

Summary
Metastatic dormancy of melanoma has not received sufficient attention, most likely because once detectable, metastasis is almost invariably fatal and, understandably, the focus has been on finding ways to prolong life of patients with overt recurrences. Nevertheless, analysis of the published clinical and experimental data on melanoma indicates that some aspect of melanoma biology imitate traits recently associated with dormancy in other solid cancers. Among them the ability of some melanomas to disseminate early during primary tumor progression and once disseminated, to remain undetected (dormant) for years. Comparison of cutaneous and uveal melanoma indicates that, in spite of being of the same origin, they differ profoundly in their clinical progression. Importantly for this discussion, between 40 and 50% of uveal melanoma remain undetected for longer than a decade, while less than 5% of cutaneous melanoma show this behavior. Both types of melanoma have activating oncogene mutations that provide autonomous pro-proliferative signals, yet the consensus is that those are not sufficient for tumor progression. If that is the case, it is possible to envision that signals from outside the tumor cell, (microenvironment) shape the fate of an individual disseminated cell, regardless of an oncogene mutation, to progress or to pause in a state of dormancy. To stimulate further debate and inquiry we describe here a few examples of potential signals that might modify the fate of disseminated cell and provide brief description of the current knowledge on dormancy in other cancers. Our hope is to convince the reader that disseminated melanoma cells do enter periods of prolonged dormancy and that finding ways to induce it, or to prolong it, might mean an extension of symptoms-free life for melanoma patients. Ultimately, understanding the biology of dormancy and the mechanisms of dormant cell survival, might allow for their specific targeting and elimination.
Keywords: residual disease, B-Raf, N-Ras, GNAQ, micrometastasis
Brief historical background
Operationally, tumor dormancy has been defined as ‘the disease free period’ between clinical ‘cure’ of the primary cancer and its subsequent local or distant recurrence/metastasis. Until recently, the prevailing theory was that metastases arise from a late-forming small sub-population of cancer cells which, through mutation and selections, become highly adapted to processes of dissemination and growth in secondary sites (Fidler, 1983; Foulds, 1957; Nowell, 2002). As such, these cells were expected to continue uninterrupted growth at the secondary sites and form overt metastases. The calculated kinetics of metastasis based on these considerations turned out to be more rapid than the actual time of appearance of clinically overt metastases (Demicheli, 2001). This suggested a possibility of a ‘pause/dormancy’ in tumor progression and also raised important questions of possible reasons of dormancy. Can dormancy be extended by interventions? Can dormant tumor cells be eliminated? Those questions have fascinated physicians and researchers for more than a century. An important very early finding by Paget (1889) with direct implications to our current understanding of dormancy was that the inherent properties of a cancer cell, but also the organ in which it lodges, determine its fate (‘seed and soil’ theory). With small, but possibly crucial modifications, this theory stood the test of time. Paget believed that metastasis will form only when a cancer cell (‘seed’) with predetermined set of qualities lodges in an organ with predetermined favorable ‘soil’; all other ‘encounters’ would be doomed to failure. We now know that both the cancer cell and the environment in which it lodges exert mutual modifying effects, that seed and soil theory is more plastic than the originally coined one and that dormancy, by definition and operationally, is a reversible process (Aguirre-Ghiso, 2007; Chambers et al., 2002). One of the fist attempts to experimentally prove the existence of dormancy was done in female rats which received intra-hepatic injection of 50 Walker carcinoma cells per rat and were found not to have liver tumors 17 weeks later, unless repeated laparoscopy was performed, (Fisher and Fisher, 1959). The authors concluded that the cancer cells were present and dormant and were forced out of dormancy by the surgical procedure. Fifty years later, the concept of cancer dormancy has been widely accepted but the mechanisms underlying both entry and especially emergence from dormancy have not yet been fully elucidated.
Current understanding of dormancy
As mentioned, metastatic dormancy in humans has been defined operationally as the ‘disease-free’ period between treatment and late recurrence. In solid tumors such as breast and gastric carcinoma, and many others, disseminated cancer cells can be found in bone marrow of patients, which have no clinical indications of disease spread. Only a proportion of these patients will go on to develop overt metastases (Aguirre-Ghiso, 2007; Braun et al., 2005; Wikman et al., 2008).
The signals that initiate and maintain dormancy of melanoma or other tumor types are poorly understood. Equally unknown are the signals that propel expansion of tumor cells after a period of dormancy. Nevertheless, clinical experience and experimental models provide several clues into the potential mechanisms at play. Tumor dormancy mechanisms can be largely grouped in two categories: dormancy of a tumor mass and dormancy of solitary tumor cells (i.e. growth arrest/quiescence). Recent reviews contain detailed description of the potential mechanisms (Aguirre-Ghiso, 2007; Marches et al., 2006; Naumov et al., 2006; Townson and Chambers, 2006; Vessella et al., 2007; Wikman et al., 2008); here we will provide a brief overview and then attempt to link this information to the available data in melanoma.
Angiogenic and immunological mechanisms of tumor mass dormancy
Angiogenic metastatic dormancy applies to the dormancy of a tumor mass. In this case a micro-metastatic lesion actively proliferates but, because it is avascular, it does not expand. The inability to recruit blood vessels is likely caused by the lack of expression of angiogenic factors such as VEGF, and/or high expression of angiogenesis inhibitors such as thrombospondin. Under such conditions the proliferating fraction and the fraction eliminated through apoptosis are in equilibrium and the overall tumor mass remains small and static (Naumov et al., 2006). In most cases, only when it emerges into a clinically detectable and/or symptomatic state, the diagnosis of previous dormant state can be made. It is unclear if these mechanisms are operational in patients with melanoma micrometastases, but Barnhill et al. (1998) have shown that human melanoma micrometastases are poorly vascularized (mean number of microvessels, 10.2), and have low rates of tumor cell proliferation (mean, 2.4%), compared with melanoma macrometastases, which have significantly greater tumor vascularity (mean number of microvessels, 18.7), higher rates of proliferation (mean, 18%). Also, some experimental evidence suggests that in animal melanoma models, and in human disease, anti-angiogenic therapies can induce tumor growth inhibition (for review of the field see Rosenblatt and Azar, 2006; Mahabeleshwar and Byzova, 2007). However, whether angiogenic dormancy is responsible for the prolonged quiescence of disseminated melanoma is still unknown.
Another possible mechanism of tumor mass dormancy is immunity dependent. It has been proposed that through a mechanism termed immuno-surveillance, or cancer editing, a state of equilibrium is reached between the immune system and the tumor (Finn, 2006; Willimsky and Blankenstein, 2005). In this scenario, a small lesion that is attempting to expand is kept at a constant mass level by active immunity. Here, like in angiogenic dormancy, tumor expansion is also prevented by cell death but in this case death is caused by the cytotoxic T-cells. Several examples in B-cell lymphoma and in models of chemical carcinogen-induced tumors provide mechanistic insight into the possible role of the counterbalance between the immune system and the tumor (Koebel et al., 2007; Rabinovsky et al., 2007).
Tumor cell quiescence
Disseminated, predominantly solitary tumor cells (DTCs), isolated from the bone marrow of patients bearing cancers of different origins, are negative for markers of proliferation such as PCNA or Ki67 (Pantel and Brakenhoff, 2004; Pantel et al., 2009; Wikman et al., 2008), suggesting that they can enter a state of cellular quiescence or dormancy. The experience from preclinical models suggests that tumor cells can enter dormancy if certain signaling pathways are malfunctioning and/or activated. The most common mechanism is a G0-G1 arrest with high expression of p21 and p27 (Ducos et al., 2000; Florenes et al., 1996; Marches et al., 1998, 1999; Pelayo et al., 2006; Sang et al., 2008 and our unpublished data). Impaired or reduced ligand-dependent signaling through adhesion molecules such as α5β1 or other β1 integrin heterodimers and adhesion signal transducers such as focal adhesion kinase (FAK) is also observed in dormant tumor cells (Aguirre-Ghiso, 2007). This mechanism was identified in human squamous carcinoma (Aguirre Ghiso, 2002; Liu et al., 2002) and more recently in mouse breast carcinoma models (Lahlou et al., 2007; Shibue and Weinberg, 2009; White et al., 2004). Also, blockade of signaling through the protease receptor uPAR leads to α5β1 integrin inactivation, decrease in the EGFR signaling and induction of dormancy (Aguirre Ghiso et al., 1999; Liu et al., 2002). All these studies suggest that the ability of tumor cells to interpret their microenvironment is critical in determining the onset of dormancy.
There is evidence (Aguirre-Ghiso et al., 2003), that downstream of the surface molecules receiving micro-environmental cues, the ratio between MAPK-ERK and SAPK-p38 is indicative of a dormant behavior. A low ERK/p38 signaling ratio predicts for dormancy while the opposite ratio predisposes to active proliferation. Targeted inhibition of EGFR with Gefitinib was shown to inhibit ERK and activate p38, suggesting that such a dormancy-inducing ERK/p38 ratio might be achieved using clinically relevant targeted therapies (Sharma et al., 2006). However, these mechanisms may be subverted in melanoma cells as they do not enter dormancy, despite high p38 signaling (Aguirre-Ghiso et al., 2003; Estrada et al., 2009). We found that in melanoma cells p38 is highly activated and it does not negatively regulate ERK activation (Aguirre-Ghiso et al., 2003). In fact, ERK activates ανβ3 expression which in turn activates p38, generating a positive loop that enhances proliferation in vivo. p38 also induces IL-8 to promote melanoma migration (Estrada et al., 2009). Thus it appears that upon progression melanoma cells turn p38 signaling to their advantage. It has been suggested that transcription factors that specifically induce quiescence while at the same time prevent terminal differentiation and/or senescence might serve as a ‘switch’ of dormancy regulation. One such transcription factor, HES-1, induced by several quiescence inducing stimuli can selectively block senescence and differentiation and maintain reversibility of quiescence (Sang et al., 2008). These authors concluded that reversion from quiescence is not passive because a brief enforced cell cycle arrest initiates spontaneous, premature, and irreversible senescence, a state which can be prevented by increased expression of HES1. Thus, according to the authors, HES1 safeguards against irreversible cell cycle exit both during normal cellular quiescence and, pathologically, in the setting of tumorigenesis (Sang et al., 2008). It can be argued that irreversible senescence is the first barrier to tumor formation during early carcinogenesis while the quiescence of dormancy is a later event. Both might be states of growth arrest but, to fulfill its function, the latter has to be reversible. Little information is available on the role of HES1 in melanoma but it is expressed in melanoma cell lines (Bhat et al., 2006) and it is a Notch signaling regulated transcriptional repressor (Jarriault et al., 1995). As Notch-1 has been implicated in melanoma progression (Liu et al., 2006; Nickoloff et al., 2005; Pinnix et al., 2009), and melanoma appears to evade senescence (Denoyelle et al., 2006), it is tempting to speculate that, during expansion and/or quiescent phases, Notch and HES-1 may protect melanoma from entering senescence. Further studies will be needed to determine whether these mechanisms are at play in disseminated tumor cells in sites such as sentinel lymph nodes in melanoma patients.
Several additional mechanisms of cancer cell dormancy have been proposed. For example, solitary melanoma disseminated tumor cells that are supported by the existing vascular system can enter dormancy by expression of metastasis suppressor genes (Bakalian et al., 2007; Ferrari et al., 2007; Nash et al., 2007). There is also evidence that dormant tumor cells, i.e. cells proliferating poorly or in a state of quiescence, might be immunogenic, suggesting that quiescence and anti-tumor immunity can contribute to dormancy of residual disease (Koebel et al., 2007; Mahnke et al., 2005). On the other hand, growth arrested tumor cells in the bone marrow are known to evade the immune system by silencing the expression of antigen presenting molecules (Saudemont and Quesnel, 2004). Thus, the role of the immune system might depend on the type of tumor and/or the state of proliferation of DTCs or micro-metastatic lesions.
