

Abstract
The systematic karyotyping of bone marrow cells was the first genomic approach used to personalize therapy for patients with leukemia. The paradigm established by cytogenetic studies in leukemia (from gene discovery to therapeutic intervention) now has the potential to be rapidly extended with the use of whole-genome sequencing approaches for cancer, which are now possible. We are now entering a period of exponential growth in cancer gene discovery that will provide many novel therapeutic targets for a large number of cancer types. Establishing the pathogenetic relevance of individual mutations is a major challenge that must be solved. However, after thousands of cancer genomes have been sequenced, the genetic rules of cancer will become known and new approaches for diagnosis, risk stratification and individualized treatment of cancer patients will surely follow.
Keywords: array CGH, cancer, comparative genomic hybridization, genomics, next-generation sequencing, SNP array
Identifying all the genetic events that lead to cancer development will expedite the discovery of novel therapeutics. Cancer genetics went through a revolutionary period with the advent of cytogenetic analysis of chromosomes 50 years ago, and we are poised to undergo another revolution in cancer gene discovery with the recent advances in DNA sequencing technology that are available today. As the discovery of genes that are mutated in cancer continues to grow with the use of unbiased genomic platforms (array-based and next-generation sequencing), many of the lessons learned from studying the genetics of leukemia over the past 50 years remain pertinent for this new era of discovery.
First ‘personalized genomics’ for cancer: karyotyping of leukemia cells
The first clue that cancer was a genetic disease came from the German biologist David von Hansemann, who observed aberrant mitotic figures in carcinoma cells in 1890 [1]. The study of chromosomal abnormalities in cancer underwent a paradigm shift when Nowell and Hungerford described the Philadelphia chromosome in patients with chronic myelogenous leukemia (CML) in 1960 [2]. The subsequent discovery that the Philadelphia chromosome was caused by a balanced translocation between chromosomes 9 and 22 (t[9;22]) not only provided a diagnostic test (a chromosomal rearrangement) that was specific for CML [3], but ultimately led to the development of targeted therapy in CML, since the fusion gene created by the translocation is the initiating mutation for the disease. The karyotyping methods used to identify t(9;22) set the stage for the landmark discoveries of other recurrent chromosomal translocations in leukemias and lymphomas in the 1970s and 1980s, which also led to the improved treatment and outcomes for many patients [4–8].
The meticulous cataloging of translocations and chromosomal abnormalities in acute myeloid leukemia (AML) over the past several decades has allowed cytogenetics to become one of the most powerful diagnostic and prognostic tools available for AML patients [9,10]. Patients can now be classified into three main cytogenetic groups: those with favorable, intermediate, or adverse risk karyotypes, which are predictive of overall survival (Figure 1) [10]. Although the cytogenetic classification system does risk-stratify patients with AML, it is not perfect; there is considerable heterogeneity in overall survival within each cytogenetic group, indicating that additional factors are relevant for defining prognosis. Some of these have recently been uncovered in candidate gene resequencing studies and genome-wide array based studies, as discussed below.
Figure 1. Cytogenetics and survival in acute myeloid leukemia.
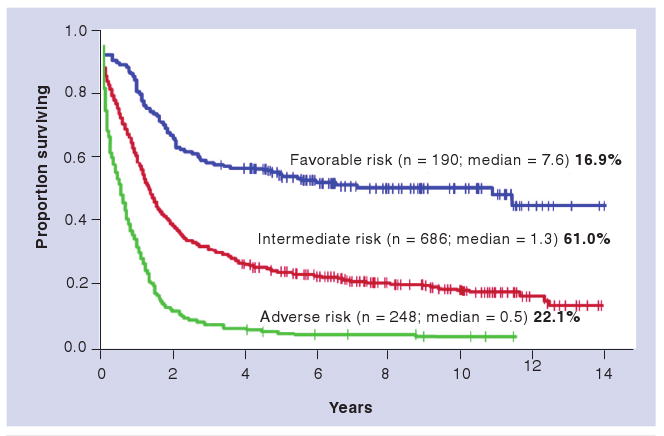
The cumulative probability of overall survival significantly differs among acute myeloid leukemia patients with favorable (t[8;21], inv[16], or t[16;16]), adverse (inv[3], t[3;3], t[6;9], t[6;11], -7, +8 sole, +8 with one other abnormality, t[11;19] [q23;p13.1], or greater than two clonal abnormalities), or intermediate (all others, including normal karyotype) cytogenetic categories, as defined by the Cancer and Leukemia Group B group.
Adapted from [10]. This research was originally published in Blood [10].
© American Society of Hematology.
Attempts to improve AML classification schemes with array-based genomic platforms
To identify small (<5 Mb in size) subcytogenetic amplifications and deletions (as well as the genes contained in those regions), array-based comparative genomic hybridization (CGH) and SNP genotyping platforms (which can also be used to deduce copy number alterations) have recently been used to screen AML genomes for acquired (somatic) copy number alterations [11–13]. The most recent of these studies, which employed the extensive validation of putative calls, suggested that there are, in fact, very few recurrent acquired copy number changes in most AML genomes. Furthermore, these data have not yet improved our ability to predict the prognosis for AML patients beyond what was known using standard cytogenetics [14,15].
