

Summary
SIRT6 is a member of a highly conserved family of NAD+-dependent deacetylases with various roles in metabolism, stress resistance and lifespan. SIRT6 deficient mice develop normally but succumb to a lethal hypoglycemia early in life; however, the mechanism underlying this hypoglycemia remained unclear. Here, we demonstrate that SIRT6 functions as a histone H3K9 deacetylase to control the expression of multiple glycolytic genes. Specifically, SIRT6 appears to function as a co-repressor of the transcription factor Hif1α, a critical regulator of nutrient stress responses. Consistent with this notion, SIRT6 deficient cells exhibit increased Hif1α activity and show increased glucose uptake with up-regulation of glycolysis and diminished mitochondrial respiration. Our studies uncover a novel role for the chromatin factor SIRT6 as a master regulator of glucose homeostasis, and may provide the basis for novel therapeutic approaches against metabolic diseases, such as diabetes and obesity.
Introduction
Adaptation to stress represents a critical cellular response for maintenance of homeostatic balance. In yeast, the founding member of the sirtuin family Sir2 was originally discovered as a silencing factor, functioning as a sensor of the metabolic activity of the cell to influence gene transcription, DNA repair, recombination and lifespan (Haigis and Guarente, 2006; Longo and Kennedy, 2006). Later studies identified Sir2 as an NAD+-dependent histone deacetylase, thereby linking chromatin silencing to cellular redox status.
In mammalian genomes, there are seven Sir2 homologs (SIRTs 1-7) (Frye, 2000). Little is known about the function of the mammalian SIRT6 protein. Our previous work has shown that SIRT6 is a nuclear, chromatin-bound protein (Mostoslavsky et al., 2006). Among the sirtuins, SIRT6 deficiency causes the most striking phenotype. SIRT6 deficient mice are born normally, and at around 3 weeks of age, they develop several acute degenerative processes, dying before one month of age. The defects include a severe metabolic imbalance, with low levels of serum IGF-1, complete loss of subcutaneous fat, lymphopenia, osteopenia, and acute onset of hypoglycemia, leading to death (Mostoslavsky et al., 2006). Furthermore, SIRT6 promotes the resistance to DNA damage and oxidative stress, and suppresses genomic instability in mouse cells, in association with a role in base excision DNA repair (BER) (Mostoslavsky et al., 2006). Recent studies have demonstrated that SIRT6 is located at the telomeres in human cells, and knock-down of SIRT6 in these cells altered the telomere structure, causing accelerated senescence and telomere-dependent genomic instability. Furthermore, SIRT6 functions as a histone deacetylase, deacetylating histone H3 lysine 9 (H3K9) specifically at telomeres (Michishita et al., 2008). New studies indicate that SIRT6 can function as a co-repressor of NF-κB, silencing NF-κB target genes through deacetylation of H3K9, and decreasing NF-κB-dependent apoptosis and senescence (Kawahara et al., 2009). Therefore, it appears that SIRT6 can function as a histone H3K9 deacetylase in a cell- and context-dependent manner. At this point, however, it remains unclear what is the molecular defect underlying the main phenotype in SIRT6 deficient mice, namely the lethal hypoglycemia. Critically, it is not known whether SIRT6 is directly or indirectly involved in the modulation of glucose metabolism.
In the presence of O2 and glucose, cells convert glucose to pyruvate, which enters the mitochondria, is converted to acetyl coenzyme A, and metabolized via the tricarboxylic acid cycle (TCA), yielding reducing equivalents that are used for oxidative phosphorylation to generate ATP. However, under hypoxic or low nutrient conditions, cells shift their metabolism from aerobic to anaerobic metabolism, converting pyruvate instead to lactate (“Pasteur effect”) (Aragones et al., 2009) (Vander Heiden et al., 2009). With this energy compensation, cells continue to generate ATP (albeit less efficiently), in an attempt to meet their metabolic demands during this period of stress. The hypoxia-inducible transcription factor Hif1α is a key mediator of this cellular adaptation to nutrient and oxygen stress (Lum et al., 2007; Seagroves et al., 2001), functioning as a direct transcriptional activator of multiple genes. On one hand, it enhances glycolytic flux by up-regulating expression of key glycolytic genes, including the glucose transporters GLUT-1 and GLUT-3, lactate dehydrogenase (LDH), phosphoglycerate kinase (PGK-1), glucose-6-phosphate isomerase (GPI) and phosphofructose kinase 1 (PFK-1) (Hu et al., 2006). On the other hand, Hif1α directly inhibits mitochondrial respiration by up-regulating expression of the pyruvate dehydrogenase kinase (PDK) gene (Kim et al., 2006; Papandreou et al., 2006). PDK in turn phosphorylates and inactivates pyruvate dehydrogenase (PDH), a rate-limiting enzyme that converts pyruvate to Acetyl-CoA to fuel the TCA cycle. Recent studies indicate that Hif1α also diminishes mitochondrial activity through inhibition of the Cytochrome Oxidase Subunit Cox4-1 and the coactivator PGC-1β (Fukuda et al., 2007; Zhang et al., 2007). Overall, Hif1α appears to modulate multiple genes in order to activate glycolysis and at the same time repress mitochondrial respiration in a coordinated fashion.
The activity of Hif1α is tightly regulated. Under normoxia, Hif1α is hydroxylated at multiple prolyl residues by the prolyl-hydroxylase-domain (PHD) proteins. Following hydroxylation, Hif1α is recognized by the von-Hippel-Lindau (VHL) ubiquitin ligase, marking Hif1α for subsequent proteasome degradation. When O2 or glucose are low, PHD proteins are inactivated, thereby stabilizing Hif1α protein levels (Aragones et al., 2009). However, even under normoxic and normoglycemic conditions, Hif1α regulates basal expression of its target genes (Carmeliet et al., 1998), suggesting that further mechanisms should be in place to ensure that this stress response is tightly regulated under normal nutrient conditions.
We now present data to demonstrate that SIRT6 deficiency causes a cell-autonomous up-regulation of glucose uptake, both in vitro and in vivo, triggering a nutrient-stress response and a switch in glucose metabolism towards glycolysis and away of mitochondrial respiration. We propose that SIRT6 functions as a co-repressor of Hif1α transcriptional activity, deacetylating H3K9 at Hif1α target gene promoters. In this way, SIRT6 maintains efficient glucose flux into the TCA cycle under normal nutrient conditions. Regulation of glucose flux by SIRT6 appears critical since SIRT6 deficiency causes a lethal hypoglycemia. In this context, it is striking that deficiency in a single chromatin factor exerts such a severe and specific metabolic phenotype, highlighting the crucial role for SIRT6 in this pathway.
Results
SIRT6 deficiency causes a cell-autonomous increase in glucose uptake
Our previous work showed that the most severe defect in SIRT6 deficient animals is lethal hypoglycemia. Although such a phenotype is typically associated with hyperinsulinemia, the mice exhibit normal pancreatic islets, and, remarkably, lower blood insulin levels, indicating that low glucose may have triggered a reduction in insulin secretion as an adaptive response (Supplemental Figure 1A-B). In addition, we found that the mice had no defects in glucose absorption in the intestine and did not exhibit increased glucose secretion by the kidney (Supplemental Figure 1C and data not shown). These observations prompted us to test whether the mice were experiencing an intrinsic increase in glucose uptake, independent of insulin levels in blood. We first took advantage of labeled glucose (1,2-13C Glucose) to trace glucose in blood. As seen in Figure 1A, SIRT6 deficient animals clear glucose from blood significantly faster than wild-type littermates. We next used 18F-fluorodeoxyglucose-positron emission tomography (FDG-PET), to determine which tissues had increased glucose uptake. We show that SIRT6 mice exhibit a pronounced increase in glucose uptake both in muscle and brown adipose tissue (Figures 1B-C), whereas no changes were observed in the liver, brain or heart (Figure 1C). Furthermore, expression of gluconeogenic genes were higher in SIRT6 deficient livers (data not shown), further suggesting that liver was trying to compensate for the hypoglycemia, rather than being a primary cause of it. This muscle- and BAT-specific increase in glucose uptake could explain the hypoglycemic phenotype of SIRT6 deficient mice.
Figure 1. Increased glucose uptake in SIRT6 deficient cells and mice.