Melanoma has long been recognized to be immunogenic, and immunotherapy of this cancer was first attempted more than a century ago. The tumor expresses antigens such as Melan A/MART-1 that can be recognized by cytotoxic T lymphocytes (CTL) in association with MHC class I and it appears that immunosurveillance might contribute to the thwarting of melanoma growth (Polak et al., 2009). More recent studies have shown that melanoma is not only capable of evading host mounted immunity through immune-editing and other mechanisms, but also to locally suppress immunity (for review see: (Polak et al., 2009). Many attempts to utilize melanoma antigens and boost immunity in various ways in a setting of therapeutic vaccines have met so far with limited success (Ralph, 2007; Terando et al., 2007). The apparent discrepancy between the ability to increase specific cytolytic T cells and the lack of clinical benefit, brought into focus local immunosuppressive melanoma functions (Gajewski, 2006). In transgenic mouse models of melanoma, however, CD8+ T-cells appear to prevent disseminated melanoma cells in the bone marrow and lymph nodes of mice from expanding (Umansky et al., 2008) suggesting that, at the minimal residual level, the disseminated systemic disease may be controlled by the immune system. Moreover, CD8+ T cells appear to have a distinct role toward cutaneous tumors and visceral metastases (Lengagne et al., 2008), suggesting that, the effect of immunity on melanoma dormancy might depend on the microenvironment.
Dormancy and tumor initiating cells
While there is a general agreement on the existence and the role of adult stem cells in most mammalian tissues, including human, the same cannot be stated about cancer stem cells. A basic unanswered question in cancer stem biology is whether the ability to colonize and form new growths at distant sites (metastasis), is encoded in most tumor cells and is stochastic in nature, or whether only a very minute fraction of tumor cells, called tumor initiating cells (TICs), have this ability. A similarly undecided issue is the question of stability of TICs versus their ability to undergo epigenetic reprogramming toward and away from the TIC phenotype (Quintana et al., 2008; Rosen and Jordan, 2009). Reversibility of the phenotype is probable because even adult differentiated cells can be reprogrammed into stem/progenitor cells when appropriate transcription factors are expressed (Aasen et al., 2008; Dimos et al., 2008; Kim et al., 2008). In analogy to the adult stem cells, which are characterized by practically unlimited self-renewal potential, enhanced survival mechanisms and the capacity for prolonged cellular quiescence, some investigators propose that TICs might display similar features (for review (Schatton et al., 2008). Fang et al. (2005) showed formation of non-adherent spheroids in approximately 20% of melanomas cultured in embryonic stem cell medium. Cells from these spheroids could be differentiated into melanocytic, adipocytic, osteocytic, and chondrocytic lineages. Compared with the adherent cells, the spheroid cells were more tumorigenic in vivo. Another group (Frank et al., 2005) identified an efflux pump in melanomas, both in tissue samples and cell lines, and showed that this pump is preferentially expressed in CD133b expressing subpopulation of melanoma, suggested previously to represent the stem cell compartment in this and other tumors. Inhibition of the ABCB5 pump significantly reversed resistance to doxorubicin. Other groups, (Klein et al., 2007; Monzani et al., 2007), identified additional markers in sub-populations of melanomas, and showed that an isolated CD133b positive population grew tumors more efficiently than CD133b negative cells. In recent studies disseminated breast cancer cells in the bone marrow were found to be enriched for markers of breast tumor stem cells: ∼65% of cells in the bone marrow were CD44+/CD24−/cytokeratin (CK)+ compared with <10% in the primary tumor (Balic et al., 2006). However, the proliferative state of these cells or their role in relapse is unknown. The existence of TICs, if definitively proven, will be of great significance to the future of anti-cancer therapy. If specific approaches could be developed to target and eliminate these quiescent cells with high survivability, eradication of cancer might become possible. In this brief chapter we do not intend to resolve this debate, although our own data in head and neck cancer cell model (Ossowski and Reich, 1980, 1983), (Aguirre-Ghiso, unpublished results), in colon carcinoma (Odoux et al., 2008) and melanoma (Quintana et al., 2008) suggest that every cell in the tumor mass has the ability to initiate and propagate new growth in vivo and that perhaps this ability can be reprogrammed (Mani et al., 2008). Several, more recent publications cast some doubt on the TICs as tumor generating entities. For example, Quintana (Quintana et al., 2008) showed that in highly immunocompromised NOD/SCID interleukin-2 receptor gamma chain null mice, the detection of tumorigenicity of individual melanoma cells is increased by several orders of magnitude, such that upon transplantation one in four melanoma cells can propagate a new tumor. Even the well established cancer stem cell notion in leukemia has been recently challenged (Kelly et al., 2007). These authors showed that when lymphomas and leukemias of mouse origin are transplanted into histocompatible mice, at least 1 in 10 tumor cells can seed tumor growth. Discussion of this debate can be found (Alison et al., 2009; Rosen and Jordan, 2009; Sabatino et al., 2009).
The relevant question is whether the mechanisms controlling metastasis dormancy, and thus its refractoriness to therapy are equivalent to, or distinct from those controlling the quiescence of tumor initiating cells (TICs). The fact that normal stem cells can be dormant for very long periods (Wilson et al., 2008) suggests that, if reinstated in TICs, this quiescence might explain tumor cell dormancy. However, despite numerous review articles proposing this mechanism, conclusive proof is still absent. An issue that further complicates this interpretation is the existence of dominant activating mutations in melanoma. To accept TICs quiescence one would have to accept the possibility that therapy, microenvironment or the stem cell-dependent reprogramming into dormancy might neutralize the action of mutant B-Raf once senescence is bypassed or more importantly antagonize mutant N-Ras signals or restore PTEN pathway function. This might have important implications for many aspects of cancer biology and most clearly for the oncogene addiction model (Weinstein, 2002). Is it possible that mechanisms that induce dormancy uncouple proliferative oncogene signaling from their survival function, or deactivate oncogene signaling altogether?
Chemotherapeutic refractoriness of advanced malignant melanoma may be attributable to several resistance mechanisms that have been implicated in TIC biology (Dean et al., 2005), including the impairment of cancer apoptotic pathways, a relative mitotic quiescence, and reduced drug accumulation because of high-expression levels of ABC drug efflux transporters (Gottesman, 2002). For suggestions on how to approach TIC eradication see review by Grichnik (2008). However, until the existence of TICs is clearly proven, and until the mechanisms that keep them quiescent are elucidated, it will be a difficult task to determine whether metastasis dormancy and TIC have a common origin.
Could the reason for dormancy of metastatic melanoma be early dissemination from the primary?
Melanoma has two distinct expansion phases, radial and vertical. It is thought that invasion and dissemination occurs during the latter phase. Because very small differences in lesion depth have significant survival consequences it is thought that more invasion by more progressed cells contribute to metastasis development. Although, this most likely happens, it is also possible that in those patients that show late metastasis, early dissemination of tumor cells that have not yet fully progressed, is a contributor to the late occurring metastasis. Also, mechanisms described in the previous chapter might contribute to this clinical outcome. While, to our knowledge, no direct information is available for this process in melanoma, experience from the clinic suggests that dissemination can occur before diagnosis for primary cancer. For example, uveal melanoma is most often diagnosed and locally cured before any signs of clinical disseminated disease are present, yet because of the high proportion of often much delayed metastases it has been classified as a systemic disease (Callejo et al., 2007; Shildkrot and Wilson, 2009). Calculation of melanoma doubling time suggested (Eskelin et al., 2000) that most uveal melanoma metastases are initiated within 5 yr before primary treatment. Also, several groups have shown that before any signs of clinically advanced disease, circulating tumor cells can be found in large proportion of patients with uveal melanoma (Callejo et al., 2007; Schuster et al., 2007). In another study, blood samples from 52 patients with clinically localized uveal melanoma and from 20 normal controls were tested for circulating tumor cells and were found to be positive in 20% of the patients (Ulmer et al., 2008). Thus, early disseminated cells might persevere as clinically dormant metastasis to give rise to late distant recurrences.
The idea of early spread, such as in the case of uveal melanoma and some very early, thin cutanoeus melanomas, does not fit well with the established paradigm which proposes that metastasis arise from rare clones that evolve in the primary tumor and acquire characteristics that allow them to disseminate to, and grow in secondary sites (Klein, 2008; Pantel and Brakenhoff, 2004). A critical aspect of this theory is that this could only happen in evolutionarily ‘late-progressed’ tumors (with multiple metastasis associated genetic alterations) and that it should not be observed in patients treated for early lesions (fewer genetic alterations) (Klein, 2008; Pantel and Brakenhoff, 2004). A major challenge to this theory came from a series of studies from the Klein lab suggesting that in breast cancer, even in lesions considered to be ‘early’ in progression, such as non-invasive atypical ductal hyperplasia (ADH) and ductal carcinoma in situ (DCIS), dissemination has already occurred (Husemann et al., 2008; Klein, 2008; Schardt et al., 2005; Schmidt-Kittler et al., 2003; Stoecklein et al., 2008). These studies proposed that the pause observed in the progression of early disseminated tumor cells may be due to a ‘lead time’(Husemann et al., 2008; Klein, 2008; Schardt et al., 2005; Schmidt-Kittler et al., 2003; Stoecklein et al., 2008). This is the time that it takes for DTCs with few genetic alterations to accumulate additional ones that will favor ectopic growth (Klein and Hölzel, 2006). This is supported by the findings that genetic alterations in DTCs detected in ADH or DCIS are very heterogeneous. In contrast, genetic anomalies in DTCs from patients carrying diagnosed metastatic disease are more homogeneous. This possibly suggests that as evolution occurs in the secondary site, the original heterogeneity in DTCs is reduced (Husemann et al., 2008; Klein, 2008; Schardt et al., 2005; Schmidt-Kittler et al., 2003; Stoecklein et al., 2008). However the kinetics driving genetic progression during this lead-time, are poorly understood, especially considering the presumed growth arrest of these cells. It is also possible that in spite of carrying genetic alterations that favor growth, the DTCs are forced into quiescence by the micro-environment, or epigenetic or therapy-derived mechanisms (Aguirre-Ghiso, 2007). In support of the microenvironment playing a role, a recent report suggests that breast cancer patients with cells disseminated to the bone marrow have longer disease free periods than patients that were negative for cells in this site but displayed metastasis in other organs (Bidard et al., 2008). Further, MMTV-Neu cells disseminated to the bone marrow of mice fail to expand unless they are transplanted into recipient mice that were irradiated (Husemann et al., 2008). This suggests that the bone microenvironment may delay cancer progression and thus prolong patient survival but also, that perturbations of this microenvironment might trigger growth. Such mechanisms might be in place in common sites of melanoma metastasis, cutaneous or uveal. In melanoma cell lines, targeting of specific integrins can curtail growth, while interaction of melanoma cells with fibrillar collagen induces cell cycle inhibitor p15INK4b expression and growth arrest (Wall et al., 2007). In addition, the crucial interaction between melanocytes and keratinocytes, lost in melanoma, can be restored by E-cadherin re-expression (Hsu et al., 2000). It was also shown that exposure of melanoma cells to matrix conditioned by human embryonic stem cells resulted in the re-expression of the melanocyte specific marker Mart-1/Melan-A as well as a reduction in invasive potential. Similarly, metastatic melanoma cells transplanted into chick embryo, close to the premigratory neural crest cells, migrated into the surrounding host tissue in a programmed manner, suggesting that they were able to respond to host neural crest migratory cues (for review see Postovit et al., 2006). On the other hand, others (Conner et al., 2003) have shown that adhesion regulated ERK1/2 activation in melanocytes is by-passed by mutation of B-Raf in malignant melanoma cells, although melanoma cells may still be dependent on integrin activated PI3K and Akt signaling for survival (Boisvert-Adamo and Aplin, 2006). Overall, more is known about factors in the microenvironment which complement aberrant genetic changes to promote rather than inhibit melanomagenesis, for review (Postovit et al., 2006). It appears that while some responses to the microenvironment are lost, new controls might emerge, and microenvironmental ‘constraints’ might lead to growth arrest and cellular dormancy.