The cytogenetic classification system has been improved by incorporating the mutation status of genes that are commonly mutated in AML (e.g., FLT3, NPM1, NRAS, CEBPA and MLL) [16,17] and the mRNA expression levels of single genes that are sometimes expressed in AML cells [17–23]. However, these classification systems are also imperfect, again suggesting that crucial genetic and/or epigenetic events in AML remain to be discovered.
‘Next generation’ of genomics: whole-genome sequencing
As noted above, the discovery of common translocations and of the genes located at translocation breakpoints ultimately led to the development and use of treatments that targeted the mutated genes (e.g., ABL in CML and RARA in acute promyelocytic leukemia) [24,25]. In the past, it took several decades from the discovery of a cytogenetic abnormality to the identification of the mutated genes. Today, the timeline for cancer gene discovery is greatly compressed owing to the development of genomic platforms that can provide nucleotide level resolution of the entire cancer genome in only a few weeks, the timeframe required to make treatment decisions. A more precise genetic classification of AML and other cancers may well be possible using next-generation sequencing approaches, which can simultaneously identify all of the structural chromosomal abnormalities and specific gene mutations in a cancer genome; it is possible that many or all of these will need to be understood to move forward with personalized therapies.
The ultimate goal of this work is to comprehensively and systematically define the critical genes that are altered in each patient with cancer and then to develop personalized therapy for that patient. The success of this approach will rely on the rigorous experimental design and cataloging of mutations in cancer genomes. This will ultimately be possible by generating the complete genetic landscape from thousands of individuals with a specific cancer, which will identify genetic subgroups of patients with a greater homogeneity than was previously possible using cytogenetics.
Why start whole-genome sequencing work with AML?
Cancer is a genetic disease, but epigenetic (see below) and non-cell intrinsic factors (e.g., angiogenesis, stromal interactions, immune responses and so on) are also important. Sequence-based studies will not capture all of this complexity, but it is the genetic factors that have so far had the greatest impact on AML diagnosis, risk stratification and tailored therapy. Therefore, there is a strong rationale for capitalizing on the recent developments in sequencing technology to completely characterize cancer genomes.
Acute myeloid leukemia was a very attractive disease for our initial studies of cancer genome sequencing for several reasons:
First, AML is a very serious cancer and is not rare. In 2008, approximately 13,000 individuals in the USA developed AML and nearly 9000 died from the disease[26].
Second, improving our ability to predict outcomes using key mutations should have an immediate impact on how we treat patients. As noted above, we already tailor our initial approach to therapy, which is based on a low-resolution genomic screen (cytogenetics). Refining this with the knowledge of all the mutations in the genome may well improve our ability to predict outcomes, and will allow us to treat our patients more precisely based on an accurate upfront risk assessment. Novel drugs will not need to be developed for these data to have an immediate impact on how we treat our patients.
Acute myeloid leukemia cells are also easy to access with nonsurgical procedures (peripheral blood sampling and/or bone marrow aspiration) and a large fraction of AML samples are only minimally contaminated with normal cells. Therefore, serial sampling of the diseased tissue is straightforward and tumor cell purification is generally not required.
In addition, nearly half of all AML genomes are cytogenetically normal. Furthermore, the study of AML genomes with array-based, high-resolution CGH approaches has revealed that many have no detectable copy number alterations at 35-kb resolution (compared with the 5-Mb resolution of cytogenetics). The study of diploid genomes using our initial whole-genome sequencing approach greatly simplifies the assessment of coverage (see below), and also simplifies the analysis and interpretation of whole-genome sequencing data. From preliminary studies performed by our group and others, it appears that cytogenetically normal genomes may contain fewer mutations than cancer genomes that are highly aneuploid (data not shown); therefore, mutations in diploid genomes may be more likely to be pathogenetically relevant.
Finally, many of the mutations found in AML genomes may also be relevant for other cancer types. While AML genomes do have some mutations that seem to be restricted to this disease (e.g., NPMc and FLT3 internal tandem duplication mutations), others (e.g., RAS and IDH1) are known to be important for other cancer types as well.
Therefore, our initial studies with whole-genome sequencing have been performed with relatively simple AML genomes, with the hope that the experience gained and mutations discovered would guide our work with aneuploid genomes (that may have many more passenger mutations, and may therefore be more difficult to understand).
Challenges of sequencing AML (& all cancer) genomes
Prior to the introduction of next-generation sequencing, the cost of sequencing whole cancer genomes was simply beyond the reach of any laboratory or institution; although large sections of the exome were successfully sequenced in breast and colorectal cancer by Velculescu and colleagues, less than 1% of the total genome was actually sequenced in this study [27]. Based on the considerations listed below, as recently as 3 years ago it would have cost nearly US$90 million to completely sequence a cancer genome and its matched normal counterpart. However, with the advent of next-generation sequencing technologies, this cost has already fallen to approximately US$100,000 and it is expected to fall further in the very near future. Therefore, until very recently, the biggest obstacle for sequencing cancer genomes was cost.