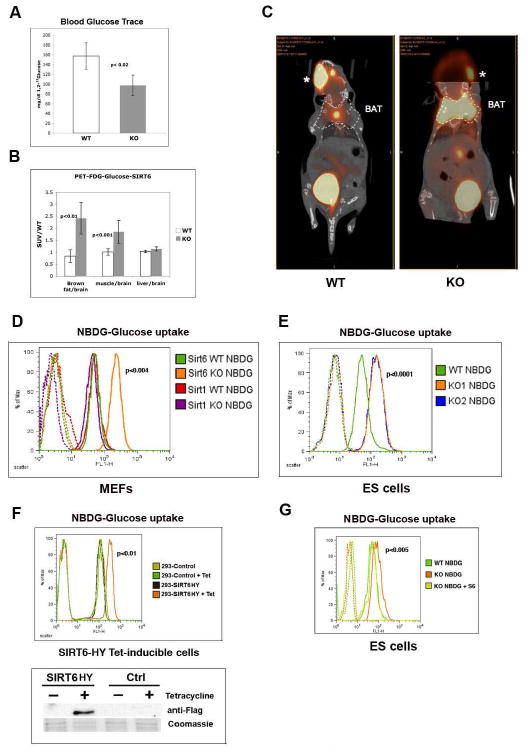
(A) [1,2 13C] labeled glucose trace assay was carried out on 16 days-old SIRT6 wild-type (WT) and knock-out (KO) mice. See Methods for details. (B) Standard Uptake Value (SUV) ratio of labeled 18FDG-Glucose incorporation in WT and KO SIRT6 mice. The different tissues analyzed are indicated. Samples were normalized against brain, which exhibit stable glucose uptake across genotypes. The experiment is an average of three mice per genotype. (C) 16-days old SIRT6 WT and KO mice were PET imaged 60 minutes following i.v. injection of 18F-glucose. Dotted lines indicate position of the brown adipose tissue (BAT). *: labeled glucose at site of injection (retro-orbital; the enhanced signal observed in the WT reflects the position of the head at this particular CT section; comparable intensity is observed in the KO on a different CT section). (D) SIRT6 WT and SIRT6 KO mouse embryonic fibroblasts (MEFs) together with SIRT1 WT and KO MEFs were grown in the presence of the fluorescent glucose analog NBDG (Invitrogen) for 1 hr., and glucose uptake was then quantified using flow cytometry (FACS). Dotted lines are controls without the fluorescent NBDG glucose analog. (E) One WT and two independently generated SIRT6 KO ES lines (KO1 and KO2) were treated as in (D), and analyzed by FACS. (F) 293T cells were stable transfected with a SIRT6 cDNA carrying a H133Y mutation (SIRT6HY) that acts as a dominant negative, under the control of the Tetracycline promoter. Lower panel: western blot showing that SIRT6 was induced specifically after tetracycline treatment (SIRT6). Empty vector was used as a control (Ctrl). Upper panel: glucose uptake was measured as in (D). (G) SIRT6 KO cells were infected with a SIRT6 expressing-lentivirus. Infected cells were sorted for GFP expression, and following expansion, cells were assayed for glucose uptake following 1 hr. incubation with NBDG. See also Figure S1.
In order to determine whether SIRT6 influences glucose uptake in a cell-autonomous fashion, we measured glucose uptake in SIRT6 wild-type (WT) and KO cells using a fluorescent glucose analog (2-NBDG) that is incorporated into cells and allows quantification of glucose uptake using flow-cytometry. Notably, both embryonic stem (ES) cells and mouse embryonic fibroblasts (MEFs) display a striking increase in glucose uptake as early as one hour following addition of the glucose analog (Figures 1D-E). Furthermore, this effect appears specific for SIRT6, since SIRT1 deficient MEFs do not show this phenotype. These results indicate that SIRT6 deficiency causes increased glucose uptake in a cell-autonomous fashion. It is still possible that this effect on glucose uptake was secondary to adaptation to chronic depletion of SIRT6. In order to rule out this possibility, we generated cells where SIRT6 can be inactivated in an acute manner. In this system, a previously characterized dominant negative form of SIRT6 (Mostoslavsky et al., 2006; Kawahara et al., 2009) is specifically induced following treatment with Tetracycline. Remarkably, 48 hr. after expression of this dominant negative mutant, cells exhibit a marked increase in glucose uptake (Figure 1F).
To definitively demonstrate that the glucose phenotype we observed was specific to the lack of SIRT6, we re-expressed SIRT6 in SIRT6 KO ES cells and MEFs, and tested whether glucose uptake was normalized. Re-expression of SIRT6 rescued the metabolic phenotype, reducing glucose uptake significantly (Figure 1G and Supplemental Figure 1D). In summary, these data show that lack of SIRT6 in multiple cell types in vivo and in vitro causes a specific and cell-autonomous increase in glucose uptake.
Increased membrane expression of the glucose transporter GLUT1 in the absence of SIRT6
Next we sought to assess whether the increased glucose uptake in SIRT6 deficient cells was associated with elevated expression of glucose transporters. The main glucose transporter in ES cells and MEFs is GLUT1, a receptor that modulates basal uptake of glucose, independent of growth factors or insulin (Pessin and Bell, 1992). Therefore, we stained cells with an antibody against GLUT1 and used confocal microscopy to determine quantitative differences in membrane expression of this receptor. We found that SIRT6 KO cells express substantially higher levels of membrane GLUT1 (Figure 2A-B), consistent with the increased glucose uptake in these cells.
Figure 2. Increased lactate production and decreased oxygen consumption in SIRT6 deficient cells.
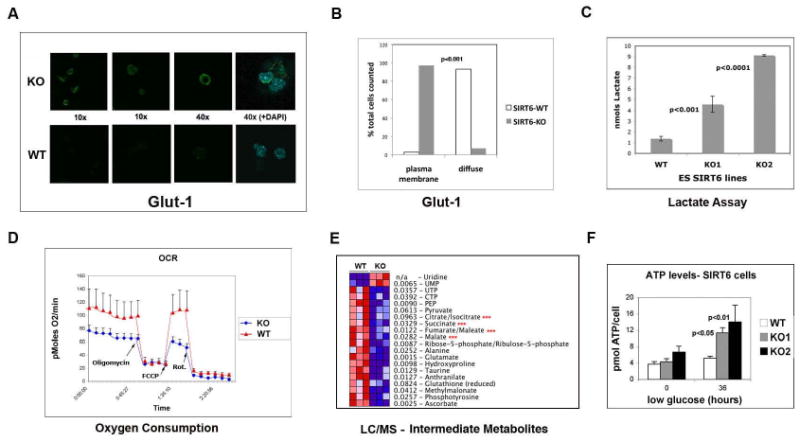
(A) Confocal immunostaining in SIRT6 WT and KO ES cells using a GLUT1 antibody. (B) Quantification of GLUT1 membrane staining in SIRT6 WT and KO cells. (C) Lactate levels in SIRT6 WT and KO ES cells (KO1 and KO2). (D) Oxygen consumption in live SIRT6 WT and KO ES cells under basal conditions, following the addition of the mitochondrial F1-F0-ATPase inhibitor oligomycin (μM), the uncoupler FCCP (1 μM) and the Complex I inhibitor rotenone (rot)(5μM) in combination with the Complex I inhibitor myxothiazol (5μM). Oxygen consumption rate (OCR) was measured using the XF24 SeaHorse Analyzer (Seahorse Bioscience). Each data point is the average of five independent measurements. Error bars indicate standard error of the mean (E) Protein lysates were purified from three independent samples of WT and KO ES cells and glucose metabolites were analyzed by liquid chromatography-mass spectrometry (LC-MS). Red asterisks: TCA intermediate metabolites. (F) ATP levels were measured using the ATP Assay Kit (SIGMA) in SIRT6 WT and KO ES cells (KO1 and KO2) that were either in regular media or in low glucose (0.5 g/L) media for 36 hours. See also Figure S2.
