

Abstract
The AIDS Clinical Trials Group designed and implemented a prospective, randomized, strategy trial in antiretroviral-experienced, HIV-infected patients, to evaluate the virologic impact of protease inhibitor dose escalation in response to therapeutic drug monitoring (TDM) with an inhibitory quotient, which integrates both drug exposure and viral drug resistance. In the process of developing this clinical trial several unique challenges were identified that required innovative solutions. The major challenge was the need to integrate resistance testing, pharmacokinetic data, medication adherence, toxicity data, clinical assessments, randomization assignment, and protocol-specified clinical management in a way that could be utilized in real-time by the protocol team, communicated promptly to the clinical sites, and transmitted accurately to the study database. In addition, the protocol team had to address the relative lack of commercially available therapeutic drug monitoring laboratories in the US that were experienced in antiretroviral drug assays, and a lack of familiarity with the principles of pharmacokinetic monitoring at participating clinical sites. This manuscript outlines the rationale for the design of this strategy trial, specific barriers to implementation that were identified, and solutions that were developed, with the hope that these experiences will facilitate the design and conduct of future trials of TDM.
Keywords: therapeutic drug monitoring, protease inhibitors, HIV drug resistance
Introduction
Management of antiretroviral-experienced HIV-infected patients continues to pose a clinical challenge. Although high rates of virologic suppression can be achieved with currently available first-line combination regimens 1, 2, the suppression rates for treatment-experienced patients have traditionally been substantially lower 3, 4, fueling interest in the use of prognostic assays, such as HIV drug resistance testing, to individualize and optimize patient management. More recently developed antiretroviral agents have demonstrated substantially greater success in this patient population 5-9. Nonetheless, optimizing and individualizing antiretroviral drug regimens will likely remain an important strategy in treatment-experienced patients.
Measurement of PI drug concentrations may prove useful to monitor treatment-experienced patients because of inter-individual variation in PI pharmacokinetics and complex drug-drug interactions. One retrospective analysis demonstrated that PI concentrations were associated with treatment outcome independent of resistance testing 10. However, such retrospective studies do not address the question of whether dose-escalation in subjects with low drug concentrations leads to improved treatment outcomes.
A prospective interventional trial of TDM in treatment-experienced patients did not show a benefit of TDM 11. There are a number of potential explanations for this result. First, this trial did not utilize an inhibitory quotient (IQ), which incorporates both drug exposure and viral drug resistance and has been shown in several studies to correlate with virologic responses in treatment-experienced patients 12-23. Second, the majority of subjects in the TDM arm did not undergo a dose adjustment 11. Third, the dose escalations, which occurred eight weeks after initiation of the salvage regimen, may have occurred too late to have an impact on virologic outcome.
A Phase II clinical trial, A5146, was designed to further study an interventional TDM strategy in PI-experienced patients. The purpose of this paper is to summarize the unique challenges that were encountered and solutions that were developed during the process of designing and implementing this TDM strategy trial.
Study Design of A5146 and its Rationale
Summary of A5146 study design
A5146 was conducted by the AIDS Clinical Trials Group (ACTG). This 48 week trial utilized an IQ normalized to a reference population (NIQ) as the parameter on which dose escalation was based; NIQs ≤ 1 were defined as potentially needing PI dose escalation. A5146 was divided into four phases (Figure 1): a screening phase, during which eligibility was assessed; an initial phase, in which study subjects initiated a new antiretroviral regimen, chosen by their primary care provider, and received results of TDM (Step 1); a subsequent phase in which subjects with low NIQs were randomized to dose escalation (“TDM”) or standard of care (“SOC”) arms (Step 2); and a final phase (Step 3) in which all subjects in Step 2 who were failing their A5146 regimen initiated a new regimen and underwent TDM monitoring and dose escalation. A subset of 50 subjects with NIQs > 1 was also eligible to enter Step 2 on an observational arm. The primary endpoint of the study was to compare the change in viral load from randomization to 20 weeks post-randomization in the TDM and SOC arms.
Figure 1. Study design of A5146.

The four phases of A5146 (screening, Steps 1, 2, and 3) are illustrated schematically. During the process of screening for study eligibility, a resistance test was obtained on the failing antiretroviral regimen, and used to design a new regimen. During Step 1, the new salvage antiretroviral regimen was begun at entry, and a PI trough sample was obtained 2 weeks later. The NIQ was calculated as outlined in the text, using the screening fold-change in IC50 for the PI(s) in the A5146 antiretroviral regimen and the week 2 trough PI concentration measured in patient plasma. Entry into Step 2 was dependent on the value of the NIQ. If it was ≤ 1, the subject was randomized to the TDM or SOC arms. If the NIQ was > 1, the subject entered Step 2 on the observational arm or discontinued study, if the accrual goal of the observational arm had been met. Subjects who developed virologic failure at or after 20 weeks post-randomization in any of the 3 arms of Step 2 could enter Step 3 to receive a repeat resistance test and TDM followed by dose escalation on a new salvage regimen. Maximum follow-up on study was 48 weeks after Step 1 entry.