Why is cancer genome sequencing so expensive? First, massive amounts of sequence data are required to discover all the mutations in a cancer genome since the human species is out-bred – each person has 3–4 million sequence variants (and hundreds of copy number variants) that need to be assessed for their possible contributions to the cancer phenotype. In addition to this, because each human genome has so many genomic variations, both the tumor genome and the genome of a matched normal tissue sample (generally skin for hematologic malignancies, or blood for solid tumors) must be sequenced from each patient in order to define whether the genomic variants are inherited or acquired. Since we expect that most somatic mutations will be heterozygous, both alleles must also be sampled at every position in the genome to obtain adequate coverage for mutation discovery (this will henceforth be referred to as ‘diploid’ coverage). While only sixfold coverage (i.e., 18 billion base pairs of sequence for a 3 billion base pair genome) is required to solve a genome's primary structure, at least four-times that (∼25-fold coverage) is required to achieve adequate diploid coverage for comprehensive mutation discovery. Finally, tumor genomes do not only contain point mutations, but also structural variations that could be relevant for pathogenesis.
Current state-of-the-art technology for sequencing cancer genomes
To discover all of the genomic alterations that could be relevant for cancer pathogenesis, a shotgun sequencing method, using ‘paired-end’ reads on massively parallel sequencing devices, is currently the method of choice in most genome centers. Transcriptome sequencing is also being performed to sample the expressed part of the genome and represents an important adjunct to whole-genome sequencing (see below). Importantly, libraries of DNA fragments from tumor samples can now be made with very small amounts of input DNA (using as little as 100 ng is now routine), which is critical since sample abundance is rate-limiting for many tumors and normal matched control samples.
Our initial work with cancer genome sequencing utilized short fragment reads (30–35 bp in length). Because of the short length of these reads, many could not be unambiguously mapped back to the reference genome. However, longer sequence reads (now up to 100 bp) from both ends of small DNA fragments (e.g., 250 bp up to several kilobases in size) are now being routinely performed in the large genome centers. The longer read lengths, coupled with paired-end reads, dramatically increase mapping efficiency and accuracy, and allow for unambiguous sequence assignment even in regions that are highly repetitive. In addition, this technology dramatically increases our ability to identify structural variants, including deletions and amplifications, translocations and inversions. Now that it is possible to make libraries with DNA fragments that are more than 1 kb in size, translocations are routinely detected. Finally, this technology requires fewer sequencing runs to achieve adequate diploid coverage for each genome, which results in additional cost savings.
DNA fragment capture techniques are also being evaluated as a strategy to reduce costs and increase the yield of sequence-based studies by focusing on the exons themselves for mutation discovery. Although there are some advantages of this approach (i.e., large numbers of patients can potentially be screened more rapidly and deep read counts for each captured exon would assure high sensitivity for mutation discovery), there are disadvantages as well. All exon capture approaches are biased towards predefined regions of the genome, and the efficiency of capture methods is still far from perfect. For the time being, whole-genome sequencing is our method of choice for cancer mutation discovery.
Challenge of understanding mutations found in cancer genomes
Over the past 2 years, we have successfully sequenced the matched tumor and normal skin genomes of two individuals with the M1 subtype of AML [28,29]. Both individuals had essentially diploid tumor genomes and we were able to estimate that each genome contained approximately 500–1000 somatic point mutations. However, current modeling estimates suggest that only five–ten mutations are required to cause most cancers [30,31]. So, why are there so many mutations in these genomes? And which ones are important?
Of all the mutations found in each tumor, only a small number were nonsynonymous; in the first AML genome we found ten mutations, and in the second, just 12. By performing deep read counts of the variant allele frequencies for each mutation, we were able to establish that all of these nonsynonymous mutations were present in virtually all of the tumor cells (assuming that the mutations were heterozygous). A total of four of these mutations were found in at least one other AML genome (out of 188 genomes tested in total), strongly suggesting that they are indeed important for pathogenesis because they are recurrent. In the second AML genome, we validated 54 mutations that were not in coding sequences, but that did fall in highly conserved regions of the genome (and/or regions with potential regulatory importance) [29]. Using deep count analysis, we demonstrated that all of these mutations were likewise present in virtually every tumor cell.
How can a tumor ‘retain’ that many acquired mutations? Are they all important? Probably not. We feel that the most likely explanation is that many of them are irrelevant mutations that were already present in the hematopoietic stem cell, which was transformed by the acquisition of one or more key initiating mutations that altered the growth and/or developmental fate of that cell. A model for this hypothesis is presented in Figure 2. We suggest that long-lived hematopoietic stem cells normally acquire a number of benign mutations that do not alter the function of these cells during the life of an individual. Even though most of them are irrelevant, they are all present in the individual cell when it acquires the critical mutation that sets the cancer in motion. Additional mutations then cause the transformed cell to progress to overt leukemia. When this transformed cell expands clonally to produce the tumor, and is sequenced with our existing technologies, we are really examining the genomic history of the initiating cell and are defining all of the mutations that may have been present in that cell from the time of transformation onwards (other potential scenarios exist to explain these data, but we currently favor this one). The greatest challenge in cancer genomics is to distinguish the benign, pre-existing ‘passenger’ mutations from those that are relevant for pathogenesis – the so-called ‘drivers’.