Enhanced glycolysis and reduced mitochondrial respiration in SIRT6 deficient cells
The above results prompted us to test how glucose is utilized in SIRT6 deficient cells. We first measured lactate production, in order to determine whether glycolysis was enhanced. Indeed, both SIRT6 deficient ES cells (Figure 2C) and MEFs (Supplemental Figure 2) display significantly higher levels of lactate when compared to WT cells. Concomitantly, lack of SIRT6 causes a reduction in oxygen consumption (Figure 2D), indicating that in SIRT6 deficient cells glucose is utilized primarily for glycolysis, whereas mitochondrial respiration is inhibited. To further validate these results, we performed mass-spectrometry based metabolic profiling. Out of 106 metabolites analyzed, 22 showed altered levels in the SIRT6 KO cells (p< 0.05). Among those, we found multiple TCA metabolites that were reduced in SIRT6 KO cells (Figure 2E), further confirming that mitochondrial respiration is inhibited in these cells. Cells switch to glycolysis in order to sustain ATP production under conditions of nutrient stress. We therefore tested whether SIRT6 deficient cells were fitter than wild-type cells when exposed to nutrient starvation. Although ATP levels were similar in both cell types when the cells were maintained in normal media, SIRT6 deficient cells produce significantly higher levels of ATP after a few hours in low-glucose (Figure 2F). Overall, these results indicate that absence of SIRT6 causes a switch towards enhanced glycolysis and reduced mitochondrial respiration, a response usually observed under conditions of nutrient/oxygen stress.
Glycolytic genes as putative SIRT6 targets in glucose metabolism
Based on the strong binding of SIRT6 to chromatin (Mostoslavsky et al., 2006), and the fact that SIRT6 is known to function as a histone H3K9 deacetylase (Kawahara et al., 2009; Michishita et al., 2008), we hypothesized that SIRT6 could influence glucose metabolism by controlling expression of key metabolic genes. We first performed comparative microarray gene expression analysis of WT and SIRT6 KO muscle and ES cells (Supplemental Figure 3). As previously reported (Kawahara et al., 2009), multiple pathways appear to be affected in the absence of SIRT6 (Supplemental Figure 3A). Notably, there is a statistically significant alteration in regulators of glucose metabolism (Supplemental Figure 3B) and clustering analysis of metabolic genes separated the samples based on genotype. When a glucose metabolic filter was applied, the highest difference was observed among key glycolytic genes, such as lactate dehydrogenase (LDH), Triose Phosphate Isomerase (TPI), Aldolase, and the rate limiting glycolytic enzyme phospho-fructo kinase (PFK1). Using real-time PCR, we validated increased expression of all these genes in independent RNA samples (Figure 3A). GLUT1 was also increased at the level of RNA, thereby explaining the increased protein levels described above. Notably, we observed higher levels of the pyruvate dehydrogenase kinase genes PDK1 and PDK4. As mentioned before, these enzymes phosphorylate and inhibit pyruvate dehydrogenase (PDH), the rate-limiting enzyme that regulates entrance of pyruvate into the TCA cycle. In brief, our results indicate that in the absence of SIRT6, expression of multiple glucose-related genes are up-regulated, causing enhanced glycolysis and in parallel, inhibition of mitochondrial respiration.
Figure 3. SIRT6 directly inhibits expression of glycolytic genes functioning as an H3K9 deacetylase.
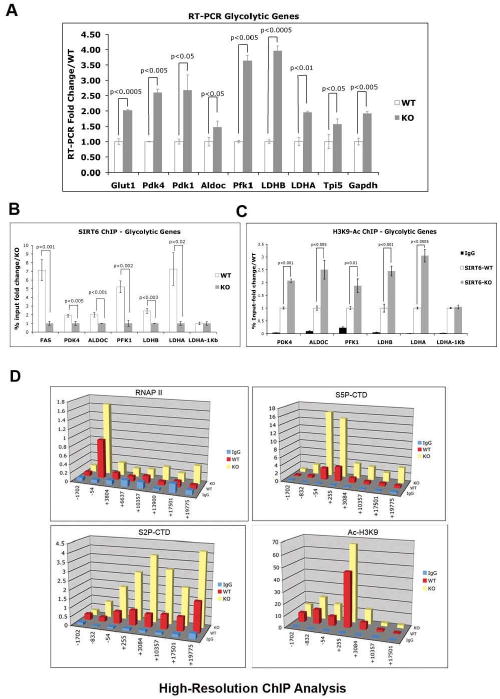
(A) RNA was purified from SIRT6 WT and KO ES cells and real-time PCR (RT-PCR) was performed with primers specific for the indicated genes. Three independent samples were averaged, keeping a threshold of 0.4 as confidence value in the threshold cycle (Ct). Values were normalized against actin. (B) Chromatin immunoprecipitation (ChIP) assays using an antibody against SIRT6 were performed on samples from SIRT6 WT and KO ES cells. Real-time PCR were carried out using primers specific for the promoter regions of the indicated glycolytic genes, except for LDHA-1kb, where primers lying 1kb downstream of the 3′ UTR of the LDHA gene were used, and served as a negative control. (C) ChIP assays were performed as described in (B), with an anti H3K9 acetylated antibody (Abcam). The LDHA-1Kb primers were used as a negative control. (D) High resolution ChIP analysis was performed in the LDHB locus using antibodies against total RNA Polymerase II (RNAP II), phosphorylated Serine 5 form of RNAP II (S5P-CTD), phosphorylated Serine 2 form of RNAP II (S2P-CTD), and acetylated H3 lysine 9. Error bars in all graphs indicate the standard error of the mean. See also Figures S3 and S4.
SIRT6 functions as a H3K9 deacetylase to regulate glucose homeostasis
In order to test whether SIRT6 directly controls expression of these genes, chromatin immuno-precipitation (ChIP) was performed using an antibody to SIRT6. As shown in Figure 3B, we find specific binding of SIRT6 to the promoter of all five of the most upregulated glycolytic genes identified in our expression analysis, strongly indicating that SIRT6 functions as a direct transcriptional repressor for these genes. Previous work has identified SIRT6 as a histone H3K9 deacetylase (Michishita et al., 2008). We have confirmed these results and further show that lack of SIRT6 causes a bulk increase in H3K9 acetylation in ES cells, suggesting that SIRT6 is one of the main H3K9 deacetylases in these cells (Supplemental Figure 4). Therefore, we tested whether SIRT6 deficient cells exhibit increased H3K9 acetylation in the promoters of these glycolytic genes. Indeed, ChIP analysis with an anti-H3K9Ac antibody clearly shows increased acetylation in all these putative targets (Figure 3C). Together, these results strongly suggest that SIRT6 directly suppresses expression of multiple glucose-metabolic genes by deacetylating H3K9 at their promoters.
To gain insight into the mechanism by which SIRT6 modulates expression of these genes, we chose one of these targets, LDHB, to perform high-resolution, quantitative ChIP analysis. Using qPCR amplicons against eight different locations within this genomic region, we analyzed the behavior of RNA polymerase II (RNAPII) in WT cells versus SIRT6 KO cells. We employed antibodies against total RNAPII as well as phospho-specific antibodies recognizing phosphorylation of Ser5 and Ser2 within the C-terminal domain (CTD) repeats of Rbp1, the largest subunit of RNAPII (Donner et al., 2007). Interestingly, this analysis revealed that in WT cells the LDHB promoter carries pre-loaded hypophosphorylated RNAPII and that SIRT6 depletion leads to increased RNAPII CTD phosphorylation concomitant with enhanced transcription elongation (Figure 3D). While RNAPII was readily detectable at the LDHB transcription start site (TSS) in WT and SIRT6-KO cells, transit throughout the intragenic region was observed only in the latter. Furthermore, total RNAPII signals at the TSS were several-fold higher than at any amplicon in the intragenic region, a hallmark of RNAPII pausing at the promoter. Typically, Ser5 phosphorylation occurs at 5′ end of genes and is associated with promoter escape by RNAPII. Accordingly, SIRT6 KO cells show significantly higher levels of this mark. Of note, the fold increase in Ser5 phosphorylation surpasses that of total RNAPII, indicating that in WT cells pre-loaded RNAPII exists in a hypophosphorylated state. The fact that LDHB transcription is stimulated at post-RNAPII recruitment steps is reinforced by analysis of Ser2-phosphorylation, a mark of actively elongating RNAPII that is increased several fold in SIRT6 KO cells. Consistent with our conventional ChIP results (Figure 3C), we also observe higher H3K9 acetylation in this assay. It is, however, of interest that this increase occurs focally, close to the TSS, without spreading to nearby regions. Overall, these results indicate that SIRT6 action represses transcription of LDHB (and arguably the other target genes) at regulatory steps downstream of RNAPII recruitment.