Experimental models of melanoma dormancy
Several interesting transgenic melanoma mouse models have been developed, including two very recent ones, in which conditionally activated B-rafV600E alone (Dhomen and Marais, 2009; Dhomen et al., 2009), or in combination with PTEN-tumor suppressor gene silencing (Dankort et al., 2009) led to melanoma generation. However, only one group has been focusing on characterizing melanoma dormancy and those studies were done on transplanted B16F10 melanoma, selected for growth in lungs. When injected into the tail vein of mice, these cells arrest preferentially in the lungs, extravasate efficiently as single cells and either develop into progressing metastases or persist as solitary, dormant cells. The authors mixed the injected cells with microspheres, which ‘permanently’ arrested in the small vessels while tumor cells arrested and extravasated into the surrounding tissue. Because their number remained roughly constant, the spheres served as a reference against which the quantification of the disseminated cells was done. At day 12–14 only ∼3.5% of the inoculated cells remained as solitary cells, of this only 3% was stained with Ki-67, a marker of proliferation, while ∼40% of cells in small, medium or large metastases stained positively for this marker. None of the solitary cells stained with markers of apoptosis suggesting that most of them were neither proliferating nor dying, and thus could be characterized as dormant. Overall, control of dormancy was largely a post-extravasation event. Regionally within the lung, cells and metastases were randomly distributed until day 4, but by day 10 preferential tumor growth was found along the lung surface and around arterial and venous vessels. Thus, trapping and early growth of injected cells was unaffected by location within the lung, whereas subsequent metastatic growth was enhanced in specific microenvironments, and prevented in restrictive microenvironment of lung tissue (Cameron et al., 2000). Because the experiments were carried out on cells propagated in tissue culture, and introduced directly into the circulation, the proportion of surviving cells and their distribution into solitary dormant cells and growing metastases might not accurately reflect the situation occurring during spontaneous dissemination in human disease. However, at the minimum these experiments confirm the ability of melanoma cells to survive and persist in a solitary quiescent state. Another group (Logan et al., 2008) injected fluorescently-labeled human uveal melanoma cells into tail vein of nude mice and imaged the cells in vivo; cells were found in multiple organs post-injection, but persisted as single cells for the duration of the experiment, solely in the liver, the site of uveal melanoma metastasis in humans. Neither of the two groups, however, answered the crucial clinical question of whether these cells were eventually eliminated or whether they emerged from dormancy. The hope is that the new awareness of the importance of melanoma dormancy and the availability of the many newly developed transgenic models of melanoma will spur wider interest and will open new approaches to therapy.
Clinical evidence of melanoma dormancy
Compared to the extensive literature on MRD and dormant disseminated cancer cells in BM and in circulation in breast cancer, and several other solid tumors (Klein, 2008), the published evidence suggesting dormancy in melanoma is modest in scope and insight; there are very limited studies examining melanoma cells in bone marrow. Because cutaneous melanoma is believed to spread mainly through the lymphatics, analysis of the sentinel lymph nodes during surgery is now routinely employed, but mostly as aid in disease staging or a guide for adjuvant therapy decisions. Effective therapy for advanced melanoma does not yet exist, but patients with LN-micro-metastases are usually offered some adjuvant therapy and small gains in overall survival have been recorded. There is also extensive literature comparing outcomes in patients in which identification of a positive sentinel LN was followed by general dissection of the LN-bed to those in which the dissection was not performed. No conclusive answers as to the benefit of the dissection have been reached, with some showing benefit of LN-dissection (Shen et al., 2000) but generally showing no benefit (Pharis, 2005). One interpretation of the fact that LNs harboring micro metastases, and left in place, do not influence disease-free or overall survival is that in some patients these cells might experience a period of dormancy. But to prove this, LN-metastatic dormancy would have to be directly investigated.
Does metastatic dormancy exist in melanoma? Clinical facts confirm the existence of dormancy. The most definitive indications of melanoma dormancy are found in patients with uveal melanoma, where the disease recurs, usually as delayed distant liver metastases in 40–50% of patients and, in some patients, distant lesions may appear very early (Eskelin et al., 2000). In cutaneous melanoma fewer opportunities exist to study metastatic dormancy. This is because the cure rate of very early cutaneous melanoma is greater than 85%, and only a very low percent (<1 to 3.5%, with one study reporting 11%) of advanced melanoma showing signs of dormancy as defined by very late recurrence after the removal of the primary. In addition to several dozen case reports showing late developing cutaneous melanomas, there are a number of studies on larger cohorts of patients. One group (Tsao et al., 1997) has examined 2766 total cases of Clark III and IV melanoma with a specific focus on ultra late recurrence (>15 yr) and found 20 patients which had ∼20 yr of disease-free interval. The authors conclude that this protracted disease free interval challenges the concept of a “cure” for melanoma. We propose that in addition, it suggests that there are mechanisms of melanoma dormancy induction, albeit yet undiscovered, which when applied therapeutically, might substantially expand the long-term overall survival of melanoma patients. Several dozens of case reports and additional studies similar to the one described by Tsao et al., have been reported. In one, (Shen et al., 2000), the authors showed that of the 1907 melanoma patients with disease-free interval (DFI) of 10 yr or more after surgical treatment of clinically localized melanoma, 217 (11%) patients had recurrences (mean DFI of ∼15 yr). They conclude that: ‘distant disease probably represents disseminated disease at the time of initial resection and that patients with early-stage cutaneous melanoma are expected to have a good prognosis, but their study shows that they are never cured of their disease and can develop fatal recurrences even after a 10-yr DFI’, further confirming the existence of melanoma dormancy. In another study (Gamel et al., 2002), the authors used a parametric survival model that incorporated a cured fraction of patients and translated these factors into specific estimates of long-term outcome. A cohort of 5837 patients treated for localized cutaneous melanoma at the Duke Comprehensive Cancer Center, were followed for up to 22 yr. For woman with 0.5 mm thick, non-ulcerated lesions on an extremity, the probability of cure was estimated at ∼80.8%, and the median tumor specific survival was 10.0 yr. This suggests that, in these patients, half of the deaths from melanoma will occur more than 10 yr after treatment. The authors' conclusion is that late recurrence, longer than a decade after treatment, is to be expected in a significant portion of patients. A group in Munich (Schmid-Wendtner et al., 2000) analyzed data of 6298 patients with cutaneous melanoma and identified 31 patients who first experienced metastatic disease 10 or more years after surgical treatment of the primary melanoma. Another group (Peters et al., 1997) examined 1015 patients who had 10 or more years of follow-up after treatment of invasive malignant melanomas (stage I and II, UICC 1978) and found 36 (3.5%) that developed late metastases; the mean DFI was 12.5 yr. The frequency of metastases (3.5%) did not vary in thickness classes from 0.76 mm to 3 mm or more. In a study of stage 1 melanoma (Gutman et al., 1989) 6 out of 94 patients, developed late relapse. In a very large study of 7104 patients with all stages of disease at Duke University (Crowley and Seigler, 1990a,b), identified 168 patients (2.4%), who experienced their first recurrence 10 or more years after diagnosis. At the time of the original diagnosis, majority of these patients had Clark's level III and IV lesions and Breslow lesions of intermediate (1.0–3.0 mm) thickness. This finding suggests that, although there is a correlation between the Clark's and Breslow characteristics of the primary lesion and the probability of spread, as indicated by the above study, some aggressive melanomas can, after dissemination, reprogram or be reprogrammed into temporary quiescence.
The mean disease-free interval for cutaneous melanomas was 14.3 yr versus 22.3 yr for ocular primary melanomas. A subsequent study by the same group showed that of 2468 melanoma patients ∼18% showed recurrences after 5–10 and >10 yr later (Crowley and Seigler, 1990a,b). The authors concluded that in cutaneous melanoma the disseminated cells can exist in a state of quiescence for prolonged periods but, once reactivated, the timing of dormancy does not influence the progression of the recurrent disease. These results further emphasize that, until effective therapy is found for recurrent disease, studying the mechanisms responsible for the extended quiescence of the residual disease, is essential. An approach that will allow therapeutic prolongation of clinically undetected (dormant) disease should have a real impact on overall survival.
An even more definitive proof that cutaneous melanoma can persist in a state of prolonged dormancy comes from a case report in which two different kidney transplant recipients, which obtained a kidney from the same donor, developed secondary melanoma near the transplanted kidneys. Examination of the records revealed that the donor had 2.6 mm thick primary melanoma excised 16 yr before donating the kidneys, but the patient was pronounced melanoma free during the 15-yr follow-up after surgery, (Mackie et al., 2003). This indicates that melanoma cells persisted in a dormant state in the kidney of the donor for 16 yr and that, upon transplantation to two different but immuno-suppressed hosts, they emerged from dormancy. In 26 additional recipients of organs from melanoma patients, 21 developed melanoma in the donated organ (Penn, 1996) (Table 1).
Table 1.
Comparison of properties with relevance to dormancy of cutaneous and uveal melanoma
| Properties | Cutaneous melanoma | Uveal melanoma | References |
|---|---|---|---|
| Mutations | B-raf, N-ras | GNAQ | Davies et al., 2002, Onken et al., 2008; Van Raamsdonk et al., 2009 |
| Predominant routes of spread | Lymphatic | Hematogenous | For review, Belmar-Lopez et al., 2008; Zapas et al., 2003 |
| Early spread | Few | Yes | Eskelin et al., 2000; Callejo et al., 2007; Shildkrot and Wilson, 2009 |
| % >10 year disease free interval | <5 | 40–50 | Crowley and Seigler, 1990a; Peters et al., 1997; Tsao et al., 1997; Shen et al., 2000; Gamel et al., 2002; Schmid-Wendtner et al., 2000; Shildkrot and Wilson, 2009 |
| Metastasis in organs | Multiple | Liver | For review, Belmar-Lopez et al., 2008 |
| Circulating tumor cells detected | Yes | Yes | Callejo et al., 2007; Schuster et al., 2007; Visús et al., 2007, Arenberger et al., 2008, Ulmer et al., 2008 |
These studies show that in both uveal and cutaneous disseminated melanoma, cells can remain dormant for longer than a decade, and when they emerge from dormancy they almost invariably are resistant to therapy and fatal. The rate of delayed recurrence is by far greater in ocular melanoma suggesting that a larger proportion of disseminated uveal melanoma cells persist in dormancy. Could understanding of the biological difference between cutaneous and uveal melanoma, both arising in the same cell of origin, provide insights into the mechanism of dormancy?