Use of an NIQ to guide PI dose escalation
NIQ is the ratio of the patient's IQ to a reference IQ derived from a population that achieved virologic success on a regimen containing the PI in question (Figure 2, Table 1). Thus, if a patient's NIQ for saquinavir is > 1, their IQ is greater than the IQ of a reference population of patients that achieved virologic success on a saquinavir-containing regimen. An advantage of NIQ is that it allows one to use fold-change in IC50 as the metric for drug resistance, rather than the absolute IC50 value. In addition, since the NIQ is the ratio of the patient and reference IQs, one does not need to make assumptions regarding the proportion of a PI that is protein-bound in patient serum versus a phenotypic resistance assay. The NIQ has been shown to correlate with outcome in recent retrospective studies 19, 23.
Figure 2. Calculation of the Normalized Inhibitory Quotient (NIQ).
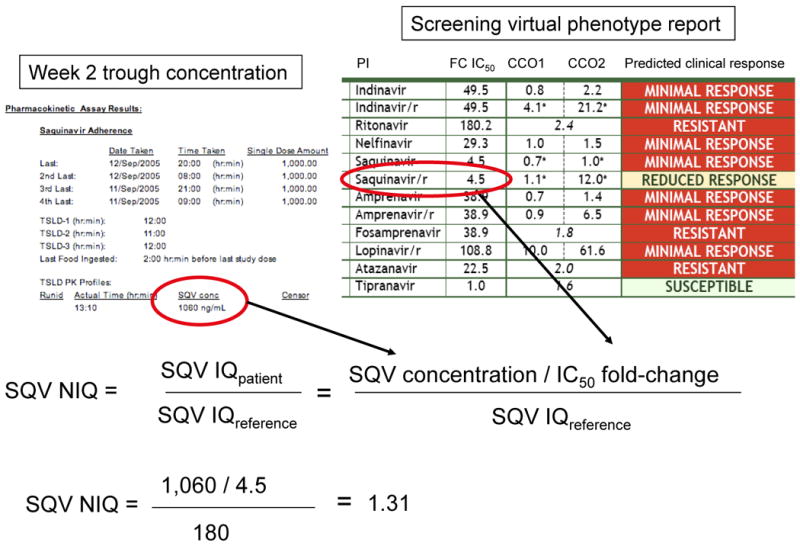
The method for calculating an NIQ is illustrated. The NIQ is the ratio of the patient's IQ to a reference IQ, which was obtained from a population of patients that was expected to achieve virologic success (180 in this example). The patient's IQ is the ratio of the week 2 plasma trough concentration of the PI being taken (in this case, saquinavir) and the fold-change in IC50 for the same PI obtained at the screening visit. In this example, the NIQ is greater than one, and the study patient would either enter the Observational arm, or discontinue the study. SQV, saquinavir; FC IC50, fold-change in the 50% inhibitory quotient for the patient's virus; CCO1, 1st clinical cutoff (threshold below which one would expect maximal virologic response); CCO2, 2nd clinical cutoff (threshold above which one would expect little or no virologic response). If the fold-change in IC50 is between CCO1 and CCO2, a reduced, but detectable virologic response would be expected). For protease inhibitors that have only a single cutoff (such as atazanavir and tipranavir in this specific example), this represents a biological cutoff for resistance, rather than a clinical cutoff.
Table 1.
Reference IQs, Dosing, and Expected Ranges for Protease Inhibitors
| Protease Inhibitor | Reference IQ | Starting Dose (protease inhibitor / ritonavir) | Highest Dose (protease inhibitor / ritonavir) | Expected ranges (ng/mL) | |
|---|---|---|---|---|---|
| Lower Limit | Upper Limit | ||||
| Atazanavir | 239 | 300 mg / 100mg QD | 300 mg / 200mg QD | 100 | 2,000 |
| Amprenavir | 420 | 600 mg /100 mg BID | 900 mg /200 mg BID | 100 | 4,105 |
| Fos-amprenavir | 420 | 700 mg / 100 mg BID | 1400 mg / 100 mg BID | 100 | 4,105 |
| Indinavir | 106 | 800 mg / 100 mg BID | 1000 mg / 200 mg BID | 100 | 2,000 |
| Lopinavir | 1430 | 400 mg /100 mg BID | 600 mg / 250 mg BID* | 200 | 10,000 |
| Nelfinavir | 350 | 1250 mg BID | 1500 mg / 200 mg BID | 200 | 4,000 |
| Saquinavir | 180 | 1000 mg / 100 mg BID | 1400 mg / 100 mg BID | 100 | 2,000 |
| Tipranavir | 9823 | 500 mg / 200 mg BID | 750 mg / 200 mg BID | 390 | 48,214 |
Highest dose for the 200 mg / 50 mg lopinavir / ritonavir tablet formulation (3 tablets given in combination with 100 mg ritonavir BID). Highest dose for the 133 mg / 33 mg lopinavir / ritonavir capsule formulation was 533 mg / 233 mg (4 capsules given in combination with 100 mg ritonavir BID).