SIRT6 functions as a co-repressor of Hif1α
Our results thus far indicate that SIRT6 might play a role in re-directing carbohydrate flux from glycolysis to mitochondrial respiration, and in the absence of SIRT6, glycolysis is enhanced and the TCA cycle inhibited, a phenotype usually observed as an adaptation against nutrient or oxygen deprivation. One of the main positive regulators of this switch is the transcription factor Hif1α. In this context, all the genes that were up-regulated in the SIRT6 deficient cells are direct targets of Hif1α. Therefore, we decided to test whether SIRT6 could function to modulate a Hif1α nutrient stress response. First, we tested whether SIRT6 could influence expression of a luciferase reporter carrying multiple Hypoxia-Responsive-Elements (HREs), the consensus binding sequence for Hif1α. This construct is specifically activated following low glucose/hypoxia, and thus it represents a direct measurement of Hif1α activation in these cells (Zimmer et al., 2008). As shown in Figure 4A, exposing the cells to 24hr. low glucose elicited robust luciferase activity and notably, SIRT6 co-expression causes significant repression. Such an effect was not observed when we over-expressed a catalytically inactive mutant of SIRT6, suggesting that the enzymatic activity of SIRT6 was required for this specific effect on the promoter.
Figure 4. SIRT6 is a co-repressor of Hif1α.
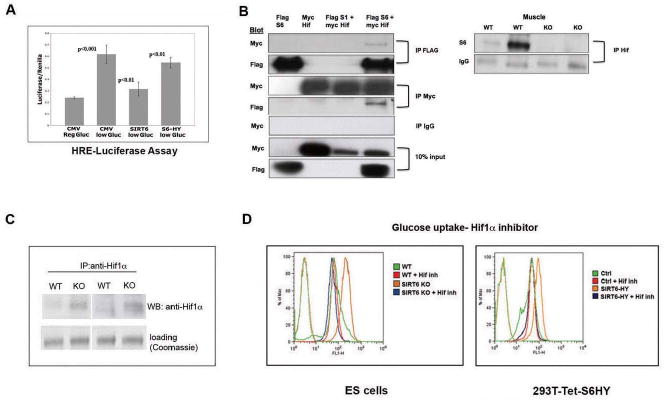
(A) A luciferase reporter gene under the regulation of 3 tandem copies of Hypoxia-Responsive Elements (HRE) was co-transfected with empty vector (CMV), SIRT6 (S6) or SIRT6-HY (catalytic dead) plasmids into 293T cells, and subjected to low-glucose (5mM) conditions for 24 hr. Extracts were analyzed for Luciferase activity. (B) Left panel: A Flag control, a SIRT6-Flag or a SIRT1-Flag proteins were either expressed alone or co-expressed with Hif1α-Myc in 293T cells, and following immunoprecipitation (IP) with either a Flag, a Myc, or an IgG antibody, extracts were analyzed by Western blot, and probed with the indicated antibodies. Right Panel: lysates were prepared from SIRT6 WT and KO muscle, and following IP with anti-Hif1α antibody, extracts were analyzed by western blot probed with anti-SIRT6 antibody. The IgG band is shown as loading control. (C) Lysates were prepared from SIRT6 WT or KO ES cells, followed by IP and western blot with a Hif1α antibody. (D) ES cells (left panel) or 293T cells stably expressing a tetracycline inducible SIRT6 dominant negative allele (S6HY)(right panel) were treated with or without the Hif1α inhibitor #77 (Zimmer et al., 2008) and glucose uptake was measured by FACS, following 1 hr. exposure to NBDG. See also Figure S5.
Since Hif1α appears to maintain basal activity even under normoglycemia (Carmeliet et al., 1998), we hypothesized that SIRT6 might bind Hif1α at the chromatin to regulate its activity. In order to test whether SIRT6 and Hif1α interact, FLAG-tagged SIRT6 was co-expressed with Myc-tagged Hif1α in 293T cells. Western analysis of the IPs revealed that Myc-Hif1α co-precipitated with SIRT6 and, likewise, FLAG-SIRT6 co precipitated with Hif1α (Figure 4B). This interaction appears specific, since other FLAG-sirtuins, like SIRT1, did not interact with Hif1α under these conditions (Figure 4B). In order to confirm that these proteins interact under physiological conditions, we immunoprecipitated Hif1α from muscle, and tested whether SIRT6 co-precipitated. As shown in Figure 4B, SIRT6 was readily detected in the Hif1α IP, clearly indicating that endogenous Hif1α and SIRT6 can interact.
Conditions of nutrient and oxygen stress cause activation of Hif1α, with increased protein levels due to both protein synthesis and stabilization of the protein (Aragones et al., 2009). Since lack of SIRT6 mimics a nutrient stress response, we determined whether SIRT6 deficient cells exhibit increased levels of Hif1α. Extracts were purified from SIRT6 wild type and KO cells grown under normoglycemic conditions, and western blot analysis was performed with an antibody specific for Hif1α. As expected for a normoglycemic condition, Hif1α was barely detected in wild-type cells. In contrast, Hif1α was readily detected in SIRT6 KO cells (Figure 4C). These results strongly indicate that under normal nutrient conditions, SIRT6 plays an important inhibitory role upon Hif1α-dependent glucose-related gene transcription, and lack of SIRT6 is sufficient to upregulate glycolytic gene-transcription.
Down-regulation of Hif1α rescues the metabolic phenotypes in SIRT6 deficient cells
The above results suggest that lack of SIRT6 triggers a Hif1α-dependent metabolic switch. In order to test whether Hif1α plays a critical role in this phenotype, we decided to inhibit Hif1α in SIRT6 KO cells and test whether we could rescue the metabolic abnormalities observed in these cells. For this purpose, we first treated SIRT6 KO ES cells with a recently described small molecule inhibitor of Hif1α/Hif2α (Zimmer et al., 2008). Treatment with this inhibitor for 24hr. was sufficient to completely revert the glucose uptake increase in SIRT6 KO cells (Figure 4D, left panel). This effect appears specific, since the compound did not affect wild-type cells. Furthermore, treatment with AKT or mTOR inhibitors, both modulators of insulin signaling and stress responses, was not able to rescue the metabolic phenotype (Supplemental Figure 5), strongly indicating that SIRT6 modulates glucose homeostasis specifically through a Hif1α-dependent pathway. To further validate these results, we performed a similar experiment in the inducible SIRT6 dominant-negative cells, where we previously showed that acute inactivation of SIRT6 leads to increase glucose uptake (Figure 1F). Similar to what we observed in the KO ES cells, treatment with the Hif1α inhibitor readily decreased glucose uptake in these cells as well (Figure 4D, right panel).
In order to confirm the role of Hif1α in this phenotype, we specifically knocked-down Hif1α in SIRT6 deficient cells. We grew multiple independent ES clones obtained following infection with a shRNA-Hif1α virus. Notably, in those clones where Hif1α was down-regulated, the increased glucose uptake was completely rescued (Figure 5A, clones #1 and #2). This effect is specific, since wild-type cells show no effect upon Hif1α knock-down. Furthermore, in those few clones where the Hif1α knock-down failed (as an example, see clone #3, Figure 5A), we observed no changes in glucose uptake. To test at the transcriptional level the critical modulators of this rescue, we purified RNA from the SIRT6KO/Hif1α-knockdown cells, and analyzed expression of the glycolytic genes previously identified. Notably, expression of most of these glycolytic genes was rescued to the levels observed in WT cells (Figure 5B). One exception is PDK1, which exhibits no statistical differences between the parental SIRT6 KO and the Hif1α knock-down cells, suggesting that in this case, the PDK4 isoform plays a more dominant role.
Figure 5. Knocking down Hif1α completely rescues the metabolic phenotype in SIRT6 deficient cells.