Immunological considerations distinguishing cutaneous and uveal melanoma
Although both uveal and cutaneous melanoma are of neural crest origin, and derived from melanocyte or their precursors, melanoblasts, they migrate to very different tissues and reside in very different micro-environmental milieu (for review, Belmar-Lopez et al., 2008). Uveal melanomas develop characteristic chromosomal abnormalities, such as loss of chromosome 3, abnormalities in chromosome 6 and, less frequently, gains in chromosome 8q. Although, there is very scarce information on the genes associated with these cytogenetic changes, they might in the future provide some clues as to the biological uniqueness of the uveal melanoma and possibly its high rate of early dissemination and metastatic dormancy. Among the many differences between uveal and cutaneous melanoma, such as hematogenous versus lymphatic dissemination, difference in blood vessel formation, organ target of metastasis and others, two that might provide clues regarding dormancy are their unique immunologic status and the difference in oncogenes that are mutated. For a comprehensive discussion on the topic of immune surveillance in melanoma the reader is referred to an excellent review (Niederkorn, 2009). It is clear that while cutaneous melanoma grows under conditions of immune-editing comprised of elimination, equilibrium, and escape (Dunn et al., 2008), the conditions in the eye, the site of uveal melanoma, are rich in immunosuppressive and anti-inflammatory factors which dampen both innate and adaptive immune responses. The unique vasculature of the eye creates a blood:ocular barrier that restricts the movement of macromolecules and leukocytes from blood vessels into the eye. Additional, membrane-bound molecules protect ocular tissues from injury inflicted by elements of both the innate and adaptive immune responses. This would suggest that uveal melanoma should progress more rapidly; however, it is believed that muted immune responses might sometimes exacerbate, rather than inhibit, tumor progression (Niederkorn, 2009). This author proposes that primary uveal melanoma contain a mixture of cells with high and low level of MHC class I antigen. However, although in contrast to cutaneous melanoma, those with low MHC are highly susceptible to elimination by NK cells, they are shielded from NK cells by the presence in the aqueous humor of the eye of MIF and TGFβ. Once the uveal melanoma cells leave the eye and enter the liver, the predominant site of metastasis, they find themselves in an organ with high concentrations of NK cells. Those expressing low MHC class I molecules can be eliminated by NK cells, those expressing high MHC class I are spared to become the dominant phenotype. It is impossible to guess whether some of the low MHC class I cells are protected from elimination by TGFβ or other growth factors in the liver and become dormant, or whether the dormant population originates from the MHC class I-high expressing cells.
Signaling pathways in cutaneous and uveal melanoma
N-Ras, B-Raf and GNAQ mutations. How do these fit in the tumor cell dormancy paradigm?
Currently, the most tangible difference between cutaneous and uveal melanoma, that might eventually explain the mechanisms that allow for prolonged quiescence (dormancy) in almost half of the uveal melanoma patients, is the difference in the mutated oncogenes. While B-Raf and N-Ras mutations, which are mutually exclusive, are predominant in cutaneous melanoma, they are almost completely absent in uveal melanoma. Instead, 46% of uveal melanoma and 83% of blue nevi carry mutations in the GNAQ gene, a Gq protein a-subunit (Onken et al., 2008; Van Raamsdonk et al., 2009) that when mutated in the 209 position has transforming capacity (Kalinec et al., 1992). So far, GNAQ status has not been linked to DFS (Bauer et al., 2009). It is possible that progression driven by this genetic variation follows different kinetics from that of B-Raf or N-ras mutation and that it maintains responsiveness to yet unidentified growth suppressive microenvironmental cues.
The direct link between the different genetic and epigenetic alterations in melanoma and tumor progression remains elusive (Dahl and Guldberg, 2007; Triozzi et al., 2008). Although, many active pathways have been identified and specific inhibitors have been synthesized, for many reasons, targeting them has not yet truly benefited melanoma patients (Dhomen and Marais, 2009; Dhomen et al., 2009; Haluska et al., 2007; Kalinsky and Haluska, 2007; Lorigan et al., 2008). Among the many reasons are the complexity and the feedback mechanisms between the oncogene-driven signaling pathways as well as their ‘rewiring’ in melanoma (Estrada et al., 2009; Lopez-Bergami et al., 2007, 2008). Similarly, the effort to precisely link specific mutations to disease free periods produced mostly inconclusive results, making extrapolation of their potential role in dormancy even more difficult. A small study (Kumar et al., 2003) showed an association between the disease-free survival periods with the presence of B-Raf and N-Ras mutations. In this study of around 40 patients, 21% had disease free periods longer than 5 yr and median DFS was 10 yr. Of the patients that showed long disease free periods an impressive 75% had B-Raf mutations (V600E or K601E); none had N-Ras mutations. Three patients (7.9%) bearing V-600E B-raf mutations were free of disease for 13–16.7 yr. Other authors have noticed no effect on DFI in patients with B-Raf mutations (Shinozaki et al., 2004). Although very limited, this study suggests that even activating mutations, considered to be responsible for cancer progression in oncogene ‘addicted’ tumors appear to, at least temporarily respond to host-imposed controls. Is it possible, as evidenced in patients, that these controls can silence tumor cells with activating mutations for a decade or longer?
In conditional transgenic models, oncogene inactivation can induce tumor cell elimination, or terminal differentiation/senescence and, as in case of Myc inactivation in hepatocellular carcinoma, induction of cancer cell dormancy (for review see Felsher, 2008a,b), suggesting that the outcome might be specific to cellular and genetic context. Can conclusions from these models be directly applied to naturally occurring metastatic dormancy and more specifically, can microenvironment exert an effect similar in nature to an effect induced by a rapidly down-regulated oncogene? At present, the answer to this question can only be speculative and complicated by the fact that as a rule in inducible systems the oncogene itself is silenced while, in the case of human disease, dormancy most likely ensues despite the presence of an active oncogene. The theory of oncogene ‘addiction’ states that a cancer cell is fully dependent on the signaling pathway generated by the activity of the specific oncogene and, further implies, that activation of the pathway is autonomous, independent of environmental influences (Weinstein, 2002). For example, as already mentioned, in melanoma with mutated B-Raf the activation of ERK was shown to be independent of integrin/matrix engagement (Conner et al., 2003). It is known that activated ERK plays a pivotal role in cell proliferation; it controls the G1-phase to S-phase transition by negatively regulating the p27/Kip1 inhibitor (Kortylewski et al., 2001) and by upregulation of c-Myc activity (Lefevre et al., 2003). Does it mean that once ERK pathway is activated through B-Raf or N-Ras mutation, its activity, and thus melanoma cell proliferation, must continue unabated? The clinical persistence of dormant disseminated melanoma cells suggests that it might not be the case. It is possible then that, although the mutant B-Raf stimulus to ERK activation is autonomous, the downstream effectors of the pathway might be dependent on the microenvironment and/or epigenetic mechanisms. There are other hints that the pathway can be counteracted. For example (Wajapeyee et al., 2008) have shown that expression of B-RafV600E mutant in primary cells leads to synthesis and secretion of IGFBP7, which through autocrine/paracrine pathways inhibits BRAF-MEK-ERK signaling and, by inducing a pro-apoptotic protein BNIP3L, leads to senescence and apoptosis. Systemically administered rIGFBP7 markedly suppresses growth of B-RafV600E-positive tumors in xenografted mice. It is theoretically possible that under some circumstances the secretion of IGFBP7 or another inhibitory protein is increased, and the oncogene pathway is inhibited.
B-Raf and GNAQ, in addition to being mutated in melanoma, are also mutated in benign nevi. Thus, it is believed that, for tumors to be formed, additional molecular alterations need to happen. In a recently developed mouse model of melanoma (Dhomen et al., 2009) conditional activation of mutant B-RafV600E was sufficient to yield melanomas but those formed only after very long latency. Another group (Dankort et al., 2009) has shown that combination of this lesion with PTEN silencing, produced aggressive melanomas with very short latency and metastasis to lungs and LNs. PTEN, tumor suppressor, which resides on human chromosome 10q23.3, has been shown to be deleted in a proportion of melanoma cell lines and in a small number of primary melanomas (Birck et al., 2000; Guldberg et al., 1997). Importantly, it was found to be epigenetically silenced by several mechanisms, including by promoter methylation, in nearly 40% of melanoma samples (Mirmohammadsadegh et al., 2006; Whiteman et al., 2002; Zhou et al., 2000), an event linked to activation of the PI3K/Akt pathway. Because most epigenetic changes in gene expression are dynamic and transient, and mostly reversible, they open a window of possibility for their modulation by environmental influences. In melanoma, expression of some 50 genes has been shown to be deregulated through aberrant DNA methylation, and/or inappropriate targeting of histone modifications and chromatin remodeling. Comprehensive reviews on melanoma epigenetics have been recently published (Dahl and Guldberg, 2007; Richards and Medrano, 2009; Rothhammer and Bosserhoff, 2007). The silencing of PTEN in melanoma allows for activation of Akt/PKB which in turn, activates the transcription of genes involved in immune activation, cell proliferation, apoptosis and cell survival. The level of activated Akt increases with melanoma progression and it is inversely correlated with overall and disease-free survival (Dai et al., 2005). The activated Akt phosphorylates and prevents nuclear localization of p27 (Liang et al., 2002), blocking inhibition of cell cycle. Akt was shown to phosphorylate Skp2 (Lin et al., 2009) and affect its relocalization to cytoplasm. Skp2, which regulates p27 proteosomal degradation, is increased in progressed melanoma, concomitantly with p27 reduction (Sanki et al., 2007). In an elegant work (Liang et al., 2007), the authors showed that during metabolic stress, LKB1–AMPK pathway regulates p27 Thr198 phosphorylation and increases its stability. They concluded that p27 level determines whether cells that become quiescent due to lack of nutrients or other stress conditions, enter the autophagy cell survival pathway or undergo rapid cell death by apoptosis. Although, in growing melanomas the mutated B-Raf overrides this pathway, its complexity suggests possibilities for control by the microenvironment.
Fibrillar collagen and TGFβ signaling as suppressors of melanoma proliferation?
It is of interest to note that in a series of manuscripts (Henriet et al., 2000; Wall et al., 2005, 2007) p27 was shown to be increased in a melanoma cell line plated on fibrillar collagen type 1. Moreover, it was the activation of Discoidin Domain Receptor 2 (DDR2) by fibrillar collagen, and increase in p15 and p21, and not the increase in p27, which was responsible for cell cycle arrest under these conditions. Thus, it is possible that in spite of active autonomous proliferation signals from N-ras and B-raf, interaction with fibrillar collagen might induce stress-like signals and cell-cycle arrest. Yet, as mentioned above, because of an increase in p27 cells can survive by activating autophagy. It is also conceivable that signals from the microenvironment restore expression of the epigenetically silenced PTEN. For example, addition of n-3 fatty acid (omega-3) increased PTEN expression and inhibited the growth of B16 melanoma in culture (Xia et al., 2006), suggesting that even nutrition might influence the fate of melanoma.