The protocol team elected to use timed trough concentrations for TDM monitoring, based on retrospective data correlating trough concentration with clinical outcomes or toxicities for many of the PIs, and on the practical consideration that the relatively stable PI concentrations at the end of a dosing interval would provide more latitude in sample timing relative to dose, compared to peak concentrations. Based on earlier studies, the protocol team did not believe that pharmacokinetic modeling using a single random sample was sufficiently robust to allow the use of randomly timed samples 24, 25. The virco®TYPE HIV-1 assay (Virco BVBA, Mechelen, Belgium) was used to measure drug resistance for the NIQ, rather than traditional phenotypic resistance testing, because previously published studies correlating IQ with outcome used Virco's virtual phenotype assay to measure drug resistance of HIV 12, 19, 26. The choice of antiretroviral regimen and calculation of NIQs at any given time were based on the then-current version of Virco's assay that was available, consistent with clinical practice. At the completion of the study, comparisons of regimen activity among the three arms will be performed using a single version of the interpretation software (VT 4.1.00).
Timing of randomization
In order to maximize the proportion of randomized subjects who were eligible for dose escalation, A5146 was designed so that randomization of each subject occurred after their PI trough concentration was obtained, and an NIQ was calculated. Two weeks after study entry, a trough blood sample for PI concentration was obtained (Figure 1). If the NIQ was ≤ 1, subjects entered Step 2, and were randomized to either receive PI dose escalations according to pre-determined algorithms (“TDM arm”) or to be followed according to current standard of care without dose escalation (“SOC arm”). Subjects whose NIQ was > 1 were assigned to an observational arm, or, once this arm was completely accrued, discontinued from study.
Subsequent timed trough samples were obtained in all subjects in Step 2 at two and six weeks after randomization, but NIQ results were only communicated to subjects on the TDM arm. If subjects on the TDM arm continued to have NIQs ≤ 1 after the first dose escalation, they were generally allowed a second dose increase, according to pre-determined algorithms.
Inclusion of specific PIs
Because A5146 was conceived as a strategy trial, all FDA-approved PIs were allowed in the study to facilitate study enrollment and to ensure that the study results would be applicable to clinical practice. Darunavir was not included in A5146 because it was FDA-approved less than two weeks before the completion of accrual. All PIs were given in protocol-specified doses, and all dose escalations occurred in a standard fashion, either by increasing the dose of the primary PI or the ritonavir used to boost its concentration (Table 1). In addition, a number of ritonavir-boosted dual PI regimens were also allowed. The choice of specific algorithms to dose-escalate individual PIs or PI combinations was based on a review of the relevant literature summarizing pharmacokinetics, toxicities, and therapeutic outcomes.
Access to TDM and dose escalation for all study subjects
Although TDM is an accepted part of HIV clinical practice in Europe, it is not widely available in the US and there are no prospective studies supporting its use to guide dose escalation in antiretroviral-experienced patients27. For this reason, the protocol team believed that A5146 should be a randomized trial, since there was equipoise with regard to the clinical utility of this monitoring strategy. In order to improve subject retention, a final phase of the study (Step 3) was designed, in which all patients in Step 2 who had confirmed virologic failure 20 weeks or later after randomization had the option to receive TDM and PI dose escalation on a new antiretroviral regimen.
Challenges to Implementation
A5146 was the first ACTG-sponsored randomized interventional TDM strategy trial that incorporated PI dose escalation based upon a NIQ. During the process of designing and conducting A5146, a number of challenges were encountered that had to be overcome to successfully implement and accrue this trial. Utilizing an individualized NIQ result as an integral component of randomization and subsequent PI dose escalation required close collaborations and real-time communications among the testing laboratories, protocol team members, clinical sites, and the statistical and data management center (SDMC; see Figure 3 for an illustration of these real-time communications). The specific issues that were raised and their solutions are summarized below. Although these problems were primarily identified and addressed during the design and early conduct of the study, the discussion below is organized according to the time line of the protocol itself.
Figure 3. Real-time communications required for Step 2 entry.

The diagram illustrates the real-time interdisciplinary communications that were needed before a study subject could enter Step 2. Dotted arrows represent communications that occurred during screening; solid arrows represent those that occurred at or near the week 2 visit; and double-lined arrows represent communications that occurred at or near the time of randomization, for those subjects with an NIQ ≤ 1. SDMC, Statistical and Data Management Center; UB PSL, University of Buffalo pharmacology support laboratory.
Screening study subjects for eligibility
Because the central question of this study was the utility of PI dose escalation guided by an IQ, it was critical to have both the resistance testing and PI trough concentration measurements performed by experienced, CLIA-certified, central laboratories. Thus, all subjects screened for this study had a plasma sample tested for a virtual phenotype (virco®TYPE HIV-1) by Virco BVBA. Electronic pdf files of each report, with assigned patient identifier number, were sent by secure email from Virco to a core group of protocol team members, and designated staff at the appropriate clinical site.
Study entry
In order to provide consistency, standardized protocol-specified starting doses and dose escalations for each PI and each ritonavir-boosted dual PI regimen were required. In order to ensure that adequate time was allotted to achieve steady state PI trough concentrations and that dose escalation occurred promptly after treatment initiation, the protocol specified that the trough sample be obtained a minimum of 10 days after initiation of the A5146 regimen, and that randomization to Step 2 occur within 3-5 weeks after study entry.
Week 2 PI trough concentration
Selection of a testing laboratory
The requirements for the centralized pharmacology laboratory for A5146 were that it be CLIA-certified, experienced in measuring PI concentrations, able to readily interface with the ACTG SDMC, and able to transfer data into the ACTG database in real-time using appropriate file formats. The SUNY at Buffalo Pharmacology Support Laboratory (UB PSL), one of six ACTG-funded pharmacology research laboratories, which was experienced in TDM of antiretroviral drugs and the pharmacokinetics of protease inhibitors 28-35, was selected by virtue of meeting these specified criteria. Following the initial implementation of A5146, the UB PSL was able to generate real-time PI trough concentration results with an average turn-around time of 7 days from specimen receipt.