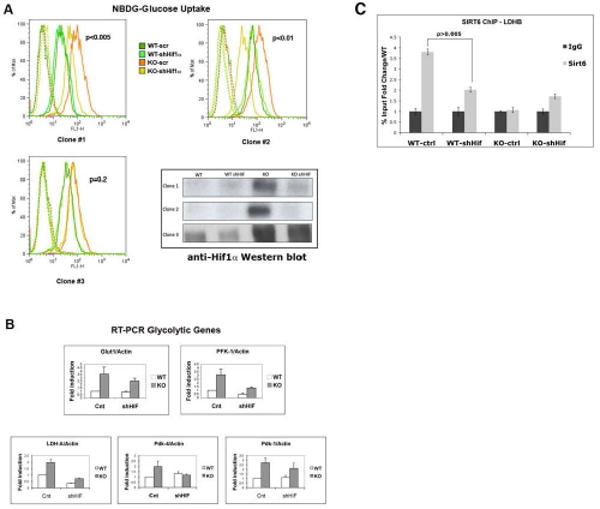
(A) SIRT6 WT and KO ES cells were infected with either a Hif1α-knockdown lentivirus (shHiflα) or vector alone (scr). Independent clones were expanded, and glucose uptake was measured using NBDG, as described before. Lower right panel: Western blot analysis of the different clones with an anti-Hif1α antibody. Note that clone #3 failed to down-regulate Hif1α, and thus it served as an internal control. (B) RNA was purified from Hif1α-KD clones and glycolytic gene expression levels were examined by RT-PCR. The different analyzed genes are indicated. Fold induction was normalized against actin. Three independent samples were averaged, keeping a threshold of 0.4 as confidence value in the threshold cycle (Ct). Error bars in all graphs indicate the standard error of the mean. (C) Hif1α recruits SIRT6 to the glycolytic promoters. ChIP was performed on wild-type control (WT-ctrl) and Hif1α knock-down cells (WT-shHif) with an antibody against SIRT6. Real-time PCR were carried out using primers specific for the promoter region of the LDHB gene. SIR6 KO cells were used as negative controls in the ChIP assay.
Taking advantage of these Hif1α knock-down cells, we tested whether Hif1α was required to recruit SIRT6 to these glycolytic-gene promoters. For this purpose, ChIP with anti-SIRT6 antibodies was performed in these cells, and SIRT6 occupancy on those promoters evaluated. As seen in Figure 5C, lack of Hif1α significantly reduced SIRT6 binding to these promoters, indicating that SIRT6 is specifically recruited to these promoters via its physical interaction with Hif1α.
Lack of SIRT6 increases both protein synthesis and stability of Hif1α
In order to gain further insight into the increase levels of Hif1α that we observed in our SIRT6 deficient cells, we first tested whether SIRT6 directly regulates Hif1α. For this purpose, we analyzed RNA levels in SIRT6 deficient cells. As shown in Figure 6A, Hif1α RNA levels were comparable between wild type and KO cells, indicating that Hif1α is not a direct transcriptional target of SIRT6. Previous studies have indicated that Hif1α could itself be acetylated (Jeong et al., 2002). However, such findings were later disputed (Arnesen et al., 2005; Murray-Rust et al., 2006). In this context, we also failed to detect Hif1α acetylation in vivo, even in SIRT6 KO cells, where total levels of Hif1α were significantly higher (Supplemental Figure 6); therefore, a direct effect for SIRT6 on Hif1α appears unlikely. We next tested whether protein stability of Hif1α was increased in the SIRT6 KO cells. For this purpose, we treated cells with the iron chelator CoCl2, which inhibits the activity of the prolyl-hydroxylases, therefore inhibiting degradation of Hif1α. While treatment with the drug robustly increase Hif1α in wild type cells, this effect was significantly diminished in the SIRT6 KO cells (Figure 6B), indicating that in these cells Hif1α is already stabilized. However, some increase was observed in the KO cells, suggesting that increased stability could only partially account for the higher levels observed in these cells. We therefore tested whether Hif1α protein synthesis was also enhanced in the absence of SIRT6. We first took advantage of a previously described luciferase reporter carrying the 5′UTR of the Hif1α gene (Bert et al., 2006). Notably, SIRT6 KO cells exhibit a significant increase in luciferase activity under basal conditions, similar to the levels observed in wild type cells following nutrient/oxygen stress (Figure 6C). As an independent assay, we purified the polysomes fraction of ribosomes, and quantified the rate of Hif1α translation in both wild type and KO cells (Serikawa et al., 2003). Consistent with the previous assay, SIRT6 KO cells exhibit a clear increase in Hif1α translation in this assay as well (Figure 6D). All together, these results indicate that both Hif1α protein synthesis and stability are increased in SIRT6 deficient cells.
Figure 6. Increased Hif1α stability and protein synthesis in SIRT6 deficient cells.
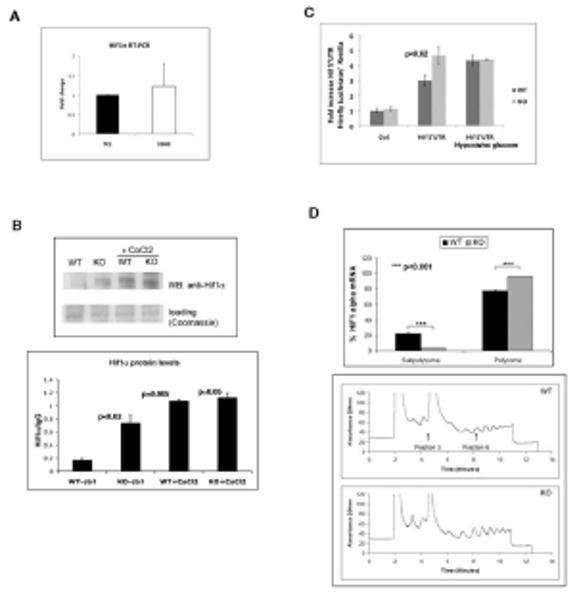
(A) RNA was purified from SIRT6 WT and KO ES cells and Hif1α expression was analyzed by RT-PCR using primers specific for the mRNA of Hif1α. Results are shown as the mean±SEM (n=6). (B) Upper panel: Lysates were prepared from SIRT6 WT or KO ES cells, followed by IP and western blot with a Hif1α antibody. Samples were either left untreated, or treated with the Hif1α stabilizer CoCl2 (150μM) for 24 hr. prior to lysate preparation. Lower panel: quantitative densitometric analysis of Hif1α levels from the upper panel blot. (C) Wild type (WT) and SIRT6 deficient (KO) cells were co-transfected with an empty 5′UTR-Luc vector or Hif1α- 5′UTR-Luciferase reporters and shifted 6hrs post-transfection to no glucose-hypoxia conditions for 24hrs for measurement of luciferase activity. (D) Polysome profile analysis of WT and Sirt6 deficient (KO) ES cells. lower panel : WT and KO cells were treated with cycloheximide (CHX) for 10 minutes before collection. The lysates were processed for polysome analysis by velocity sedimentation on sucrose gradients. Gradients were fractionated by scanning at 254 nm, and the resulting absorbance profiles are shown with sedimentation from left to right. Upper panel: Quantitative RT-PCR was performed to assess distribution of HIF1α mRNA. See also Figure S6.
Increased expression of glycolytic genes and increased lactate production in SIRT6 deficient mice
We next tested whether this glycolytic switch we observed in vitro was also responsible for the metabolic phenotype observed in vivo. Protein extracts were purified from muscle of multiple SIRT6 WT and KO mice, and westerns were performed with antibodies against the different glycolytic enzymes. Strikingly, the two rate limiting factors PFK1 and PDK1, which were barely detectable in wild-type muscle, were expressed at very high levels in SIRT6 KO muscle (Figure 7A). As well, we observed higher levels of other glycolytic genes, such as TPI1, while expression of the p53 target TIGAR, which was recently shown to inhibit glycolysis (Bensaad et al., 2006) was reduced in the KO samples. Using immunostaining, we then tested for expression of the GLUT1 transporter. Similar to what we found in ES cells, muscle but not brain from SIRT6 deficient animals expressed significantly higher levels of GLUT1 (Figure 7B). Finally, we tested whether lactate production was increased in the KO animals. As shown in Figure 7C, SIRT6 KO animals exhibit a modest but statistically significant increased in serum lactate, when compared to wild-type animals, thus supporting the argument that lack of SIRT6 in vivo promotes uncontrolled glucose uptake and a glycolytic switch, consistent with our findings in vitro.
Figure 7. Hif1α-dependent increased glycolysis and lactate production in SIRT6 deficient mice.