Another potential signaling pathway that might participate in dormancy induction in melanoma might be TGFβ. It is well established that in early stage melanoma TGFβ has decisive anti-proliferative (tumor-suppressor) effect while in advanced melanoma it is pro-invasive (for review see Hsu et al., 2005; Hussein, 2005; Javelaud et al., 2008). While the anti-proliferative pathways are understood better, and involve TGFβ induced cyclin-kinase inhibitors expression, the mechanisms of resistance to inhibition and the pro-invasive effects are less conclusive (Javelaud et al., 2008; Reed et al., 2001). There are two options to consider; in analogy to recently discovered behavior of human breast cancer, in which dissemination to distant sites is suspected to happen early in tumor development (Schardt et al., 2005), melanoma might spread before the conversion from the TGFβ-inhibitory phenotype to the pro-invasive phenotype took place. Although, because spreading is generally linked with ‘invasive’ features, the idea of early spread is somewhat counterintuitive, in breast cancer spread has been shown to take place at the stage of DCIS (Schardt et al., 2005). There is also clinical evidence of early spread of uveal melanoma and, in a smaller proportion of patients, cutaneous melanoma thinner than 0.76 mm depth (Eskelin et al., 2000; Gamel et al., 2002; Zapas et al., 2003). If that is the case, it is possible that single cells arriving at distant sites, such as liver or BM (Husemann et al., 2008) remain in cell-cycle arrest due to high levels of TGF-β and develop an invasive phenotype away and independently from the primary tumor. Alternatively, the disseminated cell is invasive, and undergoes phenotypic switching due to cues from the new microenvironment. It is premature to try and identify the stimuli and the effectors of this switch, especially as there is still no consensus on whether the resistance to TGFβ is a general blockade of TGFβ-induced transcription by inhibitors such as Ski (Deheuninck and Luo, 2009), or a specific inhibition of a cassette of genes related to proliferation (Reed et al., 2001). This protein is nuclear and cytoplasmic in primary invasive melanomas, whereas in melanoma metastasis, Ski is mostly localized in the cytoplasm and expressed at very high levels. Cytoplasmic localization of Ski prevents its associated protein Smad3 from translocation to the nucleus in response to TGF-β (Reed et al., 2001). In breast and lung cancer TGFβ treatment leads to degradation of Ski through ubiquitin/proteasome pathway and enhances tumor metastasis in vivo (Le Scolan et al., 2008). Work by others (Schlegel et al., 2009), shows that transition to increased aggressiveness in melanoma cells requires Id2 upregulation to suppress TGFβ induction of p15Ink4b and thus help to circumvent TGFβ-mediated inhibition of proliferation. Is it possible that Id2 expression is under the control of the microenvironment and thus converts a disseminated melanoma cell to one that is sensitive to TGFβ-inhibition? Similar questions can be asked about Ski and most likely many other genes in the TGFβ pathway. As already mentioned, there are very few direct experiments probing the question of phenotypic switch due to microenvironment. One exception is the study which found no gene expression patterns that correlate with activating mutations in B-Raf or N-Ras, but instead identified transcriptional signatures specific to ‘proliferative’ melanoma sensitive, and invasive melanoma resistant to TGFβ inhibition (Hoek et al., 2008). The proliferative cohort had highly activated Wnt signaling pathway with high SOX10, MITF1 and TFAP. The invasive cohort, possibly due to effects of hypoxia or inflammation, had increased Wnt-pathway inhibitors, among them CTGF (which binds Wnt co-receptor and inhibits Wnt), and others, resulting in inhibition of Wnt pathway. Relevant to the question of dormancy induction, these authors showed that, upon transplantation into nude mice, the TGFβ sensitive, ‘proliferative’ melanoma produced much more rapidly growing tumors than produced by melanoma defined by the transcriptional signature as invasive. However, when the tumors were excised and examined histochemically, both showed similar evidence of proliferative and invasive cell type, as determined by Ki67 and by Mitf expression, which is the central feature of the proliferative signature that is absent from the invasive form. The authors concluded that ‘the transcription signatures represent distinct yet interchangeable states, regulated by signaling from the microenvironment’ and that ‘melanoma cells undergo transcriptional signature switching in vivo likely regulated by local microenvironmental conditions’. Should the existence of this switch be confirmed, it will provide a starting point to identification of the environmental signals that change melanoma gene expression and phenotype, possibly leading to dormancy (Figure 1).
Figure 1.
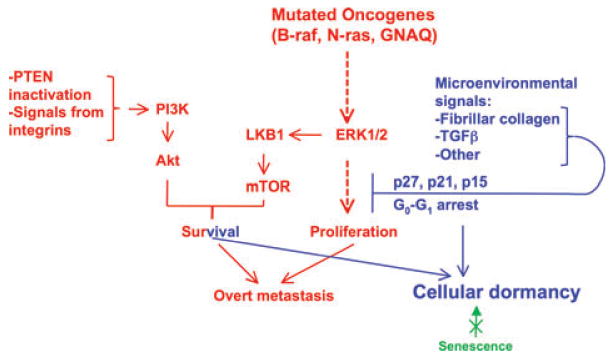
Microenvironment and dormancy in melanoma. Mutations in B-Raf or N-Ras in cooperation with PTEN silencing or deletion help melanoma cells activate pro-survival and proliferation signals. Possibly, a similar proliferative signal is relayed by the mutant Gq protein. Survival signals activating PI3K and Akt might come also from integrin interaction with extracellular matrix. Conditions of reduced ERK mediated mitogenic and survival signals, with other survival signals intact, might allow melanoma cells to persist in a quiescent state. Because senescence is thought to be an early barrier to tumor development that is no longer functional in established melanoma, it is likely that quiescence (reversible) is the cell fate that better explains the cellular dormancy of melanoma cells. Microenvironment-derived signals that limit growth might be dependent on TGFβ signaling or on fibrillar collagen-I and DDR2 signaling. These can promote the upregulation of cell cycle inhibitors that might enforce quiescence. How these signals antagonize or uncouple the proliferative signals derived from the oncogenes is unknown. Liver, the main target of uveal melanoma metastasis, remains clinically free of disease for longer than 10 yr in close to half of the patients examined. This remarkable finding suggests that in some patients the microenvironment somehow causes yet unidentified mediators to induce anti-proliferative, most likely, epigenetic changes and establish melanoma cell dormancy.
Conclusions and future approaches
There is ample clinical evidence that metastatic dormancy does exist for variable periods in melanoma. In addition to several dozens of case reports describing late appearing melanoma metastases, more than ∼20 000 patients and/or their charts have been examined and the proportion of patients with cutaneous melanoma at risk for metastases delayed by 5–10 yr was found to be 10–20%, and delayed for 10 or more years, 0.8–3.5%. A much larger proportion (∼50%) of patients with ocular melanoma were free of overt metastases for >10 yr. Most of the studies concluded that even patients with thin cutaneous melanoma, which are considered low risk for recurrence, can recur, and should have life-long follow up. To prevent recurrence, some suggest adjuvant therapy immediately following primary surgery with the hope of eliminating the disseminated cells. Most therapies assume that the disseminated cells continue to maintain their proliferative state. This might be true for tumor mass dormancy resulting from inability to induce angiogenesis. However, based on the observations in other cancers suggesting quiescence of the DTCs, (Aguirre-Ghiso, 2007; Husemann et al., 2008), the kind of therapies applied might have to be re-evaluated, and this in turn will only be possible once the molecular biology of the disseminated quiescent cells is understood.
It is our view that finding specific ways to reduce survival of dormant cells or their emergence from dormancy may significantly extend overall survival of melanoma patients. Before this happens, the identification of cellular markers, gene signatures or biomarkers indicative of the existence or potential occurrence of dormant disease would be necessary. Addressing this question will require new approaches to the clinical and basic studies. Lines of research into dormancy of other cancers revealed that specific pathways dependent on the chaperone Grp78 (Ranganathan et al., 2006), ATF6, Rheb, mTOR (Schewe and Aguirre-Ghiso, 2008) and the kinase Dyrk1B (Deng et al., 2009) might function as survival signals for quiescent cells, and as they may represent potential drug targets for elimination of dormant cells, should be tested in melanoma. It is possible that expression-profiling data will accurately predict late versus early recurrence; such signatures might allow to stage patients and/or to explore potential mechanisms for eliminating residual cells.
It will also be crucial to determine whether, like in other solid tumors, the bone marrow is a reservoir for disseminated cells or a ‘reporter’ tumor cell population for other sites like the liver. If it is, the technology applied to study these cells in other cancers (Husemann et al., 2008; Stoecklein et al., 2008) can be utilized. To better connect the microenvironment with dormancy control, further inroads into the understanding of organ target specificity of uveal melanoma metastasis to the liver will have to be made. For example are uveal melanoma cell present in other organs in a state of permanent dormancy and do pathophysiological conditions that affect the liver are predictive of recurrence of uveal melanoma in that organ? Other lines of research might need to explore whether there is a difference in the way the microenvironment cross-talks with melanoma cells bearing B-Raf versus N-Ras mutations. Answering these questions will aid in understanding of melanoma dormancy and, consequently, in improving the outcome of therapy by preventing the development of metastasis, a task perhaps more achievable than curing patients with overt metastases.
Acknowledgments
We apologize to those authors whose work we could not cite directly because of space constraints and the focus of this review. This work is supported by grants from the Samuel Waxman Cancer Research Foundation Tumor Dormancy Program, the NIH/National Cancer Institute (CA109182) and NYSTEM to J.A.A-G.
