

Abstract
Mutations in the gene encoding TDP-43—the major protein component of neuronal aggregates characteristic of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) with ubiquitin-positive inclusion bodies—have been linked to familial forms of both disorders. Aggregates of TDP-43 in cortical and spinal motorneurons in ALS, or in neurons of the frontal and temporal cortices in FTLD, are closely linked to neuron loss and atrophy in these areas. However, the mechanism by which TDP-43 mutations lead to neurodegeneration is unclear. To investigate the pathogenic role of TDP-43 mutations, we established a model of TDP-43 proteinopathies by expressing fluorescently tagged wild-type and mutant TDP-43 in primary rat cortical neurons. Expression of mutant TDP-43 was toxic to neurons, and mutant-specific toxicity was associated with increased cytoplasmic mislocalization of TDP-43. Inclusion bodies were not necessary for the toxicity and did not affect the risk of cell death. Cellular survival was unaffected by the total amount of exogenous TDP-43 in the nucleus, but the amount of cytoplasmic TDP-43 was a strong and independent predictor of neuronal death. These results suggest that mutant TDP-43 is mislocalized to the cytoplasm, where it exhibits a toxic gain-of-function and induces cell death.
Introduction
TAR-DNA binding protein of 43 kDa (TDP-43) is the major component of protein aggregates in patients with sporadic and most familial amyotrophic lateral sclerosis (ALS) and in a subclass of patients with frontotemporal lobar degeneration (FTLD) characterized by ubiquitin-positive inclusions (Arai et al., 2006; Neumann et al., 2006). The identification of TDP-43-immunopositive inclusions in the majority of FTLD and ALS cases suggested a pathogenic link between these disorders and prompted their reclassification as TDP-43-proteinopathies. Moreover, mutations in the gene encoding TDP-43 are associated with familial ALS and FTLD with motor neuron disease, strengthening the conclusion that this protein plays a central role in disease pathogenesis (Kabashi et al., 2008; Sreedharan et al., 2008; Van Deerlin et al., 2008; Yokoseki et al., 2008; Benajiba et al., 2009).
TDP-43 is a widely expressed nuclear protein that binds DNA and RNA, inhibits retroviral replication, and participates in RNA splicing and nuclear body formation (Ou et al., 1995; Buratti and Baralle, 2008). In healthy cells, TDP-43 is largely restricted to the nucleus; however, TDP-43 is also involved in nucleocytoplasmic shuttling of messenger RNA (Ayala et al., 2008; Wang et al., 2008), and TDP-43-containing cytoplasmic puncta have been detected in association with RNA granules (Elvira et al., 2006). In patients with TDP-43-proteinopathies, such as ALS and FTLD with ubiquitin inclusions, affected neurons exhibit a striking redistribution of TDP-43 from the nucleus to the cytoplasm and display insoluble TDP-43 aggregates in the nucleus, cytoplasm, or processes (Arai et al., 2006; Neumann et al., 2006).
The significance of the redistribution of TDP-43 is unclear, and despite the growing number of disease-associated mutations in TDP-43, little is known about the mechanisms by which they lead to neurodegeneration. Previous studies using conventional approaches were limited in their ability to establish causality and unable to determine whether aggregation and redistribution of TDP-43 are essential features of disease pathogenesis or epiphenomena. To further characterize TDP-43-mediated disorders, we established a model of TDP-43-proteinopathies by expressing fluorescently labeled TDP-43 in rodent primary cortical neurons. Primary culture models permit detailed genetic and biochemical manipulations in postmitotic, differentiated cells and have proven essential in studies of several neurodegenerative disorders (Saudou et al., 1998; Jacquier et al., 2009). For these investigations, we used an automated microscopy system that enables longitudinal visualization of individual neurons over extended periods. This approach permits us to quantitatively relate morphologic changes in each neuron to survival and to predict the consequences of those changes (Arrasate and Finkbeiner, 2005). Our findings reveal that mutant TDP-43 is toxic to neurons and that mislocalization of TDP-43 to the cytoplasm is critical for disease pathogenesis.