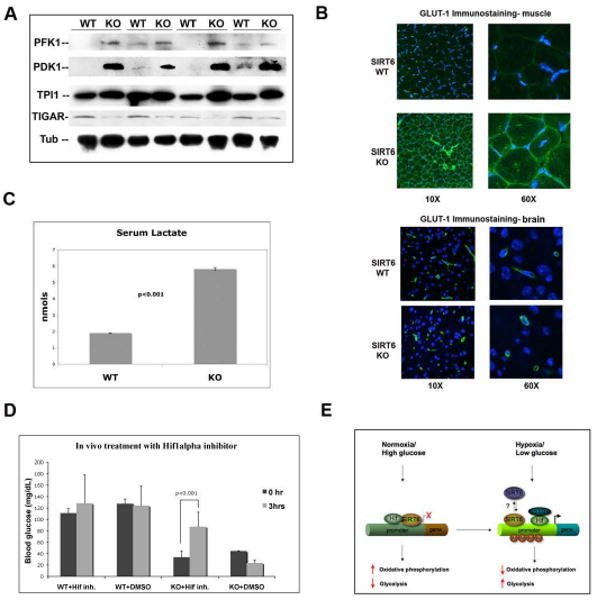
(A) Lysates were prepared from muscles of 4 littermate-pairs of SIRT6 WT and KO mice. Western analysis was carried out with antibodies against the indicated proteins. Tubulin was used as a loading control. (B) Immunostaining with a GLUT1 antibody (green) was carried out on muscles and brain from SIRT6 WT and KO mice. Nuclei were stained with DAPI (blue). Images were taken using a confocal microscope with constant laser beam for all images (KR: 39.8; IRIS: 2.0). (C) Serum was purified from SIRT6 WT and KO mice, and lactate was measured using the Lactate Assay Kit (BioVision). Error bars indicate the standard error of the mean. n=4 for each genotype. (D) A Hif1α small molecule inhibitor rescues the glucose phenotype in SIRT6 deficient mice. Hif1α inhibitor #77 (20μg/g weight) was injected i.p. in 19 days-old wild type and SIRT6 KO mice, and 30 min later blood was withdrawn for glucose measurement. 5% DMSO (dilution solution) was injected as control. (E) Model. Under normal nutrient conditions, SIRT6 inhibits expression of glycolytic genes acting as an histone deacetylase to co-repress Hif1α. This maintains proper flux of glucose to the TCA cycle. Under conditions of nutrient stress, SIRT6 is inactivated, allowing activation of Hif1α, recruitment of p300, acetylation of H3K9 at the promoters and increased expression of multiple metabolic genes, causing increased glycolysis and decreased mitochondrial respiration. See also Figure S7.
Hif1α inhibition rescues the glucose phenotype in SIRT6 deficient mice
To test whether the hypoglycemia observed in the SIRT6 deficient animals was dependent on Hif1α, as we found in vitro, we treated SIRT6 deficient animals with the Hif1α inhibitor described above. Strikingly, treatment with the drug caused a fast and specific increase in blood glucose levels specifically in the KO animals (Figure 6D). These results indicate that, similar to what we observed in our SIRT6 KO ES cells, regulation of glucose metabolism by SIRT6 depends on Hif1α in vivo as well.
Discussion
Our studies reveal that SIRT6 functions as a histone deacetylase to control glucose homeostasis by inhibiting multiple glycolytic genes in a coordinated fashion (Figure 7E). Under conditions of normal glucose availability, SIRT6 represses expression of key enzymes, diverting pyruvate towards the mitochondrial TCA cycle for efficient ATP production. At those promoters, SIRT6 competes with the transcriptional activator Hif1α to maintain proper glucose flux towards mitochondrial respiration and preventing excessive glycolysis. Several lines of evidence support this model. First, SIRT6 deficiency causes up-regulation of glycolytic genes at the level of expression, a finding that is accompanied by increased glucose uptake and a switch towards glycolysis even under normal nutrient conditions (Figures 1-3). Second, SIRT6 directly binds to the promoters of these genes, and in the absence of SIRT6, H3K9 acetylation increases specifically in those promoters (Figure 3B, 3C and 3D). Third, we find that SIRT6 influence glycolysis as a co-repressor of Hif1α, and in the absence of SIRT6, the glycolytic phenotype can be rescued by knocking-down Hif1α in these cells (Figures 4-5). Thus, SIRT6 acts as a safeguard mechanism to down-modulate basal transcription of Hif1α target genes under normal nutrient conditions (Carmeliet et al., 1998). In this context, there are two plausible scenarios. The first possibility is that SIRT6 binding to the promoters inhibits recruitment of Hif1α (accelerating its degradation). Alternatively, Hif1α could already localize to the promoters under normoglycemia, but the presence of SIRT6 would inhibit its transcriptional activity. Even though we found that SIRT6 and Hif1α can interact (Figure 4B), we were unable to perform ChIP assays with anti-Hif1α antibodies; therefore, we cannot rule out at present either possibility. However, recent studies have shown that, indeed, Hif1α occupies its target promoters even under normoxia (Xia et al., 2009), supporting our second model. Notably, early studies have demonstrated that Hif1α activates transcription through recruitment of the histone acetyl-transferase p300/CBP (Arany et al., 1996; Kallio et al., 1998); consequently, SIRT6 might compete against recruitment of p300, maintaining histones in those promoters in a hypoacetylated state. Future studies will likely address this possibility. In the context of the Hif1α knock-down experiments, it is intriguing that despite full rescue of the metabolic phenotype (see figure 5A), only a subset of glycolytic genes were rescued (Figure 5B, see for instance PDK1). These results suggest that while SIRT6 regulates multiple glycolytic genes in a coordinated fashion, only few of them play a dominant role in this glycolytic switch.
An alternative explanation to our results would be that, in addition to deacetylating histones at those putative targets, SIRT6 actually regulates Hif1α itself. In this regard, we find no changes in HIF1α RNA levels in SIRT6 deficient cells (Figure 6A), indicating that SIRT6 does not regulate expression of this factor. A second possibility would be that SIRT6 deacetylates Hif1α, and in the absence of SIRT6, Hif1α is acetylated and stabilized. However, as shown above, we also failed to detect Hif1α acetylation in vivo, even in SIRT6 KO cells, where total levels of Hif1α were significantly higher (Supplemental Figure 6A); therefore, a direct effect for SIRT6 on Hif1α appears unlikely. On the other hand, we find that lack of SIRT6 increases both protein synthesis and protein stability of Hif1α. Previous studies have shown that conditions of nutrient stress and increase in lactate production can function as a positive feedback to induce both protein synthesis and stability of Hif1α (Lu et al., 2002) (Hirota and Semenza, 2005). Although at present other possibilities cannot be ruled out, SIRT6 deficient cells experienced both nutrient stress and increased lactate production, likely explaining the increased Hif1α levels observed in these cells.
Using high-resolution quantitative ChIP mapping of the LDHB gene, we gained further insight into the molecular mechanisms of SIRT6 silencing. Our results indicate that in wild type cells, SIRT6 binding maintains low levels of H3K9 acetylation at the LDHB promoter, thereby inhibiting transcription, despite the presence of pre-loaded RNAPII. In the absence of SIRT6, transcription is activated, as indicated by robust enrichment of Ser5 and Ser2 phosphorylated RNAPII, markers of promoter escape and transcriptional elongation, respectively. These results are intriguing, suggesting that SIRT6, a histone deacetylase, might repress transcription at a stage downstream of RNAPII recruitment. Recent studies have shown that engaged but paused polymerase plays an important role on genes that need to be rapidly activated (Core and Lis, 2008). Changes in nutrient conditions could vary rapidly, and therefore, it is tempting to speculate that SIRT6 could repress transcription while maintaining an engaged polymerase, which in turn will allow rapid activation of transcription at these glycolytic genes upon changes in nutrient availability. Whether this represents a general epigenetic mode of regulation remains to be determined.
Recent studies have shown that SIRT6 can function as a co-repressor of NF-κB, modulating expression of NF-κB targets (Kawahara et al., 2009). Furthermore, RelA haploinsufficiency was able to rescue the lethality of SIRT6 deficient animals. However, glucose levels in these animals remain low for the first weeks of life. Therefore, it is unlikely that NF-κB represents the initial trigger in the hypoglycemic phenotype observed. Consistent with this notion, we do not observe changes in expression of NF-κB targets in our muscle-microarray data (Supplemental Figure 4A). Overall, these results strongly support a model where the defects in glucose metabolism observed in the absence of SIRT6 stem from its role in controlling glycolytic gene expression rather than through modulation of NF-κB targets.