References
- Aasen T, Raya A, Barrero MJ, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26:1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- Aguirre Ghiso JA. Inhibition of FAK signaling activated by urokinase receptor induces dormancy in human carcinoma cells in vivo. Oncogene. 2002;21:2513–2524. doi: 10.1038/sj.onc.1205342. [DOI] [PubMed] [Google Scholar]
- Aguirre Ghiso JA, Kovalski K, Ossowski L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol. 1999;147:89–104. doi: 10.1083/jcb.147.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834–846. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK) Cancer Res. 2003;63:1684–1695. [PubMed] [Google Scholar]
- Alison MR, Islam S, Lim SM. Number crunching in the cancer stem cell market. Breast Cancer Res. 2009;11:302. doi: 10.1186/bcr2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenberger P, Arenbergerova M, Vohradnikova O, Kremen J. Early detection of melanoma progression by quantitative real-time RT-PCR analysis for multiple melanoma markers. Keio J Med. 2008;57:57–64. doi: 10.2302/kjm.57.57. [DOI] [PubMed] [Google Scholar]
- Bakalian S, Marshall JC, Faingold D, Logan P, Antecka E, Burnier MN., Jr Expression of nm23-H1 in uveal melanoma. Melanoma Res. 2007;17:284–290. doi: 10.1097/CMR.0b013e3282eeea5a. [DOI] [PubMed] [Google Scholar]
- Balic M, Lin H, Young L, Hawes D, Giuliano A, Mcnamara G, Datar RH, Cote RJ. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin Cancer Res. 2006;12:5615–5621. doi: 10.1158/1078-0432.CCR-06-0169. [DOI] [PubMed] [Google Scholar]
- Barnhill RL, Piepkorn MW, Cochran AJ, Flynn E, Karaoli T, Folkman J. Tumor vascularity, proliferation, and apoptosis in human melanoma micrometastases and macrometastases. Arch Dermatol. 1998;134:991–994. doi: 10.1001/archderm.134.8.991. [DOI] [PubMed] [Google Scholar]
- Bauer J, Kilic E, Vaarwater J, Bastian BC, Garbe C, De Klein A. Oncogenic GNAQ mutations are not correlated with disease-free survival in uveal melanoma. Br J Cancer. 2009;101:813–815. doi: 10.1038/sj.bjc.6605226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmar-Lopez C, Mancheno-Corvo P, Saornil MA, Baril P, Vassaux G, Quintanilla M, Martin-Duque P. Uveal vs. cutaneous melanoma Origins and causes of the differences. Clin Transl Oncol. 2008;10:137–142. doi: 10.1007/s12094-008-0170-4. [DOI] [PubMed] [Google Scholar]
- Bhat KM, Maddodi N, Shashikant C, Setaluri V. Transcriptional regulation of human MAP2 gene in melanoma: role of neuronal bHLH factors and Notch1 signaling. Nucleic Acids Res. 2006;34:3819–3832. doi: 10.1093/nar/gkl476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidard FC, Vincent-Salomon A, Sigal-Zafrani B, Rodrigues M, Dieras V, Mignot L, Sastre-Garau X, Poupon MF, Pierga JY. Time to metastatic relapse and breast cancer cells dissemination in bone marrow at metastatic relapse. Clin Exp Metastasis. 2008;25:871–875. doi: 10.1007/s10585-008-9203-1. [DOI] [PubMed] [Google Scholar]
- Birck A, Ahrenkiel V, Zeuthen J, Hou-Jensen K, Guldberg P. Mutation and allelic loss of the PTEN/MMAC1 gene in primary and metastatic melanoma biopsies. J Invest Dermatol. 2000;114:277–280. doi: 10.1046/j.1523-1747.2000.00877.x. [DOI] [PubMed] [Google Scholar]
- Boisvert-Adamo K, Aplin AE. B-RAF and PI-3 kinase signaling protect melanoma cells from anoikis. Oncogene. 2006;25:4848–4856. doi: 10.1038/sj.onc.1209493. [DOI] [PubMed] [Google Scholar]
- Braun S, Vogl FD, Naume B, et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N Engl J Med. 2005;353:793–802. doi: 10.1056/NEJMoa050434. [DOI] [PubMed] [Google Scholar]
- Callejo SA, Antecka E, Blanco PL, Edelstein C, Burnier MN., Jr Identification of circulating malignant cells and its correlation with prognostic factors and treatment in uveal melanoma. A prospective longitudinal study. Eye. 2007;21:752–759. doi: 10.1038/sj.eye.6702322. [DOI] [PubMed] [Google Scholar]
- Cameron MD, Schmidt EE, Kerkvliet N, Nadkarni KV, Morris VL, Groom AC, Chambers AF, Macdonald IC. Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res. 2000;60:2541–2546. [PubMed] [Google Scholar]
- Chambers AF, Groom AC, Macdonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- Conner SR, Scott G, Aplin AE. Adhesion-dependent activation of the ERK1/2 cascade is by-passed in melanoma cells. J Biol Chem. 2003;278:34548–34554. doi: 10.1074/jbc.M305797200. [DOI] [PubMed] [Google Scholar]
- Crowley NJ, Seigler HF. Late recurrence of malignant melanoma. Analysis of 168 patients. Ann Surg. 1990a;212:173–177. doi: 10.1097/00000658-199008000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley NJ, Seigler HF. The role of elective lymph node dissection in the management of patients with thick cutaneous melanoma. Cancer. 1990b;66:2522–2527. doi: 10.1002/1097-0142(19901215)66:12<2522::aid-cncr2820661213>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Dahl C, Guldberg P. The genome and epigenome of malignant melanoma. APMIS. 2007;115:1161–1176. doi: 10.1111/j.1600-0463.2007.apm_855.xml.x. [DOI] [PubMed] [Google Scholar]
- Dai DL, Martinka M, Li G. Prognostic significance of activated Akt expression in melanoma: a clinicopathologic study of 292 cases. J Clin Oncol. 2005;23:1473–1482. doi: 10.1200/JCO.2005.07.168. [DOI] [PubMed] [Google Scholar]
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, You MJ, Depinho RA, Mcmahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–552. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- Deheuninck J, Luo K. Ski and SnoN, potent negative regulators of TGF-beta signaling. Cell Res. 2009;19:47–57. doi: 10.1038/cr.2008.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demicheli R. Tumour dormancy: findings and hypotheses from clinical research on breast cancer. Semin Cancer Biol. 2001;11:297–306. doi: 10.1006/scbi.2001.0385. [DOI] [PubMed] [Google Scholar]
- Deng X, Ewton DZ, Friedman E. Mirk/Dyrk1B maintains the viability of quiescent pancreatic cancer cells by reducing levels of reactive oxygen species. Cancer Res. 2009;69:3317–3324. doi: 10.1158/0008-5472.CAN-08-2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denoyelle C, Abou-Rjaily G, Bezrookove V, et al. Antioncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol. 2006;8:1053–1063. doi: 10.1038/ncb1471. [DOI] [PubMed] [Google Scholar]
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am. 2009;23:529–545. ix. doi: 10.1016/j.hoc.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Dhomen N, Reis-Filho JS, Da Rocha Dias S, Hayward R, Savage K, Delmas V, Larue L, Pritchard C, Marais R. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Dimos JT, Rodolfa KT, Niakan KK, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- Ducos K, Panterne B, Fortunel N, Hatzfeld A, Monier MN, Hatzfeld J. p21(cip1) mRNA is controlled by endogenous transforming growth factor-beta1 in quiescent human hematopoietic stem/progenitor cells. J Cell Physiol. 2000;184:80–85. doi: 10.1002/(SICI)1097-4652(200007)184:1<80::AID-JCP8>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Lewis JS, Jr, Sunwoo JB, Uppaluri R. Spontaneous regression of cutaneous head and neck melanoma: implications for the immunologic control of neoplasia. Head Neck. 2008;30:267–272. doi: 10.1002/hed.20701. [DOI] [PubMed] [Google Scholar]
- Eskelin S, Pyrhonen S, Summanen P, Hahka-Kemppinen M, Kivela T. Tumor doubling times in metastatic malignant melanoma of the uvea: tumor progression before and after treatment. Ophthalmology. 2000;107:1443–1449. doi: 10.1016/s0161-6420(00)00182-2. [DOI] [PubMed] [Google Scholar]
- Estrada Y, Dong J, Ossowski L. Positive crosstalk between ERK and p38 in melanoma stimulates migration and in vivo proliferation. Pigment Cell Melanoma Res. 2009;22:66–76. doi: 10.1111/j.1755-148X.2008.00520.x. [DOI] [PubMed] [Google Scholar]
- Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, Van Belle PA, Xu X, Elder DE, Herlyn M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005;65:9328–9337. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- Felsher DW. Oncogene addiction versus oncogene amnesia: perhaps more than just a bad habit. Cancer Res. 2008a;68:3081–3086. 3086. doi: 10.1158/0008-5472.CAN-07-5832. discussion. [DOI] [PubMed] [Google Scholar]
- Felsher DW. Tumor dormancy and oncogene addiction. APMIS. 2008b;116:629–637. doi: 10.1111/j.1600-0463.2008.01037.x. [DOI] [PubMed] [Google Scholar]
- Ferrari D, Lombardi M, Ricci R, Michiara M, Santini M, De Panfilis G. Dermatopathological indicators of poor melanoma prognosis are significantly inversely correlated with the expression of NM23 protein in primary cutaneous melanoma. J Cutan Pathol. 2007;34:705–712. doi: 10.1111/j.1600-0560.2006.00692.x. [DOI] [PubMed] [Google Scholar]
- Fidler IJ. The Ernst W. Bertner Memorial Award lecture: the evolution of biological heterogeneity in metastatic neoplasms. Symp Fundam Cancer Res. 1983;36:5–26. [PubMed] [Google Scholar]
- Finn OJ. Human tumor antigens, immunosurveillance, and cancer vaccines. Immunol Res. 2006;36:73–82. doi: 10.1385/IR:36:1:73. [DOI] [PubMed] [Google Scholar]
- Fisher B, Fisher ER. Experimental evidence in support of the dormant tumor cell. Science. 1959;130:918–919. doi: 10.1126/science.130.3380.918. [DOI] [PubMed] [Google Scholar]
- Florenes VA, Bhattacharya N, Bani MR, Ben-David Y, Kerbel RS, Slingerland JM. TGF-beta mediated G1 arrest in a human melanoma cell line lacking p15INK4B: evidence for cooperation between p21Cip1/WAF1 and p27Kip1. Oncogene. 1996;13:2447–2457. [PubMed] [Google Scholar]
- Foulds L. Tumor progression. Cancer Res. 1957;17:355–356. [PubMed] [Google Scholar]
- Frank NY, Margaryan A, Huang Y, Schatton T, Waaga-Gasser AM, Gasser M, Sayegh MH, Sadee W, Frank MH. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res. 2005;65:4320–4333. doi: 10.1158/0008-5472.CAN-04-3327. [DOI] [PubMed] [Google Scholar]
- Gajewski TF. Identifying and overcoming immune resistance mechanisms in the melanoma tumor microenvironment. Clin Cancer Res. 2006;12:2326s–2330s. doi: 10.1158/1078-0432.CCR-05-2517. [DOI] [PubMed] [Google Scholar]
- Gamel JW, George SL, Edwards MJ, Seigler HF. The long-term clinical course of patients with cutaneous melanoma. Cancer. 2002;95:1286–1293. doi: 10.1002/cncr.10813. [DOI] [PubMed] [Google Scholar]
- Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- Grichnik JM. Melanoma, nevogenesis, and stem cell biology. J Invest Dermatol. 2008;128:2365–2380. doi: 10.1038/jid.2008.166. [DOI] [PubMed] [Google Scholar]
- Guldberg P, Thor Straten P, Birck A, Ahrenkiel V, Kirkin AF, Zeuthen J. Disruption of the MMAC1/PTEN gene by deletion or mutation is a frequent event in malignant melanoma. Cancer Res. 1997;57:3660–3663. [PubMed] [Google Scholar]
- Gutman M, Klausner JM, Inbar M, Rozin RR. Late recurrence of stage I malignant melanoma. J Surg Oncol. 1989;42:96–98. doi: 10.1002/jso.2930420206. [DOI] [PubMed] [Google Scholar]