Obtaining accurate trough samples
Before beginning enrollment, and at periodic intervals during the trial, the study team and pharmacology laboratory personnel conducted a series of conference calls and face-to-face meetings with clinical site personnel to provide the necessary background for managing study patients. Topics reviewed included a summary of retrospective studies correlating PI IQ to virologic outcome, the definition of IQ and NIQ, and examples of week 2 NIQ reports that would be generated in A5146. In addition, the ACTG network conducted prospective educational initiatives to summarize the pharmacokinetic principles that underlie effective TDM programs. Ultimately, the ACTG developed a tutorial for study coordinators that was a requirement for enrolling patients into pharmacology-oriented studies, including A5146. These educational sessions, which were initiated during the early phase of enrollment into A5146, proved helpful to the sites, and resulted in fewer errors in obtaining the PI trough specimens 36.
Optimizing Adherence
The clinical site personnel conducted adherence support interviews during visits at which trough samples were to be obtained, to review dosing instructions and barriers to adherence, and to provide specific interventions to improve adherence. Telephone contact was also made approximately 48 hours before the scheduled trough sample to reinforce adherence.
Assessment of specimen quality by the testing laboratory
In addition to assessing the physical integrity of the PI trough sample, the UB PSL also verified the presence and accuracy of required data, including the timing and dose of PIs and a list of all other medications. These data were summarized in a pharmacokinetic case report form filled out by the clinical site staff and sent together with the sample to the UB PSL.
Generation of the NIQ report
Initial assessment of the PI trough concentration report by the study chairs
The study chairs reviewed the PI trough concentration report from the testing laboratory, including time intervals between PI doses and sample collection, and concomitant medications, to identify any factors that might affect the results. In addition, the chairs identified reports in which the PI trough concentration was outside the expected ranges for patients on standard dosing regimens, and contacted sites to request a reassessment and reinforcement of adherence, followed by a repeat trough sample. If an undetectable or unexpectedly high trough concentration was verified on repeat testing, the study subject was allowed to proceed to Step 2 entry and randomization, if otherwise eligible.
Generation of the NIQ report
The NIQ is the ratio of the patient's IQ to a reference IQ, obtained from a population of patients who received the same PI and achieved virologic suppression. The patient IQ was the ratio of the patient trough PI concentration to the patient's virus' fold-change in IC50 for that PI, obtained at screening (Figure 2). The reference IQ for each PI was calculated using the mean trough concentration divided by the mean fold-change in IC50 for clinical cohorts that had high rates of virologic suppression 12, 13, 26, 37, 38 (Table 1).
The NIQ report that was distributed to the site staff and investigators was sent by the protocol chairs following team sign-off via email and included the PI trough concentration, fold-change in IC50, the NIQ value, and recommendations to the clinical site regarding assessment of toxicities and eligibility for Step 2 entry. Figure 3 summarizes the communication of real-time data among the protocol team, clinical site, and testing laboratories that was required to generate an NIQ report and determine eligibility for Step 2 entry.
Randomization
Assessment of toxicities before randomization
The protocol team determined that subjects experiencing toxicities or intolerance should not undergo dose escalation for safety reasons. The primary concerns were liver function tests, for subjects receiving tipranavir; and EKG conduction abnormalities for all subjects. In order to assure appropriate and timely management of the first PI dose escalation, direct communications were established between the clinical sites and the protocol team for each subject approaching randomization.
Complicating the assessment of toxicities was the introduction of monitoring for EKG abnormalities in June 2004, after atazanavir was added to the study. This additional monitoring was incorporated because of reports that higher trough concentrations of atazanavir were associated with prolongation of the P-R interval 39. There had also been anecdotal reports of prolonged QT intervals in some patients receiving other PI regimens 40. For safety reasons, EKG monitoring was therefore instituted at week 2, and at the time of each trough sample, for all subjects in Step 2. EKGs were interpreted at each site, with conduction intervals communicated to the team via email. A scheme for grading the adverse events of prolonged PR and QTc intervals was also created for the Adverse Event Database and protocol management.
Dose escalation for subjects randomized to the TDM arm
Because a dosing recommendation from the protocol team was required after randomization to the TDM arm, direct communications between the site and the protocol team were relied on to facilitate prompt dose escalation. In general, dose escalations were provided to the sites within 1-2 hours after randomization.
On-study follow-up
When designing this trial, it was not known what the distribution of NIQs would be, and whether a single dose escalation would be optimal for improving virologic responses. The trial was therefore designed to allow up to two dose escalations, with a repeat trough concentration obtained two weeks after randomization, followed by dose escalation within two weeks of sampling if the NIQ remained ≤ 1. At the time each NIQ was calculated, the study team also monitored whether the previously recommended dose escalation had been implemented. Case report forms were designed to capture dosing information for all subjects, to allow assessment of whether subjects and their care providers complied with the assigned treatment strategy.