Our model for SIRT6 function predicts that under conditions of nutrient stress, SIRT6 would be inactivated, triggering a Hif1α-dependent glycolytic switch, similar to what we observed in our SIRT6 deficient cells (Figure 7E). In this regard, we do not observe changes in total levels nor in localization of SIRT6 protein following glucose deprivation (Supplemental Figure 7). One possibility is that SIRT6 activity is controlled at a post-transcriptional level, an alternative that is under current investigation. It is interesting that nutrient deprivation has been shown to increase levels of another mammalian sirtuin, SIRT1 (Cohen et al., 2004; Nemoto et al., 2004), indicating that these proteins might have evolved to function in contrasting scenarios. In this context, while SIRT1 activators have been shown to protect against metabolic diseases such as type-II diabetes, as published (Baur et al., 2006; Lagouge et al., 2006; Milne et al., 2007), in the case of SIRT6, inhibition rather than activation might prove beneficial to lower blood glucose in metabolic diseases.
The increased glycolytic capacity and reduced oxidative phosphorylation we observe in SIRT6 deficient cells are reminiscent to the “Warburg effect” described by Otto Warburg several decades ago (Warburg, 1956). Such a phenomenon describes the peculiarity that most cancer and highly proliferative cells rely on aerobic glycolysis rather than respiration for their energy and metabolic needs (Vander Heiden et al., 2009). Consistent with our observations in SIRT6 deficient cells, recent studies indicate that aerobic glycolysis requires Hif1α as well (Lum et al., 2007), and Hif1α confers resistance to apoptosis in cancer cells under hypoxic conditions in a GLUT-1 dependent manner (Kilic et al., 2007). Based on this analogy, one could predict that lack of SIRT6 should provide an advantage for tumorigenic growth. In this context, SIRT6 deficient ES cells exhibit increased resistance to apoptosis when exposed to hypoxia/hypoglycemia (Supplemental Figure 6B); however, we are currently unable to test this hypothesis in vivo, since SIRT6 deficient animals die early in life. While it remains unclear what is the trigger that allows the switch from oxidative phosphorylation to aerobic glycolysis (Vander Heiden et al., 2009), our results indicate that inhibition of SIRT6 might be an important player.
Previous studies in yeast, worms and flies have linked Sir2 proteins to the regulation of longevity (Finkel et al., 2009; Longo and Kennedy, 2006; Yu and Auwerx, 2009). Whether such a role is conserved in mammals remains unclear. However, multiple lines of evidence indicate a critical role for some of these mammalian sirtuins in regulating metabolic homeostasis (Canto et al., 2009). Since changes in calorie intake and metabolic balance has been previously linked to lifespan regulation in mammals (Yu and Auwerx, 2009) (Barzilai and Bartke, 2009), our new results with SIRT6 place this chromatin deacetylase as a potential candidate among sirtuins to influence ageing and age-related diseases. Notably, two recent articles have shown that Hif1α can modulate lifespan in C. Elegans (Chen et al., 2009; Mehta et al., 2009). Whether this is the case in mammals remains unknown, however our results suggest that sirtuins and Hif1α may function in a coordinated fashion to modulate metabolic homeostasis in higher eukaryotes.
Overall, our studies have demonstrated a novel role for the histone deacetylase SIRT6 in controlling glucose homeostasis. The severe metabolic phenotypes we observed indicate that among the mammalian sirtuins, SIRT6 appears to play a dominant role in regulating energy balance. It also suggests that while a glycolytic switch might be an important acute adaptive response in situations of nutrient stress or cancer growth, chronic and sustained activation of this switch (as in the case of SIRT6 deficiency) is rather detrimental.
Experimental Procedures
Western and Immunostaining Analysis
Western analysis was carried out as previously described (Cheng et al., 2003). The antibodies used are: anti-SIRT6 (Abcam), anti-Hif1α (Novus), anti-Flag (Sigma), anti-PFK1 (Abcam), anti-PDK1 (Abcam), anti-TPI1 (Proteintech), anti-TIGAR (Abcam), anti-Tubulin (Sigma). For immunostaining of ES cells, cells were grown on coverslips and fixed with 4% paraformaldehyde and permabilized with 0.1% Triton X-100 in PBS as described (Bassing et al., 2002). Cells were stained with anti-Glut1 antibody (Alpha Diagnostic). Images were taken using a confocal microscope with constant laser beam for all images (KR:39.8; IRIS:2.0).
Lactate and Oxygen Consumption Assays
Lactate concentration was determined with the Lactate Assay Kit (BioVision). O.D. was measured at 570nm, 30 min. after addition of substrate. For oxgen consumption, 4×105 SIRT6 WT and KO ES cells were seeded, and 24 hours oxygen consumption rate was measured with the Seahorse XF24 instrument (Seahorse Bioscience), as published (Liu et al., 2009).
Metabolite Analysis
SIRT6 wild type and KO cells were grown under normal nutrient conditions, and methanol-fixed proteins were analyzed by liquid chromatography-mass spectrometry (LC-MS), as described (Lewis et al., 2008).
Glucose Uptake Assays in mice and in cells
For the assessment of in vivo glucose disposal, [1,2-13C] glucose was IP injected (2 mg/gm body weight) into mice (n=4). Blood samples were collected 30 min after, and processed for GC/MS analysis as previously described (Xu et al., 2004). For the glucose uptake assay in mice, 16-days old SIRT6 WT and KO mice were imaged using a Siemens Inveon PET-CT 45 minutes post injection of approximately 500 uCi of FDG, as published elsewhere (Boiselle et al., 1998). For glucose uptake assays in cells, cells were grown under normal conditions for 24 hours and 100 μM 2-NBDG (Invitrogen) was added to the media for 2 hours. Fluorescence was measured in a FACSCalibur Analyzer (BD).
ATP Concentration Assays
SIRT6 WT or KO ES cells were grown in low glucose (0.5 g/L) media for 24 hours and ATP concentration was measured by Adenosine 5′-triphosphate (ATP) bioluminescent somatic cell assay kit (Sigma) per manufacturer instructions.
Luciferase Reporter Assays
1×105 293T cells were transfected using Trans-IT 293 (Mirus) with 1μg of the following plasmids as described in the text: pGL3∷HRE4, pCMV-3xF-SIRT6 and pCMV-3xF-SIRT6HY. 24 hours after transfection, cells were harvested and luciferase activity was determined using the Dual-Luciferase Reporter Assay system (Promega).
ChIPs and Q-RT-PCR
ChIP and Q-RT-PCR were performed as previously described (Donner et al., 2007). The antibodies and the primers' sequences for all the RT-PCRs are included in Supplemental Experimental Procedures and Supplemental Table 1.
Retroviral Infection
SIRT6-WT cDNA was amplified by PCR and cloned into the pHAGE2-EF1a-dsRed-IRES-tomato vector. Hif1α shRNA lentivirus vectors were obtained from The RNAi Consortium Library (MGH). SIRT6-WT and SIRT6-KO ES cells were infected by incubating with virus and 10 μg/ml polybrene. 48 hours later, cells were selected in 2.5 g/ml puromycin and single colonies were picked and plated for various experiments.
5′UTR assays
SIRT6 WT and KO ES cells were transfected using Lipofectamine 2000 with a luciferase vector lacking 5′UTR as control and a vector with a partial HIF1α 5′UTR (kindly provided by Gregory J. Goodall) co-transfected with the Renilla Luciferase plasmid. 6hr post-transfection media was changed to normal or no glucose and incubated in 1% Oxigen chamber for 24hrs. Protein was extracted as detailed in the promega kit for Dual Luciferase Assay per manufacturer instructions. Luminescence was read at 500nm.
Polysome Profiling Analysis
Cytoplasmic extracts were purified from cycloheximide-treated SIRT6 WT and KO ES cells. The lysates were processed for polysome analysis by velocity sedimentation on sucrose gradients. Quantitative RT-PCR was performed on purified mRNA to assess distribution of HIF1α mRNA, which was normalized to 18S mRNA. For details, see Supplemental Experimental procedures.