- Haluska F, Pemberton T, Ibrahim N, Kalinsky K. The RTK/RAS/BRAF/PI3K pathways in melanoma: biology, small molecule inhibitors, and potential applications. Semin Oncol. 2007;34:546–554. doi: 10.1053/j.seminoncol.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Henriet P, Zhong ZD, Brooks PC, Weinberg KI, Declerck YA. Contact with fibrillar collagen inhibits melanoma cell proliferation by up-regulating p27KIP1. Proc Natl Acad Sci USA. 2000;97:10026–10031. doi: 10.1073/pnas.170290997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek KS, Eichhoff OM, Schlegel NC, Dobbeling U, Kobert N, Schaerer L, Hemmi S, Dummer R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008;68:650–656. doi: 10.1158/0008-5472.CAN-07-2491. [DOI] [PubMed] [Google Scholar]
- Hsu MY, Meier FE, Nesbit M, Hsu JY, Van Belle P, Elder DE, Herlyn M. E-cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. Am J Pathol. 2000;156:1515–1525. doi: 10.1016/S0002-9440(10)65023-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu MY, Rovinsky S, Penmatcha S, Herlyn M, Muirhead D. Bone morphogenetic proteins in melanoma: angel or devil? Cancer Metastasis Rev. 2005;24:251–263. doi: 10.1007/s10555-005-1575-y. [DOI] [PubMed] [Google Scholar]
- Husemann Y, Geigl JB, Schubert F, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13:58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Hussein MR. Transforming growth factor-beta and malignant melanoma: molecular mechanisms. J Cutan Pathol. 2005;32:389–395. doi: 10.1111/j.0303-6987.2005.00356.x. [DOI] [PubMed] [Google Scholar]
- Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- Javelaud D, Alexaki VI, Mauviel A. Transforming growth factor-beta in cutaneous melanoma. Pigment Cell Melanoma Res. 2008;21:123–132. doi: 10.1111/j.1755-148X.2008.00450.x. [DOI] [PubMed] [Google Scholar]
- Kalinec G, Nazarali AJ, Hermouet S, Xu N, Gutkind JS. Mutated alpha subunit of the Gq protein induces malignant transformation in NIH 3T3 cells. Mol Cell Biol. 1992;12:4687–4693. doi: 10.1128/mcb.12.10.4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinsky K, Haluska FG. Novel inhibitors in the treatment of metastatic melanoma. Expert Rev Anticancer Ther. 2007;7:715–724. doi: 10.1586/14737140.7.5.715. [DOI] [PubMed] [Google Scholar]
- Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science. 2007;317:337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- Kim JB, Zaehres H, Wu G, et al. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature. 2008;454:646–650. doi: 10.1038/nature07061. [DOI] [PubMed] [Google Scholar]
- Klein CA. The direct molecular analysis of metastatic precursor cells in breast cancer: a chance for a better understanding of metastasis and for personalised medicine. Eur J Cancer. 2008;44:2721–2725. doi: 10.1016/j.ejca.2008.09.035. [DOI] [PubMed] [Google Scholar]
- Klein CA, Hölzel D. Systemic cancer progression and tumor dormancy: mathematical models meet single cell genomics. Cell Cycle. 2006;5:1788–1798. doi: 10.4161/cc.5.16.3097. [DOI] [PubMed] [Google Scholar]
- Klein WM, Wu BP, Zhao S, Wu H, Klein-Szanto AJ, Tahan SR. Increased expression of stem cell markers in malignant melanoma. Mod Pathol. 2007;20:102–107. doi: 10.1038/modpathol.3800720. [DOI] [PubMed] [Google Scholar]
- Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–907. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- Kortylewski M, Heinrich PC, Kauffmann ME, Bohm M, Mackiewicz A, Behrmann I. Mitogen-activated protein kinases control p27/Kip1 expression and growth of human melanoma cells. Biochem J. 2001;357:297–303. doi: 10.1042/0264-6021:3570297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Angelini S, Czene K, Sauroja I, Hahka-Kemppinen M, Pyrhonen S, Hemminki K. BRAF mutations in metastatic melanoma: a possible association with clinical outcome. Clin Cancer Res. 2003;9:3362–3368. [PubMed] [Google Scholar]
- Lahlou H, Sanguin-Gendreau V, Zuo D, Cardiff RD, Mclean GW, Frame MC, Muller WJ. Mammary epithelial-specific disruption of the focal adhesion kinase blocks mammary tumor progression. Proc Natl Acad Sci USA. 2007;104:20302–20307. doi: 10.1073/pnas.0710091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Scolan E, Zhu Q, Wang L, Bandyopadhyay A, Javelaud D, Mauviel A, Sun L, Luo K. Transforming growth factor-beta suppresses the ability of Ski to inhibit tumor metastasis by inducing its degradation. Cancer Res. 2008;68:3277–3285. doi: 10.1158/0008-5472.CAN-07-6793. [DOI] [PubMed] [Google Scholar]
- Lefevre G, Calipel A, Mouriaux F, Hecquet C, Malecaze F, Mascarelli F. Opposite long-term regulation of c-Myc and p27Kip1 through overactivation of Raf-1 and the MEK/ERK module in proliferating human choroidal melanoma cells. Oncogene. 2003;22:8813–8822. doi: 10.1038/sj.onc.1207099. [DOI] [PubMed] [Google Scholar]
- Lengagne R, Graff-Dubois S, Garcette M, Renia L, Kato M, Guillet JG, Engelhard VH, Avril MF, Abastado JP, Prevost-Blondel A. Distinct role for CD8 T cells toward cutaneous tumors and visceral metastases. J Immunol. 2008;180:130–137. doi: 10.4049/jimmunol.180.1.130. [DOI] [PubMed] [Google Scholar]
- Liang J, Zubovitz J, Petrocelli T, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–1160. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- Liang J, Shao SH, Xu ZX, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- Lin HK, Wang G, Chen Z, et al. Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of Skp2 by Akt/PKB. Nat Cell Biol. 2009;11:420–432. doi: 10.1038/ncb1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Aguirre Ghiso J, Estrada Y, Ossowski L. EGFR is a transducer of the urokinase receptor initiated signal that is required for in vivo growth of a human carcinoma. Cancer Cell. 2002;1:445–457. doi: 10.1016/s1535-6108(02)00072-7. [DOI] [PubMed] [Google Scholar]
- Liu ZJ, Xiao M, Balint K, Smalley KS, Brafford P, Qiu R, Pinnix CC, Li X, Herlyn M. Notch1 signaling promotes primary melanoma progression by activating mitogen-activated protein kinase/phosphatidylinositol 3-kinase-Akt pathways and up-regulating N-cadherin expression. Cancer Res. 2006;66:4182–4190. doi: 10.1158/0008-5472.CAN-05-3589. [DOI] [PubMed] [Google Scholar]
- Logan PT, Fernandes BF, Di Cesare S, Marshall JC, Maloney SC, Burnier MN., Jr Single-cell tumor dormancy model of uveal melanoma. Clin Exp Metastasis. 2008;25:509–516. doi: 10.1007/s10585-008-9158-2. [DOI] [PubMed] [Google Scholar]
- Lopez-Bergami P, Huang C, Goydos JS, et al. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell. 2007;11:447–460. doi: 10.1016/j.ccr.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bergami P, Fitchman B, Ronai Z. Understanding signaling cascades in melanoma. Photochem Photobiol. 2008;84:289–306. doi: 10.1111/j.1751-1097.2007.00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorigan P, Eisen T, Hauschild A. Systemic therapy for metastatic malignant melanoma–from deeply disappointing to bright future? Exp Dermatol. 2008;17:383–394. doi: 10.1111/j.1600-0625.2007.00673.x. [DOI] [PubMed] [Google Scholar]
- Mackie RM, Reid R, Junor B. Fatal melanoma transferred in a donated kidney 16 years after melanoma surgery. N Engl J Med. 2003;348:567–568. doi: 10.1056/NEJM200302063480620. [DOI] [PubMed] [Google Scholar]
- Mahabeleshwar GH, Byzova TV. Angiogenesis in melanoma. Semin Oncol. 2007;34:555–565. doi: 10.1053/j.seminoncol.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahnke YD, Schwendemann J, Beckhove P, Schirrmacher V. Maintenance of long-term tumour-specific T-cell memory by residual dormant tumour cells. Immunology. 2005;115:325–336. doi: 10.1111/j.1365-2567.2005.02163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marches R, Scheuermann RH, Uhr JW. Cancer dormancy: role of cyclin-dependent kinase inhibitors in induction of cell cycle arrest mediated via membrane IgM. Cancer Res. 1998;58:691–697. [PubMed] [Google Scholar]
- Marches R, Hsueh R, Uhr JW. Cancer dormancy and cell signaling: induction of p21(waf1) initiated by membrane IgM engagement increases survival of B lymphoma cells. Proc Natl Acad Sci USA. 1999;96:8711–8715. doi: 10.1073/pnas.96.15.8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marches R, Scheuermann RH, Uhr J. Cancer dormancy from mice to man: a review. Cell Cycle. 2006;5:1772–1778. doi: 10.4161/cc.5.16.2995. [DOI] [PubMed] [Google Scholar]
- Mirmohammadsadegh A, Marini A, Nambiar S, Hassan M, Tannapfel A, Ruzicka T, Hengge UR. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res. 2006;66:6546–6552. doi: 10.1158/0008-5472.CAN-06-0384. [DOI] [PubMed] [Google Scholar]
- Monzani E, Facchetti F, Galmozzi E, et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur J Cancer. 2007;43:935–946. doi: 10.1016/j.ejca.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Nash KT, Phadke PA, Navenot JM, et al. Requirement of KISS1 secretion for multiple organ metastasis suppression and maintenance of tumor dormancy. J Natl Cancer Inst. 2007;99:309–321. doi: 10.1093/jnci/djk053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumov GN, Akslen LA, Folkman J. Role of angiogenesis in human tumor dormancy: animal models of the angiogenic switch. Cell Cycle. 2006;5:1779–1787. doi: 10.4161/cc.5.16.3018. [DOI] [PubMed] [Google Scholar]
- Nickoloff BJ, Hendrix MJ, Pollock PM, Trent JM, Miele L, Qin JZ. Notch and NOXA-related pathways in melanoma cells. J Investig Dermatol Symp Proc. 2005;10:95–104. doi: 10.1111/j.1087-0024.2005.200404.x. [DOI] [PubMed] [Google Scholar]
- Niederkorn JY. Immune escape mechanisms of intraocular tumors. Prog Retin Eye Res. 2009;28:329–347. doi: 10.1016/j.preteyeres.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell PC. Tumor progression: a brief historical perspective. Semin Cancer Biol. 2002;12:261–266. doi: 10.1016/s1044-579x(02)00012-3. [DOI] [PubMed] [Google Scholar]
- Odoux C, Fohrer H, Hoppo T, Guzik L, Stolz DB, Lewis DW, Gollin SM, Gamblin TC, Geller DA, Lagasse E. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res. 2008;68:6932–6941. doi: 10.1158/0008-5472.CAN-07-5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onken MD, Worley LA, Long MD, Duan S, Council ML, Bowcock AM, Harbour JW. Oncogenic mutations in GNAQ occur early in uveal melanoma. Invest Ophthalmol Vis Sci. 2008;49:5230–5234. doi: 10.1167/iovs.08-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski L, Reich E. Loss of malignancy during serial passage of human carcinoma in culture and discordance between malignancy and transformation parameters. Cancer Res. 1980;40:2310–2315. [PubMed] [Google Scholar]
- Ossowski L, Reich E. Changes in malignant phenotype of a human carcinoma conditioned by growth environment. Cell. 1983;33:323–333. doi: 10.1016/0092-8674(83)90414-2. [DOI] [PubMed] [Google Scholar]
- Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;1:571–573. [PubMed] [Google Scholar]
- Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004;4:448–456. doi: 10.1038/nrc1370. [DOI] [PubMed] [Google Scholar]
- Pantel K, Alix-Panabieres C, Riethdorf S. Cancer micrometastases. Nat Rev Clin Oncol. 2009;6:339–351. doi: 10.1038/nrclinonc.2009.44. [DOI] [PubMed] [Google Scholar]
- Pelayo R, Miyazaki K, Huang J, Garrett KP, Osmond DG, Kincade PW. Cell cycle quiescence of early lymphoid progenitors in adult bone marrow. Stem Cells. 2006;24:2703–2713. doi: 10.1634/stemcells.2006-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn I. Malignant melanoma in organ allograft recipients. Transplantation. 1996;61:274–278. doi: 10.1097/00007890-199601270-00019. [DOI] [PubMed] [Google Scholar]