Figure 2. Model for evolution of genetic changes in acute myeloid leukemia.

A hypothetical model in which nonpathogenic somatic mutations (1–3) acquired over the lifespan of a stem cell are propagated in the malignant clone after it acquires a critical initiating mutation (4). Mutation 5 is a progression mutation that cooperates with the AML-initiating mutation 4 to contribute to AML development. Other mutations (represented by 6 and 7) do not cooperate with the AML-initiating mutation 4, and do not contribute to AML development. These subclones are lost, or fail to expand to the limit of detection by sequencing studies. AML: Acute myeloid leukemia; HSC: Hematopoietic stem cell.
Finding the relevant mutations in a sea of noise
Since hundreds to thousands of mutations will be present in any given cancer genome, the prospect of assigning relevance to each is a daunting task. Furthermore, assembling all of the potentially relevant mutations in a test cell or organism to assess the role of each ‘in context’ is an enormous challenge.
How can we begin to define the truly relevant mutations in any cancer genome? A number of strategies can be employed, and together they should provide powerful clues that will be helpful in the short term.
First and most importantly, recurring mutations in a gene, or at an individual nucleotide position in the genome, are likely to be important for pathogenesis. The likelihood of having an identical mutation at the same position in 188 genomes (the number chosen for our initial studies given the statistical and cost constraints) with a mutation frequency of 1 in 4 million bp is only approximately 1 × 10-9; therefore, the vast majority of recurring mutations will be important. To identify these recurring mutations, tens of thousands of cancer genomes will need to be sequenced over the next several years. Initially, much of the sequencing will be performed with highly annotated samples in the academic sector. However, in the future, many cancer genomes will probably be sequenced by commercial entities. We feel that it will be critical to capture data generated by these commercial sources for cancer genome databases, so that recurring mutations can be more rapidly identified.
In nongenic regions of the genome that are minimally annotated (i.e., most of the genome), finding recurring mutations may be extremely helpful in identifying functional regions that are currently of unknown significance. In addition, some studies have suggested that large portions of intergenic regions are transcribed, adding to their potential to be functionally relevant [32]. For example, we found a recurring point mutation in a conserved (but nongenic) region of chromosome 10 in two AML cases. Finding recurring mutations in nonannotated parts of the genome may greatly improve our understanding of the genome in general, and will undoubtedly uncover new mechanisms for cancer pathogenesis.
Needless to say, the gold standard for assessing the importance of mutations will continue to be functional validation in tissue culture cells, or in a model organism. However, assembling multiple mutations into a single cell or model organism will probably be required to discern the importance of individual mutations for cancer pathogenesis. This represents an extraordinarily daunting challenge that will require new approaches for its execution.
The analysis of cancer genomes from mice and other model organisms may also be very useful for identifying potentially relevant human cancer mutations. Since mice have a relatively short lifespan, they may accumulate fewer somatic mutations in the time it takes to develop a cancer. Since many laboratory mice are maintained as homozygous strains, the DNA sequence coverage required to identify acquired mutations will be far less than that required for human genomes, and matched normal DNA may not need to be fully sequenced for each mouse tumor analyzed. In inbred strains of the worm Caenorhabditis elegans, only 8× coverage was required to routinely detect somatic mutations [33], and similar sequence coverage should be reasonable for identifying mutations in inbred mouse genomes. Furthermore, mutations found in genes that are associated with cancers in both mice and humans will almost certainly be pathogenetically relevant.
Epigenetics & cancer
Currently, there are several array-based platforms that are utilized to assess the epigenetic contributions to cancer pathogenesis. Expression studies are routinely performed on arrays. Chromatin immunoprecipitation (ChIP) studies and methylation studies are now routinely performed using array-based technologies as well. Although these studies are relatively robust, it is clear that next-generation sequencing technologies will further increase the quality of data generated by these strategies.
Next-generation sequencing can be used to quantitate mRNA abundance by sequencing cDNA libraries derived from tumors. The read counts of different mRNA species can be used to obtain a highly accurate digital representation of mRNA abundance in a tumor sample. Furthermore, since primary sequence data are generated as well, point mutations and fusion transcripts can also be identified [34–36]. Although transcriptome analysis with next-generation sequencing is powerful, this technology will miss mutations that inactivate genes (such as deletions, mutations that cause nonsense-mediated decay and regulatory mutations) and these would be found with whole-genome sequencing. Many aspects of this technology are still evolving, but it will certainly be an important adjunct to whole-genome sequencing for the foreseeable future.
Chromatin immunoprecipitation is a powerful tool for determining the DNA sequences with which proteins interact in the genome. This technology has been greatly aided by array-based approaches, but most arrays do not represent the entire genome and are therefore biased to varying extents. By coupling ChIP with next-generation sequencing techniques (ChIP-Seq) [37,38], an even greater level of precision will be obtained and will complement and extend ChIP studies in the future. Likewise, the evaluation of methylated regions of the genome may be highly relevant for understanding cancer pathogenesis. Whole-genome methylation studies have been successfully performed using array-based technologies, but the sequencing of methylated regions of the genome on next-generation platforms (Methyl-Seq) will improve the quality of data.