Accrual
Accrual to A5146 occurred over an extended period of time, from October 2002 until June 2006 (Figure 4). There were a number of factors that contributed to the initial slow accrual, primarily restrictive entry criteria and lack of site familiarity with TDM. The initial protocol version had specified that subjects be failing their second, third, or fourth PI-containing regimen to be eligible for A5146. Because this strict requirement reduced screening efficiency, the entry criteria were then liberalized with a revised protocol on October 29, 2003, which allowed subjects who had failed at least one PI-containing regimen to enter the study. After liberalization of the entry criteria, and as the site investigators and personnel became more familiar with the protocol, there was a significant upsurge in accrual, with 77% of the step 2 accrual occurring after February 2004.
Figure 4. A5146 accrual.
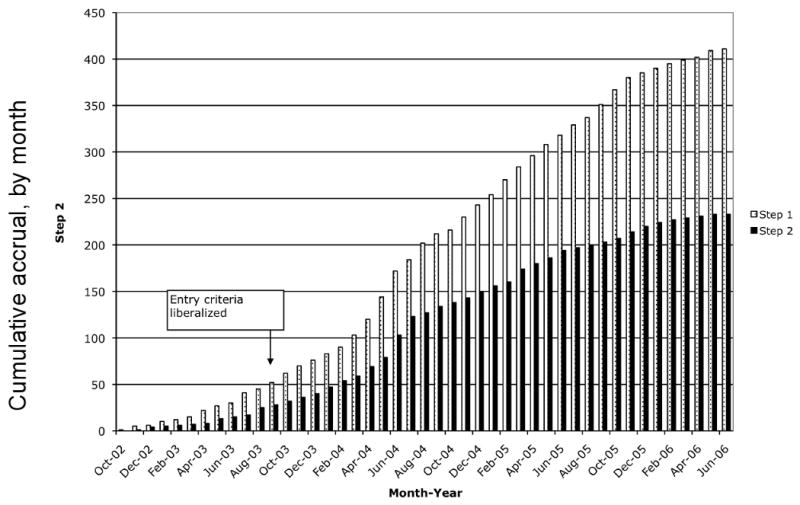
The graph illustrates the accrual, by month, of study subjects into A5146. X-axis, month of accrual; Y-axis, number of study subjects. Gray bars, Step 1 accrual; black bars, Step 2 accrual. Arrow points to the month in which the revised protocol with liberalized entry criteria was released to the sites. The lag in time to increased rates of accrual was due in large part to the time needed for each site to obtain IRB approval.
Response to Changes in Clinical Practice
In part because of the extended duration of enrollment, the A5146 team was faced with the need to adapt to a number of changes in clinical practice. Most significant was the incorporation of two new FDA-approved PIs: atazanavir (allowed after 6/2/04) and tipranavir (allowed after 1/7/05). This process required the development, validation, and regulatory approval of an assay for each new PI; the formulation of a reference IQ, which was based on presented and published data, as well as communications with the appropriate pharmaceutical companies; the development of PI dosing algorithms; and revisions to the monitoring and management of toxicities.
The protocol team also dealt with the incorporation of new PI formulations, including fos-amprenavir, the saquinavir (Invirase) tablet, and the lopinavir/ritonavir (Kaletra) tablet; and the discontinuation of the amprenavir (Agenerase®) capsule in December 2005. New formulations did not require new reference IQs or trough concentration assays, but did require changes in dose escalation schemes and case report forms.
Discussion
There were a number of factors that complicated the design and execution of this prospective, randomized, IQ-based TDM trial. Because of the need to obtain accurate trough concentrations and reliable resistance testing, and the desire to maximize the proportion of study subjects requiring an intervention, this trial was designed to have a lead-in phase (Step 1) in which a new regimen was initiated, a trough concentration was obtained after allowing sufficient time to achieve steady-state concentrations, and a NIQ was generated using the trough concentration and the screening resistance test obtained on the prior failing regimen. This trial was unusual in that the intervention, and therefore randomization, occurred approximately four weeks after study entry. Since accrual goals were based on the number of subjects in the three Step 2 arms, accrual into both Steps 1 and 2 was monitored. Because the TDM-prompted randomized intervention occurred at approximately four weeks after study entry, the primary endpoint was timed relative to randomization, rather than study entry.
One important consequence of this study design is that all subjects and their health care providers received information on PI trough concentration and NIQ before randomization. This information was necessary to ensure eligibility for randomization and correct implementation of the different treatment strategies, but could have encouraged dose escalation by providers of patients assigned to the SOC arm. We implemented careful monitoring of dosing records to assess how often, if at all, deviations from the assigned treatment strategy occurred in each randomized arm, and did not distribute dose escalation algorithms to the clinical sites. In addition, information on drug exposure at week 2 could potentially have influenced a patient's adherence to their treatment regimen, further confounding the ability to detect an impact of TDM; for this reason, adherence was monitored at the time of each trough concentration sample. The inclusion of an “open-label” phase of the trial in which all subjects could receive TDM and dose escalation provided incentive for subjects randomized to the SOC arm to continue study follow-up through the primary study endpoint.
A second consequence of the study design was that the four week time interval needed to establish steady-state concentrations and measure trough concentrations could have reduced a positive impact of the TDM strategy on virologic responses to the new regimen. Although this time interval could potentially be shortened by further reducing the turn-around time of the NIQ report, one would still need a ten to 14 day period to allow steady-state concentrations to be achieved.