- Peters A, Lippold A, Hundeiker M. First melanoma metastases after 10 years and more of remission. Hautarzt. 1997;48:311–317. doi: 10.1007/s001050050588. [DOI] [PubMed] [Google Scholar]
- Pharis DB. Cutaneous melanoma: therapeutic lymph node and elective lymph node dissections, lymphatic mapping, and sentinel lymph node biopsy. Dermatol Ther. 2005;18:397–406. doi: 10.1111/j.1529-8019.2005.00046.x. [DOI] [PubMed] [Google Scholar]
- Pinnix CC, Lee JT, Liu ZJ, et al. Active Notch1 confers a transformed phenotype to primary human melanocytes. Cancer Res. 2009;69:5312–5320. doi: 10.1158/0008-5472.CAN-08-3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak ME, Borthwick NJ, Jager MJ, Cree IA. Melanoma vaccines: the problems of local immunosuppression. Hum Immunol. 2009;70:331–339. doi: 10.1016/j.humimm.2009.01.017. [DOI] [PubMed] [Google Scholar]
- Postovit LM, Seftor EA, Seftor RE, Hendrix MJ. A three-dimensional model to study the epigenetic effects induced by the microenvironment of human embryonic stem cells. Stem Cells. 2006;24:501–505. doi: 10.1634/stemcells.2005-0459. [DOI] [PubMed] [Google Scholar]
- Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovsky R, Uhr JW, Vitetta ES, Yefenof E. Cancer dormancy: lessons from a B cell lymphoma and adenocarcinoma of the prostate. Adv Cancer Res. 2007;97:189–202. doi: 10.1016/S0065-230X(06)97008-0. [DOI] [PubMed] [Google Scholar]
- Ralph SJ. An update on malignant melanoma vaccine research: insights into mechanisms for improving the design and potency of melanoma therapeutic vaccines. Am J Clin Dermatol. 2007;8:123–141. doi: 10.2165/00128071-200708030-00001. [DOI] [PubMed] [Google Scholar]
- Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006;66:1702–1711. doi: 10.1158/0008-5472.CAN-05-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JA, Bales E, Xu W, Okan NA, Bandyopadhyay D, Medrano EE. Cytoplasmic localization of the oncogenic protein Ski in human cutaneous melanomas in vivo: functional implications for transforming growth factor beta signaling. Cancer Res. 2001;61:8074–8078. [PubMed] [Google Scholar]
- Richards HW, Medrano EE. Epigenetic marks in melanoma. Pigment Cell Melanoma Res. 2009;22:14–29. doi: 10.1111/j.1755-148X.2008.00534.x. [DOI] [PubMed] [Google Scholar]
- Rosen JM, Jordan CT. The increasing complexity of the cancer stem cell paradigm. Science. 2009;324:1670–1673. doi: 10.1126/science.1171837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt MI, Azar DT. Anti-angiogenic therapy: prospects for treatment of ocular tumors. Semin Ophthalmol. 2006;21:151–160. doi: 10.1080/08820530500350787. [DOI] [PubMed] [Google Scholar]
- Rothhammer T, Bosserhoff AK. Epigenetic events in malignant melanoma. Pigment Cell Res. 2007;20:92–111. doi: 10.1111/j.1600-0749.2007.00367.x. [DOI] [PubMed] [Google Scholar]
- Sabatino M, Stroncek DF, Klein H, Marincola FM, Wang E. Stem cells in melanoma development. Cancer Lett. 2009;279:119–125. doi: 10.1016/j.canlet.2008.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang L, Coller HA, Roberts JM. Control of the reversibility of cellular quiescence by the transcriptional repressor HES1. Science. 2008;321:1095–1100. doi: 10.1126/science.1155998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanki A, Li W, Colman M, Karim RZ, Thompson JF, Scolyer RA. Reduced expression of p16 and p27 is correlated with tumour progression in cutaneous melanoma. Pathology. 2007;39:551–557. doi: 10.1080/00313020701684409. [DOI] [PubMed] [Google Scholar]
- Saudemont A, Quesnel B. In a model of tumor dormancy, long-term persistent leukemic cells have increased B7-H1 and B7.1 expression and resist CTL-mediated lysis. Blood. 2004;104:2124–2133. doi: 10.1182/blood-2004-01-0064. [DOI] [PubMed] [Google Scholar]
- Schardt JA, Meyer M, Hartmann CH, Schubert F, Schmidt-Kittler O, Fuhrmann C, Polzer B, Petronio M, Eils R, Klein CA. Genomic analysis of single cytokeratin-positive cells from bone marrow reveals early mutational events in breast cancer. Cancer Cell. 2005;8:227–239. doi: 10.1016/j.ccr.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Schatton T, Murphy GF, Frank NY, et al. Identification of cells initiating human melanomas. Nature. 2008;451:345–349. doi: 10.1038/nature06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schewe DM, Aguirre-Ghiso JA. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci USA. 2008;105:10519–10524. doi: 10.1073/pnas.0800939105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel NC, Eichhoff OM, Hemmi S, Werner S, Dummer R, Hoek KS. Id2 suppression of p15 counters TGF-beta-mediated growth inhibition of melanoma cells. Pigment Cell Melanoma Res. 2009;22:445–453. doi: 10.1111/j.1755-148X.2009.00571.x. [DOI] [PubMed] [Google Scholar]
- Schmidt-Kittler O, Ragg T, Daskalakis A, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci USA. 2003;100:7737–7742. doi: 10.1073/pnas.1331931100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid-Wendtner MH, Baumert J, Schmidt M, Konz B, Holzel D, Plewig G, Volkenandt M. Late metastases of cutaneous melanoma: an analysis of 31 patients. J Am Acad Dermatol. 2000;43:605–609. doi: 10.1067/mjd.2000.107234. [DOI] [PubMed] [Google Scholar]
- Schuster R, Bechrakis NE, Stroux A, Busse A, Schmittel A, Scheibenbogen C, Thiel E, Foerster MH, Keilholz U. Circulating tumor cells as prognostic factor for distant metastases and survival in patients with primary uveal melanoma. Clin Cancer Res. 2007;13:1171–1178. doi: 10.1158/1078-0432.CCR-06-2329. [DOI] [PubMed] [Google Scholar]
- Sharma SV, Gajowniczek P, Way IP, Lee DY, Jiang J, Yuza Y, Classon M, Haber DA, Settleman J. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell. 2006;10:425–435. doi: 10.1016/j.ccr.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen P, Guenther JM, Wanek LA, Morton DL. Can elective lymph node dissection decrease the frequency and mortality rate of late melanoma recurrences? Ann Surg Oncol. 2000;7:114–119. doi: 10.1007/s10434-000-0114-x. [DOI] [PubMed] [Google Scholar]
- Shibue T, Weinberg RA. Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci USA. 2009;106:10290–10295. doi: 10.1073/pnas.0904227106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shildkrot Y, Wilson MW. Update on posterior uveal melanoma: treatment of the eye and emerging strategies in the prognosis and treatment of metastatic disease. Curr Opin Ophthalmol. 2009;20:504–510. doi: 10.1097/ICU.0b013e328330b549. [DOI] [PubMed] [Google Scholar]
- Shinozaki M, Fujimoto A, Morton DL, Hoon DS. Incidence of BRAF oncogene mutation and clinical relevance for primary cutaneous melanomas. Clin Cancer Res. 2004;10:1753–1757. doi: 10.1158/1078-0432.ccr-1169-3. [DOI] [PubMed] [Google Scholar]
- Stoecklein NH, Hosch SB, Bezler M, et al. Direct genetic analysis of single disseminated cancer cells for prediction of outcome and therapy selection in esophageal cancer. Cancer Cell. 2008;13:441–453. doi: 10.1016/j.ccr.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Terando AM, Faries MB, Morton DL. Vaccine therapy for melanoma: current status and future directions. Vaccine. 2007;25(Suppl 2):B4–B16. doi: 10.1016/j.vaccine.2007.06.033. [DOI] [PubMed] [Google Scholar]
- Townson JL, Chambers AF. Dormancy of solitary metastatic cells. Cell Cycle. 2006;5:1744–1750. doi: 10.4161/cc.5.16.2864. [DOI] [PubMed] [Google Scholar]
- Triozzi PL, Eng C, Singh AD. Targeted therapy for uveal melanoma. Cancer Treat Rev. 2008;34:247–258. doi: 10.1016/j.ctrv.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Tsao H, Cosimi AB, Sober AJ. Ultra-late recurrence (15 years or longer) of cutaneous melanoma. Cancer. 1997;79:2361–2370. [PubMed] [Google Scholar]
- Ulmer A, Beutel J, Susskind D, et al. Visualization of circulating melanoma cells in peripheral blood of patients with primary uveal melanoma. Clin Cancer Res. 2008;14:4469–4474. doi: 10.1158/1078-0432.CCR-08-0012. [DOI] [PubMed] [Google Scholar]
- Umansky V, Abschuetz O, Osen W, Ramacher M, Zhao F, Kato M, Schadendorf D. Melanoma-specific memory T cells are functionally active in Ret transgenic mice without macroscopic tumors. Cancer Res. 2008;68:9451–9458. doi: 10.1158/0008-5472.CAN-08-1464. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O'brien JM, Simpson EM, Barsh GS, Bastian BC. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessella RL, Pantel K, Mohla S. Tumor cell dormancy: an NCI workshop report. Cancer Biol Ther. 2007;6:1496–1504. doi: 10.4161/cbt.6.9.4828. [DOI] [PubMed] [Google Scholar]
- Visús C, Andres R, Mayordomo JI, et al. Prognostic role of circulating melanoma cells detected by reverse transcriptase-polymerase chain reaction for tyrosinase mRNA in patients with melanoma. Melanoma Res. 2007;73:83–89. doi: 10.1097/CMR.0b013e3280a60878. [DOI] [PubMed] [Google Scholar]
- Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–374. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall SJ, Werner E, Werb Z, Declerck YA. Discoidin domain receptor 2 mediates tumor cell cycle arrest induced by fibrillar collagen. J Biol Chem. 2005;280:40187–40194. doi: 10.1074/jbc.M508226200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall SJ, Zhong ZD, Declerck YA. The cyclin-dependent kinase inhibitors p15INK4B and p21CIP1 are critical regulators of fibrillar collagen-induced tumor cell cycle arrest. J Biol Chem. 2007;282:24471–24476. doi: 10.1074/jbc.M702697200. [DOI] [PubMed] [Google Scholar]
- Weinstein IB. Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- White DE, Kurpios NA, Zuo D, Hassell JA, Blaess S, Mueller U, Muller WJ. Targeted disruption of beta1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell. 2004;6:159–170. doi: 10.1016/j.ccr.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Whiteman DC, Zhou XP, Cummings MC, Pavey S, Hayward NK, Eng C. Nuclear PTEN expression and clinicopathologic features in a population-based series of primary cutaneous melanoma. Int J Cancer. 2002;99:63–67. doi: 10.1002/ijc.10294. [DOI] [PubMed] [Google Scholar]
- Wikman H, Vessella R, Pantel K. Cancer micrometastasis and tumour dormancy. APMIS. 2008;116:754–770. doi: 10.1111/j.1600-0463.2008.01033.x. [DOI] [PubMed] [Google Scholar]
- Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437:141–146. doi: 10.1038/nature03954. [DOI] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Oser G, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- Xia S, Lu Y, Wang J, He C, Hong S, Serhan CN, Kang JX. Melanoma growth is reduced in fat-1 transgenic mice: impact of omega-6/omega-3 essential fatty acids. Proc Natl Acad Sci USA. 2006;103:12499–12504. doi: 10.1073/pnas.0605394103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapas JL, Coley HC, Beam SL, Brown SD, Jablonski KA, Elias EG. The risk of regional lymph node metastases in patients with melanoma less than 1.0 mm thick: recommendations for sentinel lymph node biopsy. J Am Coll Surg. 2003;197:403–407. doi: 10.1016/S1072-7515(03)00432-0. [DOI] [PubMed] [Google Scholar]
- Zhou XP, Gimm O, Hampel H, Niemann T, Walker MJ, Eng C. Epigenetic PTEN silencing in malignant melanomas without PTEN mutation. Am J Pathol. 2000;157:1123–1128. doi: 10.1016/S0002-9440(10)64627-5. [DOI] [PMC free article] [PubMed] [Google Scholar]