Another challenge: data integration across platforms
As noted above, cancer genomics requires an understanding of not only the structure of primary cancer genomes, but also of their function. One of the greatest technical challenges facing the field is the integration of different kinds of genomic data (generated on different platforms) into a seamless whole. When this is routine, an even greater intellectual challenge will exist: how to integrate alterations occurring at the level of genes versus alterations occurring at the levels of pathways. Based on the analysis of the first few cancer genomes, it seems possible that the combination of mutations associated with a given cancer may be extremely large; however, the pathways affected by these mutations may be limited and obey a set of rules that remain to be defined. Integrating mutational and pathway data remains an enormous challenge for the field, but this problem will hopefully be solved when thousands of cancer genomes have been successfully sequenced.
Understanding inherited variants that affect AML susceptibility
Cancer genome sequencing will reveal all of the inherited variants present in each individual with cancer. It is highly likely that some of these inherited variants may provide a critical ‘substrate’ for the acquired mutations that have been the focus of this perspective thus far. However, the role of inherited variants will ultimately need to be integrated into the sequencing analysis in order to fully understand the cancer genome. Characterizing these inherited factors is not only of great interest scientifically, but it will also have relevance for genetic counseling and screening, and potentially, for cancer prevention.
Inherited variants with established roles in AML susceptibility occur in two genes associated with rare familial leukemia syndromes and several others that cause more general cancer predisposition syndromes. The best-characterized familial leukemia syndrome is the familial platelet disorder with a predisposition to AML (FDP/AML), which is caused by inherited mutations in RUNX1 [39]. A smaller number of families with germline mutations in CEBPA have also been reported [40,41]. These individuals develop AML with nearly complete penetrance. The RUNX1 and CEBPA pedigrees display features that are typical of single-gene cancer predisposition syndromes, including multiple affected first-degree relatives, early-onset of disease compared with sporadic cases and low prevalence. Other cases of familial leukemia have been described in which linkage to RUNX1 and CEBPA have been excluded, implying that there are additional susceptibility loci that have not yet been identified [42,43].
Several classical cancer predisposition syndromes are associated with an increased risk of AML in addition to other tumors. The incidence of leukemia is increased in Li Fraumeni families (caused by mutations in TP53), although myelodysplastic syndromes (MDS) or AML probably occur in fewer than 5% of affected individuals [44]. Children with neurofibromatosis Type I (caused by NF1 mutations) have a greatly increased risk of developing MDS/AML [45]. Other cancer predisposition syndromes associated with excess leukemia cases include ataxia telangiectasia (exclusively associated with ALL) and Wiskott–Aldrich Syndrome [46,47].
Several genetic syndromes causing bone marrow failure are also associated with a predisposition to MDS/AML. In some of these, the genetic lesion causes loss of genomic integrity that presumably contributes directly to tumor formation (e.g., Fanconi's anemia, Bloom's syndrome and Dyskeratosis congenita). In others, the mechanism of leukemia predisposition is less clear but may be an indirect consequence of chronic bone marrow ‘stress’ (e.g., severe congenital neutropenia, Diamond–Blackfan anemia or Schwachman–Diamond syndrome).
Perhaps most importantly for the general population, sporadic AML may also have an inherited component. From twin studies, the inherited contribution to leukemia susceptibility was estimated to be approximately 20% [48]. The cumulative impact of all of the syndromes discussed above falls far short of this figure, implying that most of the inherited factors that are important for leukemia susceptibility have not yet been discovered. In nonsyndromic AML, familial aggregation of cases is rare. This suggests that inherited susceptibility alleles have modest effects, that they may arise spontaneously in affected individuals (de novo mutations), or that AML in the general population does not have a significant inherited component. This will be an extremely challenging problem to resolve, given the large number of polymorphisms that have been detected in the small number of genomes that have been systematically analyzed to date (∼3.5 million SNPs and hundreds of copy number variants per genome). Genome-wide association studies using array-based platforms will fail to detect de novo or rare variants that are not in linkage disequilibrium with markers included in the standard panels. As consensus builds around the idea that AML, like other sporadic cancers, may not conform to the ‘common variant, common disease’ paradigm (i.e., AML may not be a consequence of polymorphisms that occur frequently in the general population) [49,50], it will become increasingly important to examine these rare variants in association studies.