A potential limitation to the way TDM was implemented in this study was the avoidance of dose escalations in patients experiencing toxicities. We believe that this is the most prudent approach, pending further information on the associations between PI trough concentrations and toxicities. If toxicities were identified that were clearly not associated with PI concentrations, this information would increase the number of patients that could potentially benefit from TDM.
The most significant problem identified in implementing this trial was the need to integrate a number of different types of data from different sources, including resistance testing, trough PI concentrations, toxicities, adherence, randomization assignment, and dosing recommendations. These pieces of data needed to be integrated in real-time to impact on patient management, and therefore, the traditional approaches to data entry and management were often not sufficient for the needs of this protocol. Two other critical requirements for implementation were the identification of a CLIA-certified TDM laboratory providing accurate PI concentrations, and education of clinical site personnel regarding implementation of TDM and PI dose escalation.
Addressing the problem of integrating different sources of data in real-time required a number of interdisciplinary collaborations among the study chairs, statisticians, pharmacologists, the SDMC, site personnel, pharmacology laboratory personnel, and pharmaceutical companies; as well as new approaches to tracking patients and patient data. The difficulties inherent in rapidly linking clinical information and decision-making with the SDMC and study database resulted in the protocol team monitoring aspects of patient management more closely than would ordinarily occur in a phase II clinical trial. Adapting clinical trials database management and structure to allow incorporation of clinical recommendations into randomization schemes would greatly simplify the execution of a trial such as this, in which dosing recommendations for drugs not provided by study were linked to randomization assignment. This issue is important for any trial in which strategies, rather than new antiretroviral agents, are studied using drugs prescribed through clinical practice.
A final critical component of implementing this trial was our intensive efforts to educate site personnel regarding the underlying pharmacokinetic principles of the requirements for obtaining reliable PI trough concentrations, as well as providing assistance in designing practical solutions for implementing TDM at individual sites. These educational interventions were instrumental in increasing the interest, enthusiasm, and ability of sites to accrue into this study and accurately implement the strategy being studied.
This trial has now completed follow-up, and analysis of the outcome of this TDM intervention is currently in progress. Independent of the outcome of this study, the critical components of the design and implementation of this real-time TDM strategy trial that are summarized here, should facilitate the design and conduct of future studies evaluating TDM interventions in HIV-infected patients.
Acknowledgments
This work was supported in part by NIH grants U01-AI-069511, N01-AI-38858, N01-AI-68636, U01 AI-069472, U01 AI-38855, U01 AI-068634, and M01-RR00044. Virco provided resistance testing, and supported the performance of drug concentration testing for the study. We would also like to acknowledge the other members of the A5146 protocol team: Carrie Dykes, Ph.D., Thorner Harris, Bernadette Jarocki, Karin Klingman, M.D., Jennifer Nowak, Nancy Reynolds, Ph.D., David Rusin, and Margie Vasquez, R.N.
References
- 1.Gulick RM, Ribaudo HJ, Shikuma CM, et al. Triple-nucleoside regimens versus efavirenz-containing regimens for the initial treatment of HIV-1 infection. N Engl J Med. 2004 Apr 29;350(18):1850–1861. doi: 10.1056/NEJMoa031772. [DOI] [PubMed] [Google Scholar]
- 2.Walmsley S, Bernstein B, King M, et al. Lopinavir-ritonavir versus nelfinavir for the initial treatment of HIV infection. N Engl J Med. 2002 Jun 27;346(26):2039–2046. doi: 10.1056/NEJMoa012354. [DOI] [PubMed] [Google Scholar]
- 3.Lalezari JP, Henry K, O'Hearn M, et al. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N Engl J Med. 2003 May 29;348(22):2175–2185. doi: 10.1056/NEJMoa035026. [DOI] [PubMed] [Google Scholar]
- 4.Lazzarin A, Clotet B, Cooper D, et al. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N Engl J Med. 2003 May 29;348(22):2186–2195. doi: 10.1056/NEJMoa035211. [DOI] [PubMed] [Google Scholar]
- 5.Katlama C, Esposito R, Gatell JM, et al. Efficacy and safety of TMC114/ritonavir in treatment-experienced HIV patients: 24-week results of POWER 1. AIDS. 2007 Feb 19;21(4):395–402. doi: 10.1097/QAD.0b013e328013d9d7. [DOI] [PubMed] [Google Scholar]