Materials and Methods
Plasmids.
All TDP-43-bearing plasmids consisted of human TDP-43 fused at the C terminus to enhanced green fluorescent protein (EGFP) (Fig. 1A). A linker region of 14 aa separates the C terminus of TDP-43 from the N terminus of enhanced green fluorescent protein (EGFP), and six histidine residues were added to the C terminus of EGFP to facilitate purification of the translated protein. The A315T mutation was created by site-directed mutagenesis of the adenine base at position 943 to guanine. The nuclear localization signal (NLS) was mutated by PCR as described previously (Winton et al., 2008) with the following primers: 5′-CAACTATCCAAAAGATAACGCAGCAGCAATGGATGAGACAGATGC-3′ and 5′-GCATCTGTCTCATCCATTGCTGCTGCGTTATCTTTTGGATAGTTG-3′. The nuclear export signal (NES) was disrupted similarly with the following primers: 5′-GCAGTCTCTTTGTGGAGAGGACGGAGGGGGAAAAGGAATAGCGTTCATATATC-3′ and 5′-GATATATGAACGCTGATTCCTTTTCCCCCTCCGTCCTCTCCACAAAGAGACTC-3′. Mutant huntingtin, mCherry, and EGFP were cloned into pGW1-CMV vectors as described previously (Arrasate et al., 2004). Sequence information for all plasmids is available at http://gind-db.ucsf.edu:8000/cgi-bin/Plasmid/main_menu2.cgi.
Figure 1.

TDP43-EGFP is localized to the nucleus when expressed in primary cortical neurons. A, Schematic of TDP43-EGFP showing the RNA-recognition motifs (RRMs) 1 and 2, as well as the glycine-rich domain (GRD). EGFP is fused to the C terminus of the protein. The red line denotes the approximate location of the A315T mutation. B–G, Neurons expressing TDP43(WT)-EGFP (B–D) or TDP43(A315T)-EGFP (E–G) demonstrate nuclear localization of the protein when imaged by fluorescence microscopy. B, E, EGFP fluorescence. C, F, mCherry fluorescence. D, G, Merged images with EGFP fluorescence in green, mCherry in red, and overlap in yellow. Scale bar, 10 μm.
Cell culture and transfection.
Rat cortical neurons were isolated from embryonic d 20–21 pups, cultured at 0.6 × 106 cells/ml for 6 d in vitro, and transfected with calcium phosphate as described previously (Saudou et al., 1998) (specific protocol information is available at http://www.gladstone.ucsf.edu/gladstone/site/finkbeiner/section/1193). For survival analysis, neurons in 24 well plates were cotransfected with TDP-43-containing plasmids and pGW1-mCherry in a 1:1 molar ratio (1 μg of DNA per well) and cultured in serum-free medium for the remainder of the analysis. For confocal fluorescence microscopy, neurons were transfected with selected plasmids and visualized 24 h later. For evaluation of the detergent-resistance of inclusion bodies (IBs), neurons were fixed with 1% paraformaldehyde in PBS for 20 min at 37°C, rinsed twice in PBS, and treated with 5% Triton X-100 and 5% SDS for 20 min at 37°C. Neurons were then rinsed twice more in PBS and imaged by fluorescence microscopy (Kazantsev et al., 1999).
Immunocytochemistry.
Rat cortical neurons were isolated and plated onto 12 mm glass coverslips coated with laminin and poly-d-lysine. At 6 d in vitro, neurons were transfected with TDP-43-containing plasmids as described above. Immunocytochemistry was performed as described previously (Saudou et al., 1998) with rabbit anti-TDP-43 polyclonal antibodies (Proteintech; 1:1000) or anti-myc (Santa Cruz Biotechnology; 1:2500) as primary antibodies and anti-rabbit Cy3-labeled secondary antibodies (Jackson ImmunoResearch Laboratories; 1:200). Nuclear staining was accomplished by incubating fixed and permeabilized cells briefly with Hoechst dye diluted to a final concentration of 10 μm in PBS.