Comprehensive analysis of DNA from individuals with cancer using next-generation sequencing approaches provides an unbiased view of all variants present in these genomes. One of the important lessons from our studies of somatic mutations in AML is that the sequence of a paired sample from nonmalignant tissue (e.g., skin) obtained from each patient is an essential component of the analytical pipeline. This strategy allows for the unambiguous classification of nucleotide variants detected in the tumor sample as acquired versus inherited mutations. When the goal of the study is to identify acquired mutations, those changes detected in both the tumor and nonmalignant sample are ‘discarded’ as inherited. For studies of susceptibility, the complete complement of germline variants (e.g., single nucleotide variants, insertion/deletions and copy number variants) must be considered. What must occur next is a comparison between the genomes of affected (with AML) and unaffected (without AML) individuals from similar genetic backgrounds. The power of such a study could, in principle, be improved by choosing cases with a higher a priori probability of harboring novel susceptibility alleles (e.g., early disease onset, exclusion of known predisposing factors, excess first-degree relatives with AML, other hematologic malignancy, or other cancers). Discovery of alleles that play a role in AML susceptibility will require a comparison with thousands of cases to appropriately matched controls. As daunting as this task may seem, these studies will be feasible in the near future, and should lead to novel prevention and control strategies for AML.
Conclusion
Next-generation sequencing technologies have the potential to revolutionize our understanding of cancer. The basic rules for using whole-genome screens to understand cancer pathogenesis were established with the use of karyotyping (a low-resolution genomic screen) in AML cases more than 30 years ago. By using many of the paradigms established in those studies, the scientific community should be able to use whole-genome sequencing data (and associated epigenetic data) to establish a clear picture of the mutations that cause each cancer in the not too distant future. This will require sequencing tens of thousands of cancer genomes and the careful analysis of these genomes for recurrent mutations that impact the pathways relevant for cancer pathogenesis.
In our view, there is no question that these data will dramatically change our understanding of cancer and lead to new ways to diagnose and classify this large group of diseases. Some of the information will have an immediate impact on the care of patients, and some will lead to the development of novel drugs and strategies for the treatment of individual patients in the future. Regardless, this revolutionary technology will permanently change our understanding of cancer by providing a complete picture of the mutations that cause it. Only then can we realistically hope to find the novel approaches that will lead to increased therapeutic success.
Future perspective
Whole-genome sequencing (structural genomics) and epigenomic studies (functional genomics) are important new tools for understanding cancer and improving its treatment. When the cost of these studies reaches a critical threshold (probably a few thousand dollars per patient), we suggest that most, if not all, cancer patients should have high resolution genomic studies performed as part of their initial evaluation. It is highly likely that many genomic studies will be performed by commercial entities, with interpretation provided by trained pathologists and oncologists. In the short term, we believe that data from individual patients will be used to provide more accurate diagnostic and prognostic information, and that it will heavily influence treatment decisions.
Although many great challenges remain, the information gained from next-generation sequencing platforms should ultimately lead to the discovery of novel drugs that more effectively target the key genes and pathways that cause cancer. The marriage of genomics and targeted therapies will hopefully lead to therapeutic successes akin to that achieved for chronic myelogenous leukemia patients with imatinib, where the drug directly targets the initiating oncogene. Clearly, this is the ultimate goal for all of this work.
Executive summary.
Cytogenetics provided the first tool for the personalization of acute myeloid leukemia (AML) therapy and has remained a powerful predictor of survival.
SNP array, array comparative genomic hybridization (CGH) and candidate gene resequencing studies in de novo AML have identified very few acquired DNA changes per genome, suggesting that many mutations remain to be discovered.
mRNA profiling has identified genes that may have prognostic value in AML and validation studies are ongoing.
Next-generation sequencing of cancer genomes is now feasible. To determine which sequence changes represent somatic mutations, a matched normal genome must be sequenced from the same patient for comparison.
Sequence coverage must be adequate to identify both copies of every nucleotide across the genome. Several strategies will be required to define the mutations in a cancer genome that are the most important for pathogenesis.
Inherited variants that contribute to cancer susceptibility will be discovered using whole-genome sequencing.
A complete understanding of cancer genomes will require integration across many genomic platforms (cytogenetics, array CGH, expression analysis, next-generation sequencing [including RNA seq, Chip-Seq and Methylome-Seq]) for thousands of patients.
It is likely that thousands of genomes will be sequenced by academic and commercial centers in the near future, and that the identification of recurrent mutations may revolutionize our understanding of cancer.
