.1161/hc0302.102142. [DOI] [PubMed] [Google Scholar]
- 15.Silvestre JS, Bergaya S, Tamarat R, Duriez M, Boulanger CM, Levy BI. Proangiogenic effect of angiotensin-converting enzyme inhibition is mediated by the bradykinin B(2) receptor pathway. Circ Res. 2001;89:678–683. doi: 10.1161/hh2001.097691. [DOI] [PubMed] [Google Scholar]
- 16.Parenti A, Morbidelli L, Ledda F, Granger HJ, Ziche M. The bradykinin/B1 receptor promotes angiogenesis by up-regulation of endogenous FGF-2 in endothelium via the nitric oxide synthase pathway. FASEB J. 2001;15:1487–1489. [PubMed] [Google Scholar]
- 17.Emanueli C, Salis MB, Linthout SV, Meloni M, Desortes E, Silvestre J, Clergue M, Figueroa CD, Gadau S, Condorelli G, Madeddu P. Akt/ protein kinase B and endothelial nitric oxide synthase mediate muscular neovascularization induced by tissue kallikrein gene transfer. Circulation. 2004;110:1638–1644. doi: 10.1161/01.CIR.0000142051.36244.83. [DOI] [PubMed] [Google Scholar]
- 18.Ferreira HH, Medeiros MV, Lima CS, Flores CA, Sannomiya P, Autunes E, Nucci GD. Inhibition of eosinophil chemotaxis by chronic blockade of nitric oxide biosynthesis. Eur J Pharmacol. 1996;310:201–207. doi: 10.1016/0014-2999(96)00379-2. [DOI] [PubMed] [Google Scholar]
- 19.Madeddu P, Kraenkel N, Barcelos LS, Siragusa M, Campagnolo P, Oikawa A, Caporali A, Herman A, Azzolino O, Barberis L, Perino A, Damilano F, Emanueli C, Hirsch E. Phosphoinositide 3-kinase γ gene knockout impairs post-ischemic neovascularization and endothelial progenitor cell functions. Arterioscler Thromb Vasc Biol. 2008;28:68–76. doi: 10.1161/ATVBAHA.107.145573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chavakis E, Carmona G, Urbich C, Göttig S, Henschler R, Penninger JM, Zeiher AM, Chavakis T, Dimmeler S. Phosphatidylinositol-3-kinase-gamma is integral to homing functions of progenitor cells. Circ Res. 2008;102:942–949. doi: 10.1161/CIRCRESAHA.107.164376. [DOI] [PubMed] [Google Scholar]
- 21.Oh IY, Yoon CH, Hur J, Kim JH, Kim TY, Lee CS, Park KW, Chae IH, Oh BH, Park YB, Kim HS. Involvement of E-selectin in recruitment of endothelial progenitor cells and angiogenesis in ischemic muscle. Blood. 2007;110:3891–3899. doi: 10.1182/blood-2006-10-048991. [DOI] [PubMed] [Google Scholar]
- 22.Niu J, Azfer A, Zhelyabovska O, Fatma S, Kolattukudy PE. Monocyte chemoattractant protein (MCP)-1 promotes angiogenesis via a novel transcription factor, MCP-1-induced protein (MCPIP) J Biol Chem. 2008;283:14542–14551. doi: 10.1074/jbc.M802139200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawamoto A, Asahara T. Role of progenitor endothelial cells in cardiovascular disease and upcoming therapies. Catheter Cardiovasc Interv. 2007;70:477–484. doi: 10.1002/ccd.21292. [DOI] [PubMed] [Google Scholar]
- 24.van Weel V, Seghers L, de Vries MR, Kuiper EJ, Schlingemann RO, Bajema IM, Lindeman JHN, Diemen PMD, van Hinsbergh VWM, van Bockel JH, Quax PHA. Expression of vascular endothelial growth factor, stromal cell-derived factor-1, and CXCR4 in human limb muscle with acute and chronic ischemia. Arterioscler Thromb Vasc Biol. 2007;27:1426–1432. doi: 10.1161/ATVBAHA.107.139642. [DOI] [PubMed] [Google Scholar]
- 25.Walter DH, Haendeler J, Reinhold J, Rochwalsky U, Seeger F, Honold J, Hoffmann J, Urbich C, Lehmann R, Arenzana-Seisdesdos F, Aicher A, Heeschen C, Fichtlscherer S, Zeiher AM, Dimmeler S. Impaired CXCR4 signaling contributes to the reduced neovascularization capacity of endothelial progenitor cells from patients with coronary artery disease. Circ Res. 2005;97:1142–1151. doi: 10.1161/01.RES.0000193596.94936.2c. [DOI] [PubMed] [Google Scholar]
- 26.Rafii S, Avecilla S, Shmelkov S, Shido K, Tejada R, Moore MAS, Heissig B, Hattori K. Angiogenic factors reconstitute hematopoiesis by recruiting stem cells from bone marrow microenvironment. Ann N Y Acad Sci. 2003;996:49–60. doi: 10.1111/j.1749-6632.2003.tb03232.x. [DOI] [PubMed] [Google Scholar]
- 27.Tschöpe C, Walther T, Escher F, Spillmann F, Du J, Altmann C, Schimke I, Bader M, Sanchez-Ferrer CF, Schultheiss HP, Noutsias M. Transgenic activation of the kallikrein-kinin system inhibits intramyocardial inflammation, endothelial dysfunction and oxidative stress in experimental diabetic cardiomyopathy. FASEB J. 2005;19:2057–2059. doi: 10.1096/fj.05-4095fje. [DOI] [PubMed] [Google Scholar]