- 6.Lalezari J, Goodrich J, Dejesus E, et al. Efficacy and safety of maraviroc plus optimized background therapy in viremic antiretroviral-experienced patients infected with CCR5-tropic HIV-1: 24 week results of a Phase 2b/3 study in the US and Canada. 14th Conference on Retroviruses and Opportunistic Infections; 2007. p. 104bLB. [Google Scholar]
- 7.Nelson M, Fatkenheuer G, Konourina I, et al. Efficacy and safety of maraviroc plus optimized background therapy in viremic antiretroviral-experienced patients infected with CCR5-tropic HIV-1 in Europe, Australia, and North America: 24 week results. 14th Conference on Retroviruses and Opportunistic Infections; 2007. p. 104aLB. [Google Scholar]
- 8.Cooper D, Gatell J, Rockstroh J, et al. Results of BENCHMRK-1, a Phase III study evaluating the efficacy and safety of MK-0518, a novel HIV-1 integrase inhibitor, in patients with triple-class resistant virus. 14th Conference on Retroviruses and Opportunistic Infections; 2006. p. 105aLB. [Google Scholar]
- 9.Steigbigel R, Kumar P, Eron J, et al. Results of BENCHMRK-2, a Phase III study evaluating the efficacy and safety of MK-0518, a novel HIV-1 integrase inhibitor, in patients with triple-class resistant virus. 14th Conference on Retroviruses and Opportunistic Infections; 2007. p. 105bLB. [Google Scholar]
- 10.Durant J, Clevenbergh P, Garraffo R, et al. Importance of protease inhibitor plasma levels in HIV-infected patients treated with genotypic-guided therapy: pharmacological data from the Viradapt Study. Aids. 2000;14(10):1333–1339. doi: 10.1097/00002030-200007070-00005. [DOI] [PubMed] [Google Scholar]
- 11.Clevenbergh P, Garraffo R, Durant J, Dellamonica P. PharmAdapt: a randomized prospective study to evaluate the benefit of therapeutic monitoring of protease inhibitors: 12 week results. Aids. 2002 Nov 22;16(17):2311–2315. doi: 10.1097/00002030-200211220-00011. [DOI] [PubMed] [Google Scholar]
- 12.Shulman N, Zolopa A, Havlir D, et al. Virtual inhibitory quotient predicts response to ritonavir boosting of indinavir-based therapy in human immunodeficiency virus-infected patients with ongoing viremia. Antimicrob Agents Chemother. 2002 Dec;46(12):3907–3916. doi: 10.1128/AAC.46.12.3907-3916.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu A, Isaacson J, Brun S, et al. Pharmacokinetic-pharmacodynamic analysis of lopinavir-ritonavir in combination with efavirenz and two nucleoside reverse transcriptase inhibitors in extensively pretreated human immunodeficiency virus-infected patients. Antimicrob Agents Chemother. 2003 Jan;47(1):350–359. doi: 10.1128/AAC.47.1.350-359.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marcelin AG, Lamotte C, Delaugerre C, et al. Genotypic inhibitory quotient as predictor of virological response to ritonavir-amprenavir in human immunodeficiency virus type 1 protease inhibitor-experienced patients. Antimicrob Agents Chemother. 2003 Feb;47(2):594–600. doi: 10.1128/AAC.47.2.594-600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Casado JL, Moreno A, Sabido R, et al. Individualizing salvage regimens: the inhibitory quotient (Ctrough/IC50) as predictor of virological response. Aids. 2003 Jan 24;17(2):262–264. doi: 10.1097/01.aids.0000050800.28043.4d. [DOI] [PubMed] [Google Scholar]
- 16.Taburet AM, Raguin G, Le Tiec C, et al. Interactions between amprenavir and the lopinavir-ritonavir combination in heavily pretreated patients infected with human immunodeficiency virus. Clin Pharmacol Ther. 2004 Apr;75(4):310–323. doi: 10.1016/j.clpt.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 17.De Luca A, Baldini F, Cingolani A, Di Giambenedetto S, Hoetelmans RM, Cauda R. Deep salvage with amprenavir and lopinavir/ritonavir: correlation of pharmacokinetics and drug resistance with pharmacodynamics. J Acquir Immune Defic Syndr. 2004 Apr 1;35(4):359–366. doi: 10.1097/00126334-200404010-00005. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez de Requena D, Gallego O, Valer L, Jimenez-Nacher I, Soriano V. Prediction of virological response to lopinavir/ritonavir using the genotypic inhibitory quotient. AIDS Res Hum Retroviruses. 2004 Mar;20(3):275–278. doi: 10.1089/088922204322996509. [DOI] [PubMed] [Google Scholar]
- 19.Castagna A, Gianotti N, Galli L, et al. The NIQ of lopinavir is predictive of a 48-week virological response in highly treatment-experienced HIV-1-infected subjects treated with a lopinavir/ritonavir-containing regimen. Antivir Ther. 2004 Aug;9(4):537–543. [PubMed] [Google Scholar]
- 20.Barrios A, Rendon AL, Gallego O, et al. Predictors of virological response to atazanavir in protease inhibitor-experienced patients. HIV Clin Trials. 2004 Jul-Aug;5(4):201–205. doi: 10.1310/3HL3-HHBD-WKLR-XELL. [DOI] [PubMed] [Google Scholar]