Footnotes
Financial & competing interests disclosure: The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Bibliography
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.von Hansemann D. Ueber asymmetrische Zelltheilung in epithel Krebsen und deren biologische Bedeutung. Virchow's Arch Path Anat. 1890;119:299. [Google Scholar]
- 2.Nowell PC, Hungerford DA. A minute chromosome in human granulocytic leukemia. Science. 1960;132:1497. [Google Scholar]
- 3.Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243(5405):290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 4.Zech L, Haglund U, Nilsson K, Klein G. Characteristic chromosomal abnormalities in biopsies and lymphoid-cell lines from patients with Burkitt and non-Burkitt lymphomas. Int J Cancer. 1976;17(1):47–56. doi: 10.1002/ijc.2910170108. [DOI] [PubMed] [Google Scholar]
- 5.Rowley JD. Identificaton of a translocation with quinacrine fluorescence in a patient with acute leukemia. Ann Genet. 1973;16(2):109–112. [PubMed] [Google Scholar]
- 6.Le Beau MM, Larson RA, Bitter MA, Vardiman JW, Golomb HM, Rowley JD. Association of an inversion of chromosome 16 with abnormal marrow eosinophils in acute myelomonocytic leukemia. A unique cytogenetic-clinicopathological association. N Engl J Med. 1983;309(11):630–636. doi: 10.1056/NEJM198309153091103. [DOI] [PubMed] [Google Scholar]
- 7.Larson RA, Kondo K, Vardiman JW, Butler AE, Golomb HM, Rowley JD. Evidence for a 15;17 translocation in every patient with acute promyelocytic leukemia. Am J Med. 1984;76(5):827–841. doi: 10.1016/0002-9343(84)90994-x. [DOI] [PubMed] [Google Scholar]
- 8.Fukuhara S, Rowley JD, Variakojis D, Golomb HM. Chromosome abnormalities in poorly differentiated lymphocytic lymphoma. Cancer Res. 1979;39(8):3119–3128. [PubMed] [Google Scholar]
- 9.Grimwade D, Walker H, Oliver F, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood. 1998;92(7):2322–2333. [PubMed] [Google Scholar]
- 10.Byrd JC, Mrozek K, Dodge RK, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461) Blood. 2002;100(13):4325–4336. doi: 10.1182/blood-2002-03-0772. [DOI] [PubMed] [Google Scholar]
- 11.Rucker FG, Bullinger L, Schwaenen C, et al. Disclosure of candidate genes in acute myeloid leukemia with complex karyotypes using microarray-based molecular characterization. J Clin Oncol. 2006;24(24):3887–3894. doi: 10.1200/JCO.2005.04.5450. [DOI] [PubMed] [Google Scholar]
- 12.Suela J, Alvarez S, Cifuentes F, et al. DNA profiling analysis of 100 consecutive de novo acute myeloid leukemia cases reveals patterns of genomic instability that affect all cytogenetic risk groups. Leukemia. 2007;21(6):1224–1231. doi: 10.1038/sj.leu.2404653. [DOI] [PubMed] [Google Scholar]
- 13.Tyybakinoja A, Elonen E, Piippo K, Porkka K, Knuutila S. Oligonucleotide array-CGH reveals cryptic gene copy number alterations in karyotypically normal acute myeloid leukemia. Leukemia. 2007;21(3):571–574. doi: 10.1038/sj.leu.2404543. [DOI] [PubMed] [Google Scholar]
- 14.Walter MJ, Payton JE, Ries RE, et al. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc Natl Acad Sci USA. 2009;106(31):12950–12955. doi: 10.1073/pnas.0903091106. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪ A copy number alteration study in adult de novo acute myeloid leukemia (AML) that identified an average of 2.34 changes per genome using paired normal and tumor samples with secondary validation of altered loci.
- 15.Radtke I, Mullighan CG, Ishii M, et al. Genomic analysis reveals few genetic alterations in pediatric acute myeloid leukemia. Proc Natl Acad Sci USA. 2009;106(31):12944–12949. doi: 10.1073/pnas.0903142106. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪ Pediatric AML copy number alteration study that identified an average of 2.38 alterations per genome.
- 16.Schlenk RF, Dohner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358(18):1909–1918. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]; ▪ Landmark study demonstrating that the mutational status of NPM1, FLT3, CEBPA, and MLL in cytogenetically normal AML are associated with treatment outcome.
- 17.Baldus CD, Mrozek K, Marcucci G, Bloomfield CD. Clinical outcome of de novo acute myeloid leukaemia patients with normal cytogenetics is affected by molecular genetic alterations: a concise review. Br J Haematol. 2007;137(5):387–400. doi: 10.1111/j.1365-2141.2007.06566.x. [DOI] [PubMed] [Google Scholar]
- 18.Bullinger L, Dohner K, Bair E, et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350(16):1605–1616. doi: 10.1056/NEJMoa031046. [DOI] [PubMed] [Google Scholar]; ▪ Landmark study using gene-expression profiling to identify specific subgroups of AML that had prognostic significance.
- 19.Heuser M, Beutel G, Krauter J, et al. High meningioma 1 (MN1) expression as a predictor for poor outcome in acute myeloid leukemia with normal cytogenetics. Blood. 2006;108(12):3898–3905. doi: 10.1182/blood-2006-04-014845. [DOI] [PubMed] [Google Scholar]
- 20.Langer C, Radmacher MD, Ruppert AS, et al. High BAALC expression associates with other molecular prognostic markers, poor outcome, and a distinct gene-expression signature in cytogenetically normal patients younger than 60 years with acute myeloid leukemia: a Cancer and Leukemia Group B (CALGB) study. Blood. 2008;111(11):5371–5379. doi: 10.1182/blood-2007-11-124958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lugthart S, van Drunen E, van Norden Y, et al. High EVI1 levels predict adverse outcome in acute myeloid leukemia: prevalence of EVI1 overexpression and chromosome 3q26 abnormalities underestimated. Blood. 2008;111(8):4329–4337. doi: 10.1182/blood-2007-10-119230. [DOI] [PubMed] [Google Scholar]
- 22.Marcucci G, Maharry K, Whitman SP, et al. High expression levels of the ETS-related gene, ERG, predict adverse outcome and improve molecular risk-based classification of cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B Study. J Clin Oncol. 2007;25(22):3337–3343. doi: 10.1200/JCO.2007.10.8720. [DOI] [PubMed] [Google Scholar]
- 23.Valk PJ, Verhaak RG, Beijen MA, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350(16):1617–1628. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]; ▪ Landmark study using gene-expression profiling to identify specific subgroups of AML that had prognostic significance.