- 28.Emanueli C, Minasi A, Zacheo A, Chao J, Chao L, Salis MB, Straino S, Tozzi MG, Smith R, Gaspa L, Bianchini G, Stillo F, Capogrossi MC, Madeddu P. Local delivery of human tissue kallikrein gene accelerates spontaneous angiogenesis in mouse model of hindlimb ischemia. Circulation. 2001;103:125–132. doi: 10.1161/01.cir.103.1.125. [DOI] [PubMed] [Google Scholar]
- 29.Porcu P, Emanueli C, Desortes E, Marongiu GM, Piredda F, Chao L, Chao J, Madeddu P. Circulating tissue kallikrein levels correlate with severity of carotid atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:1104–1110. doi: 10.1161/01.ATV.0000128126.57688.a9. [DOI] [PubMed] [Google Scholar]
- 30.Spillmann F, Graiani G, Linthout SV, Meloni M, Campesi I, Lagrasta C, Westermann D, Tschöpe C, Quaini F, Emanueli C, Madeddu P. Regional and global protective effects of tissue kallikrein gene delivery to the peri-infarct myocardium. Regen Med. 2006;1:235–254. doi: 10.2217/17460751.1.2.235. [DOI] [PubMed] [Google Scholar]
- 31.Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, Zeiher AM, Dimmeler S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9:1370–1376. doi: 10.1038/nm948. [DOI] [PubMed] [Google Scholar]
- 32.Yu J, deMuinck ED, Zhuang Z, Drinane M, Kauser K, Rubanyi GM, Qian HS, Murata T, Escalante B, Sessa WC. Endothelial nitric oxide synthase is critical for ischemic remodeling, mural cell recruitment, and blood flow reserve. Proc Natl Acad Sci U S A. 2005;102:10999–11004. doi: 10.1073/pnas.0501444102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ju H, Venema VJ, Marrero MB, Venema RC. Inhibitory interactions of the bradykinin B2 receptor with endothelial nitric-oxide synthase. J Biol Chem. 1998;273:24025–24029. doi: 10.1074/jbc.273.37.24025. [DOI] [PubMed] [Google Scholar]
- 34.Murga C, Fukuhara S, Gutkind JS. A novel role for phosphatidylinositol 3-kinase beta in signaling from G protein-coupled receptors to Akt. J Biol Chem. 2000;275:12069–12073. doi: 10.1074/jbc.275.16.12069. [DOI] [PubMed] [Google Scholar]
- 35.Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J, Cutillas PR, Smith AJ, Ridley AJ, Ruhrberg C, Gerhardt H, Vanhaesebroeck B. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature. 2008;453:662–666. doi: 10.1038/nature06892. [DOI] [PubMed] [Google Scholar]
- 36.Miura S, Matsuo Y, Saku K. Transactivation of KDR/Flk-1 by the B2 receptor induces tube formation in human coronary endothelial cells. Hypertension. 2003;41:1118–1123. doi: 10.1161/01.HYP.0000064345.33807.57. [DOI] [PubMed] [Google Scholar]
- 37.Bartunek J, Vanderheyden M, Vandekerckhove B, Mansour S, De Bruyne B, De Bondt P, Van Haute I, Lootens N, Heyndrickx G, Wijns W. Intracoronary injection of CD133-positive enriched bone marrow progenitor cells promotes cardiac recovery after recent myocardial infarction: feasibility and safety. Circulation. 2005;112(suppl I):I-178–I-183. doi: 10.1161/CIRCULATIONAHA.104.522292. [DOI] [PubMed] [Google Scholar]
- 38.Ahmadi H, Baharvand H, Ashtiani SK, Soleimani M, Sadeghian H, Ardekani JM, Mehrjerdi NZ, Kouhkan A, Namiri M, Madani-Civi M, Fattahi F, Shahverdi A, Dizaji AV. Safety analysis and improved cardiac function following local autologous transplantation of CD133(+) enriched bone marrow cells after myocardial infarction. Curr Neurovasc Res. 2007;4:153–160. doi: 10.2174/156720207781387141. [DOI] [PubMed] [Google Scholar]
- 39.Vöö S, Eggermann J, Dunaeva M, Ramakers-van Oosterhoud C, Waltenberger J. Enhanced functional response of CD133+ circulating progenitor cells in patients early after acute myocardial infarction. Eur Heart J. 2008;29:241–250. doi: 10.1093/eurheartj/ehm542. [DOI] [PubMed] [Google Scholar]
- 40.Wojakowski W, Tendera M, Michalowska A, Majka M, Kucia M, Maślankiewicz K, Wyderka R, Ochala A, Ratajczak MZ. Mobilization of CD34/CXCR4+, CD34/CD117+, c-met+ stem cells, and mononuclear cells expressing early cardiac, muscle, and endothelial markers into peripheral blood in patients with acute myocardial infarction. Circulation. 2004;110:3213–3220. doi: 10.1161/01.CIR.0000147609.39780.02. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.