- 21.Marcelin AG, Dalban C, Peytavin G, et al. Clinically relevant interpretation of genotype and relationship to plasma drug concentrations for resistance to saquinavir-ritonavir in human immunodeficiency virus type 1 protease inhibitor-experienced patients. Antimicrob Agents Chemother. 2004 Dec;48(12):4687–4692. doi: 10.1128/AAC.48.12.4687-4692.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marcelin AG, Cohen-Codar I, King MS, et al. Virological and pharmacological parameters predicting the response to lopinavir-ritonavir in heavily protease inhibitor-experienced patients. Antimicrob Agents Chemother. 2005 May;49(5):1720–1726. doi: 10.1128/AAC.49.5.1720-1726.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winston A, Hales G, Amin J, van Schaick E, Cooper DA, Emery S. The normalized inhibitory quotient of boosted protease inhibitors is predictive of viral load response in treatment-experienced HIV-1-infected individuals. AIDS. 2005 Sep 2;19(13):1393–1399. doi: 10.1097/01.aids.0000181009.77632.36. [DOI] [PubMed] [Google Scholar]
- 24.DiCenzo R, Forrest A, Smith PF, et al. Comparing Intensive and Sparse Sampling for Estimating the Population Pharmacokinetics (PK) of Indinavir (IDV) in Efavirenz (EFV)-Containing Regimens. 8th Conference on Retroviruses and Opportunistic Infections; Chicago, IL. 2001. Abstract # 751. [Google Scholar]
- 25.DiCenzo R, Forrest A, Squires K, et al. Designing Sparse Sampling Approaches to Optimize Indinavir (IDV) Sampling Times. 41st ICAAC; Chicago, IL. 2001. Abstract #487. [Google Scholar]
- 26.Hoetelmans RM, Reijers MH, Weverling GJ, et al. The effect of plasma drug concentrations on HIV-1 clearance rate during quadruple drug therapy. AIDS. 1998;12(11):F111–115. doi: 10.1097/00002030-199811000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Boffito M, Acosta E, Burger D, et al. Current status and future prospects of therapeutic drug monitoring and applied clinical pharmacology in antiretroviral therapy. Antivir Ther. 2005;10(3):375–392. [PubMed] [Google Scholar]
- 28.Demeter LM, Meehan PM, Morse G, et al. Phase I study of atevirdine mesylate (U-87201E) monotherapy in HIV-1- infected patients. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;19(2):135–144. doi: 10.1097/00042560-199810010-00006. [DOI] [PubMed] [Google Scholar]
- 29.Morse GD, Reichman RC, Fischl MA, et al. Concentration-targeted phase I trials of atevirdine mesylate in patients with HIV infection: dosage requirements and pharmacokinetic studies. The ACTG 187 and 199 study teams. Antiviral Res. 2000 Jan;45(1):47–58. doi: 10.1016/s0166-3542(99)00073-x. [DOI] [PubMed] [Google Scholar]
- 30.Para MF, Meehan P, Holden-Wiltse J, et al. ACTG 260: a randomized, phase I-II, dose-ranging trial of the anti- human immunodeficiency virus activity of delavirdine monotherapy. Antimicrob Agents Chemother. 1999;43(6):1373–1378. doi: 10.1128/aac.43.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friedland GH, Pollard R, Griffith B, et al. Efficacy and safety of delavirdine mesylate with zidovudine and didanosine compared with two-drug combinations of these agents in persons with HIV disease with CD4 counts of 100 to 500 cells/mm3 (ACTG 261). ACTG 261 Team. J Acquir Immune Defic Syndr. 1999 Aug 1;21(4):281–292. doi: 10.1097/00126334-199908010-00005. [DOI] [PubMed] [Google Scholar]
- 32.Okusanya O, Forrest A, DiFrancesco R, et al. Compartmental pharmacokinetic analysis of oral amprenavir with secondary peaks. Antimicrob Agents Chemother. 2007 May;51(5):1822–1826. doi: 10.1128/AAC.00570-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keil K, Hochreitter J, DiFrancesco R, et al. Integration of atazanavir into an existing liquid chromatography UV method for protease inhibitors: validation and application. Ther Drug Monit. 2007 Feb;29(1):103–109. doi: 10.1097/FTD.0b013e3180318ef3. [DOI] [PubMed] [Google Scholar]
- 34.DiFrancesco R, Fischl MA, Donnelly J, et al. Buprenorphine assay and plasma concentration monitoring in HIV-infected substance users. J Pharm Biomed Anal. 2007 May 9;44(1):188–195. doi: 10.1016/j.jpba.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 35.Keil K, Difrancesco R, Morse GD. Determination of tipranavir in human plasma by reverse phase liquid chromatography with UV detection using photodiode array. Ther Drug Monit. 2006 Aug;28(4):512–516. doi: 10.1097/00007691-200608000-00005. [DOI] [PubMed] [Google Scholar]
- 36.DiFrancesco R, Rosenkranz SL, Craft J, Morse GD. Tutorial reduces protocol deviations in multicenter ACTG trials with pharmacology endpoints. HIV Clin Trials. 2006;7(4):203–209. doi: 10.1310/hct0704-203. [DOI] [PubMed] [Google Scholar]
- 37. [August 4, 2007]; www.fda.gov/cder/foi/nda/2003/021567_reyataz_toc.htm.
- 38. [August 4, 2007]; www.fda.gov/cder/foi/label/2005/021814lbl.pdf.
- 39.Busti AJ, Tsikouris JP, Peeters MJ, et al. A prospective evaluation of the effect of atazanavir on the QTc interval and QTc dispersion in HIV-positive patients. HIV Medicine. 2006;7(5):317–322. doi: 10.1111/j.1468-1293.2006.00382.x. [DOI] [PubMed] [Google Scholar]
- 40.Anson BD, Weaver JG, Ackerman MJ, et al. Blockade of HERG channels by HIV protease inhibitors. Lancet. 2005 Feb 19-25;365(9460):682–686. doi: 10.1016/S0140-6736(05)17950-1. [DOI] [PubMed] [Google Scholar]