Subcellular fractionation.
For fractionation experiments, HEK293 cells were plated onto 15 cm Petri dishes and transfected after 24 h in vitro. The cells were rinsed once with ice-cold saline, and harvested in 5 ml of chilled Buffer 1 (containing, in mm: 15 Tris-HCl, pH 7.5, 60 KCl, 15 NaCl, 5 MgCl20, 1 CaCl2, 2 Na3VO4, 0.25 m sucrose, 1 PMSF; 1 DTT). After a 5 min incubation on ice, an equal volume of Buffer 1 with 0.6% NP-40 was added to the solution, and the sample was incubated for an additional 5 min on ice, and stirred occasionally. The suspension was then centrifuged at 1000 × g for 10 min at 4°C. The supernatant was removed, and set aside as the cytosolic fraction. The pellet was gently resuspended in Buffer 1, and centrifuged as before. The supernatant was removed, and the pellet resuspended in Buffer 2 (containing, in mm: 10 HEPES, pH 7.9, 10 KCl, 1.5 MgCl2, 0.1 EGTA, 0.4 NaCl, 5% glycerol, 0.1 DTT, 0.5 PMSF) by trituration with a 20G needle and syringe. The suspension was incubated on ice for 30 min, with occasional mixing. After a final centrifugation, the supernatant was removed and saved as the nuclear fraction. The equivalent of 5 μg of protein from each sample was separated using SDS-PAGE. Proteins were detected by Western blotting with antibodies specific for TDP-43 (rabbit anti-TDP-43 polyclonal antibodies from Proteintech, 1:1000), histone H1 (mouse anti-histone H1.0 monoclonal antibodies from Abcam, 1:000) and α-tubulin (mouse B512 anti-α-tubulin monoclonal antibodies from Sigma, 1:2000) diluted in 5% nonfat dry milk in Tris-buffered saline with 0.1% Tween 20, and incubated overnight at 4°C. Secondary antibody incubations were performed at room temperature for 30 min using HRP-conjugated anti-mouse or anti-rabbit IgG antibodies, diluted 1:2000 in 5% nonfat dry milk in Tris-buffered saline with 0.1% Tween 20. Proteins were then visualized with the ECL system (Pierce).
Despite taking specific measures to accurately isolate nuclear and cytosolic fractions, we found that the initial steps of biochemical fractionation can disrupt cellular gradients of some ectopically targeted proteins. In such cases, immunocytochemical analysis has proven to be a more accurate indicator of localization in live cells.
Robotic microscopy.
For neuronal survival analysis, we used a robotic imaging system described by Arrasate and Finkbeiner (2005). Briefly, images were obtained with an inverted Nikon microscope (TE300 Quantum) equipped with a 20× objective lens (numerical aperture 0.45) and a Hamamatsu Orca II 12/14-bit, digital, cooled charge-coupled device. Illumination was provided by a xenon lamp (300 W) with a liquid light guide to maximize the signal-noise ratio. All movements of the stage were controlled with electrical stepper motors. Coordination of fluorescence excitation and emission filters, stage movements, focusing, and imaging acquisition was accomplished with custom-designed and commercially available programs.
Image analysis and statistical comparisons.
Digitized images were analyzed with MetaMorph and original programs written with MatLab and Visual C software. Protein expression levels were estimated by determining the fluorescence intensity of the marker to which the protein is fused, as described previously (Arrasate et al., 2004). Cell bodies of transfected neurons were identified by mCherry fluorescence, and TDP-43-dependent fluorescence in one or more cellular compartments was determined by measuring fluorescence intensity in a defined region of interest. Background fluorescence from neighboring regions of interest was subtracted before further data analysis.