- 24.Huang ME, Ye YC, Chen SR, et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72(2):567–572. [Google Scholar]
- 25.Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344(14):1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 26.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer Statistics, 2009. CA Cancer J Clin. 2009;59(4):225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 27.Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 28.Ley TJ, Mardis ER, Ding L, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456(7218):66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪ First report of a completely sequenced cancer genome.
- 29.Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪▪ Describes the complete sequence of a second cancer genome and is particularly notable for the discovery of a highly recurrent mutation that was not previously detected in this disease.
- 30.Schinzel AC, Hahn WC. Oncogenic transformation and experimental models of human cancer. Front Biosci. 2008;13:71–84. doi: 10.2741/2661. [DOI] [PubMed] [Google Scholar]
- 31.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Birney E, Stamatoyannopoulos JA, Dutta A, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarin S, Prabhu S, O'Meara MM, Pe'er I, Hobert O. Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat Methods. 2008;5(10):865–867. doi: 10.1038/nmeth.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shah SP, Kobel M, Senz J, et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N Engl J Med. 2009;360(26):2719–2729. doi: 10.1056/NEJMoa0902542. [DOI] [PubMed] [Google Scholar]
- 35.Campbell PJ, Stephens PJ, Pleasance ED, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;40(6):722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪ Landmark study using paired-end next-generation sequencing to detect copy number alterations (insertions, deletions, inversions and translocations) in cancer cells.
- 36.Maher CA, Palanisamy N, Brenner JC, et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc Natl Acad Sci USA. 2009;106(30):12353–12358. doi: 10.1073/pnas.0904720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robertson G, Hirst M, Bainbridge M, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4(8):651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 38.Mardis ER. ChIP-seq: welcome to the new frontier. Nat Methods. 2007;4(8):613–614. doi: 10.1038/nmeth0807-613. [DOI] [PubMed] [Google Scholar]
- 39.Song WJ, Sullivan MG, Legare RD, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23(2):166–175. doi: 10.1038/13793. [DOI] [PubMed] [Google Scholar]
- 40.Smith ML, Cavenagh JD, Lister TA, Fitzgibbon J. Mutation of CEBPA in familial acute myeloid leukemia. N Engl J Med. 2004;351(23):2403–2407. doi: 10.1056/NEJMoa041331. [DOI] [PubMed] [Google Scholar]
- 41.Pabst T, Eyholzer M, Haefliger S, Schardt J, Mueller BU. Somatic CEBPA mutations are a frequent second event in families with germline CEBPA mutations and familial acute myeloid leukemia. J Clin Oncol. 2008;26(31):5088–5093. doi: 10.1200/JCO.2008.16.5563. [DOI] [PubMed] [Google Scholar]
- 42.Owen C, Barnett M, Fitzgibbon J. Familial myelodysplasia and acute myeloid leukaemia – a review. Br J Haematol. 2008;140(2):123–132. doi: 10.1111/j.1365-2141.2007.06909.x. [DOI] [PubMed] [Google Scholar]
- 43.Sellick GS, Pritchard-Jones K, Shepherd V, Swansbury J, Catovsky D, Houlston RS. Loci other than 21q22.12 (RUNX1) and 16q21–23.2 cause familial AML. Leukemia. 2005;19(3):465–466. doi: 10.1038/sj.leu.2403648. [DOI] [PubMed] [Google Scholar]
- 44.Li FP, Fraumeni JF, Jr, Mulvihill JJ, et al. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48(18):5358–5362. [PubMed] [Google Scholar]
- 45.Side L, Taylor B, Cayouette M, et al. Homozygous inactivation of the NF1 gene in bone marrow cells from children with neurofibromatosis Type 1 and malignant myeloid disorders. N Engl J Med. 1997;336(24):1713–1720. doi: 10.1056/NEJM199706123362404. [DOI] [PubMed] [Google Scholar]
- 46.Taylor AM, Metcalfe JA, Thick J, Mak YF. Leukemia and lymphoma in ataxia telangiectasia. Blood. 1996;87(2):423–438. [PubMed] [Google Scholar]
- 47.Perry GS, 3rd, Spector BD, Schuman LM, et al. The Wiskott–Aldrich syndrome in the United States and Canada (1892–1979) J Pediatr. 1980;97(1):72–78. doi: 10.1016/s0022-3476(80)80133-8. [DOI] [PubMed] [Google Scholar]
- 48.Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer – analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343(2):78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 49.Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360(17):1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 50.Pharoah PD, Dunning AM, Ponder BA, Easton DF. Association studies for finding cancer-susceptibility genetic variants. Nat Rev Cancer. 2004;4(11):850–860. doi: 10.1038/nrc1476. [DOI] [PubMed] [Google Scholar]