Supplementary Material
Acknowledgments
This work was supported by the V Foundation (R.M.), the Sidney Kimmel Cancer Research Foundation (R.M.), a New Investigator Grant from AFAR (R.M.), and Grant P30 DK57521 from the Metabolic Physiology Core (BADERC). Y.D. is supported by JDRF. J.M.E. is supported by NIH-RO1 CA117907 and the Howard Hughes Medical Institute. We thank Magali Silberman, Daniel Holoch, Pere Puigserver, John Dominy, Matthew Vander Heiden, Mike Zimmer, David Lombard, Bjoern Schwer, Fred Alt, Katrin Chua and members of the Bardeesy, Hochedlinger, Hock and Ramaswamy labs for reagents and helpful discussions. We thank Kelly Shay, Laura Prickett-Rice and Kate Folz-Donahue for technical assistance. We are also thankful to Nabeel Bardeesy and Hanno Hock for critically reading the manuscript and Jose Polo for assistance with the ChIP assays.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aragones J, Fraisl P, Baes M, Carmeliet P. Oxygen sensors at the crossroad of metabolism. Cell metabolism. 2009;9:11–22. doi: 10.1016/j.cmet.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, Bunn HF, Livingston DM. An essential role for p300/CBP in the cellular response to hypoxia. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:12969–12973. doi: 10.1073/pnas.93.23.12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnesen T, Kong X, Evjenth R, Gromyko D, Varhaug JE, Lin Z, Sang N, Caro J, Lillehaug JR. Interaction between HIF-1 alpha (ODD) and hARD1 does not induce acetylation and destabilization of HIF-1 alpha. FEBS letters. 2005;579:6428–6432. doi: 10.1016/j.febslet.2005.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilai N, Bartke A. Biological approaches to mechanistically understand the healthy life span extension achieved by calorie restriction and modulation of hormones. J Gerontol A Biol Sci Med Sci. 2009;64:187–191. doi: 10.1093/gerona/gln061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Bert AG, Grepin R, Vadas MA, Goodall GJ. Assessing IRES activity in the HIF-1alpha and other cellular 5′ UTRs. RNA. 2006;12:1074–1083. doi: 10.1261/rna.2320506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiselle PM, Patz EF, Jr, Vining DJ, Weissleder R, Shepard JA, McLoud TC. Imaging of mediastinal lymph nodes: CT, MR, and FDG PET. Radiographics. 1998;18:1061–1069. doi: 10.1148/radiographics.18.5.9747607. [DOI] [PubMed] [Google Scholar]
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- Chen D, Thomas EL, Kapahi P. HIF-1 modulates dietary restriction-mediated lifespan extension via IRE-1 in Caenorhabditis elegans. PLoS genetics. 2009;5:e1000486. doi: 10.1371/journal.pgen.1000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Mostoslavsky R, Saito Si, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua aKF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proceedings of the National Academy of Sciences. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science (New York), NY. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- Core LJ, Lis JT. Transcription regulation through promoter-proximal pausing of RNA polymerase II. Science (New York), NY. 2008;319:1791–1792. doi: 10.1126/science.1150843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donner AJ, Szostek S, Hoover JM, Espinosa JM. CDK8 is a stimulus-specific positive coregulator of p53 target genes. Molecular cell. 2007;27:121–133. doi: 10.1016/j.molcel.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochemical and biophysical research communications. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes & development. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- Hirota K, Semenza GL. Regulation of hypoxia-inducible factor 1 by prolyl and asparaginyl hydroxylases. Biochemical and biophysical research communications. 2005;338:610–616. doi: 10.1016/j.bbrc.2005.08.193. [DOI] [PubMed] [Google Scholar]
- Hu CJ, Iyer S, Sataur A, Covello KL, Chodosh LA, Simon MC. Differential regulation of the transcriptional activities of hypoxia-inducible factor 1 alpha (HIF-1alpha) and HIF-2alpha in stem cells. Mol Cell Biol. 2006;26:3514–3526. doi: 10.1128/MCB.26.9.3514-3526.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong JW, Bae MK, Ahn MY, Kim SH, Sohn TK, Bae MH, Yoo MA, Song EJ, Lee KJ, Kim KW. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell. 2002;111:709–720. doi: 10.1016/s0092-8674(02)01085-1. [DOI] [PubMed] [Google Scholar]
- Kallio PJ, Okamoto K, O'Brien S, Carrero P, Makino Y, Tanaka H, Poellinger L. Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. Embo J. 1998;17:6573–6586. doi: 10.1093/emboj/17.22.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, McCord RA, Ongaigui KC, Boxer LD, Chang HY, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 2009;136:62–74. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilic M, Kasperczyk H, Fulda S, Debatin KM. Role of hypoxia inducible factor-1 alpha in modulation of apoptosis resistance. Oncogene. 2007;26:2027–2038. doi: 10.1038/sj.onc.1210008. [DOI] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell metabolism. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Lewis GD, Wei R, Liu E, Yang E, Shi X, Martinovic M, Farrell L, Asnani A, Cyrille M, Ramanathan A, et al. Metabolite profiling of blood from individuals undergoing planned myocardial infarction reveals early markers of myocardial injury. The Journal of clinical investigation. 2008;118:3503–3512. doi: 10.1172/JCI35111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C, Liu H, Keyvanfar K, Chen H, Cao LY, et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459:387–392. doi: 10.1038/nature08040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD, Kennedy BK. Sirtuins in aging and age-related disease. Cell. 2006;126:257–268. doi: 10.1016/j.cell.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. The Journal of biological chemistry. 2002;277:23111–23115. doi: 10.1074/jbc.M202487200. [DOI] [PubMed] [Google Scholar]
- Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, Simon MC, Thompson CB. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes & development. 2007;21:1037–1049. doi: 10.1101/gad.1529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta R, Steinkraus KA, Sutphin GL, Ramos FJ, Shamieh LS, Huh A, Davis C, Chandler-Brown D, Kaeberlein M. Proteasomal regulation of the hypoxic response modulates aging in C. elegans. Science (New York), NY. 2009;324:1196–1198. doi: 10.1126/science.1173507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Murray-Rust TA, Oldham NJ, Hewitson KS, Schofield CJ. Purified recombinant hARD1 does not catalyse acetylation of Lys532 of HIF-1alpha fragments in vitro. FEBS letters. 2006;580:1911–1918. doi: 10.1016/j.febslet.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science (New York), NY. 2004;306:2105–2108. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell metabolism. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Pessin JE, Bell GI. Mammalian facilitative glucose transporter family: structure and molecular regulation. Annual review of physiology. 1992;54:911–930. doi: 10.1146/annurev.ph.54.030192.004403. [DOI] [PubMed] [Google Scholar]
- Seagroves TN, Ryan HE, Lu H, Wouters BG, Knapp M, Thibault P, Laderoute K, Johnson RS. Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol Cell Biol. 2001;21:3436–3444. doi: 10.1128/MCB.21.10.3436-3444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serikawa KA, Xu XL, MacKay VL, Law GL, Zong Q, Zhao LP, Bumgarner R, Morris DR. The transcriptome and its translation during recovery from cell cycle arrest in Saccharomyces cerevisiae. Mol Cell Proteomics. 2003;2:191–204. doi: 10.1074/mcp.D200002-MCP200. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (New York), NY. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science (New York), NY. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Xia X, Lemieux ME, Li W, Carroll JS, Brown M, Liu XS, Kung AL. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:4260–4265. doi: 10.1073/pnas.0810067106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Chang V, Joseph SB, Trujillo C, Bassilian S, Saad MF, Lee WN, Kurland IJ. Peroxisomal proliferator-activated receptor alpha deficiency diminishes insulin-responsiveness of gluconeogenic/glycolytic/pentose gene expression and substrate cycle flux. Endocrinology. 2004;145:1087–1095. doi: 10.1210/en.2003-1173. [DOI] [PubMed] [Google Scholar]
- Yu J, Auwerx J. The role of sirtuins in the control of metabolic homeostasis. Ann N Y Acad Sci. 2009;1173 1:E10–19. doi: 10.1111/j.1749-6632.2009.04952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer cell. 2007;11:407–420. doi: 10.1016/j.ccr.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Zimmer M, Ebert BL, Neil C, Brenner K, Papaioannou I, Melas A, Tolliday N, Lamb J, Pantopoulos K, Golub T, et al. Small-molecule inhibitors of HIF-2a translation link its 5′UTR iron-responsive element to oxygen sensing. Molecular cell. 2008;32:838–848. doi: 10.1016/j.molcel.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.