In survival analyses, neuronal death was determined by cell membrane rupture, blebbing, or loss of mCherry fluorescence. Kaplan-Meier and cumulative risk of death curves were generated with StatView software, and the statistical significance of differences between cohorts of neurons was determined with the log-rank test. Cox proportional hazards analysis was used to evaluate the effects of expression level, IB formation, and protein localization on the risk of death. Differences in continuous variables (e.g., expression level) between two groups were assessed with the unpaired t test. For nonparametric comparisons involving more than two groups, significance was determined with the log-rank test; for parametric comparisons, Tukey's multiple-comparison test was used. Statistical comparisons and plots were generated with GraphPad or Statview software.
Results
An in vitro model of TDP-43 proteinopathies
To generate a model of TDP-43-mediated neurodegeneration, we transfected rat primary cortical neurons with constructs encoding EGFP-tagged wild-type (WT) TDP-43 or TDP-43 bearing a mutation (A315T) associated with familial ALS (Kabashi et al., 2008) (Fig. 1A). The pathology of ALS is characterized by motoneuron degeneration in the spinal cord and motor cortex (Kato, 2008), and FTLDu by atrophy in the frontal and temporal cortices (Graff-Radford and Woodruff, 2007). Therefore, we chose primary cortical neurons as the system in which to assess the ability of mutant TDP-43 to induce neurodegeneration.
Similar to the endogenous TDP-43 protein, WT (Fig. 1B–D) and mutant TDP43-EGFP (Fig. 1E–G) were largely restricted to the nucleus. In some cells, however, TDP43-EGFP was distributed throughout the cytoplasm and the nucleus (Fig. 2A–C). Twenty-four hours after transfection, TDP-43-immunoreactive protein aggregates, a characteristic feature of neurodegeneration in FTLD and ALS (Arai et al., 2006; Neumann et al., 2006), were detected in 10–15% of neurons expressing WT or mutant TDP43-EGFP (Fig. 2D–F). We noted colocalization of ubiquitin and aggregated TDP43-EGFP in cells cotransfected with TDP43-EGFP and epitope-tagged ubiquitin (data not shown), suggesting that these deposits are ubiquitinated in cultured neurons. EGFP fluorescence in mutant TDP43-EGFP aggregates was resistant to detergent treatment, consistent with a densely packed structure such as an IB (Arrasate et al., 2004), but fluorescence from WT TDP43-EGFP aggregates was destroyed (Fig. 2G–J). Thus, mutant TDP43-EGFP forms ubiquitinated and detergent-resistant IB typical of both ALS and FTLD (Arai et al., 2006; Neumann et al., 2006).
Figure 2.

Expression of TDP43-EGFP in primary cortical neurons recapitulates key features of TDP43-proteinopathies. A–C, Diffuse cytoplasmic TDP43-EGFP (arrow) was noted in a fraction of neurons expressing either WT or mutant TDP43-EGFP. D–F, Aggregates (arrow) were observed in a small proportion of neurons transfected with WT or mutant TDP43-EGFP. G–J, Detergent-resistance assay. Cells displaying aggregates were fixed and imaged using fluorescence microscopy before (G, I) and after (H, J) detergent treatment. EGFP fluorescence from TDP43(WT)-EGFP aggregates (G, H) was destroyed by detergent treatment; fluorescence from TDP43(A315T)-EGFP aggregates (I, J) was resistant. A, D, G–J, EGFP fluorescence. B, E, TDP-43 immunofluorescence. C, F, Merged images with Hoechst nuclear staining in blue, EGFP fluorescence in green, TDP-43 immunofluorescence in red, and overlap in yellow. The neurons in A–C and G and H were transfected with TDP43(WT)-EGFP; cells in D–F, I, and J were transfected with TDP43(A315T)-EGFP. Scale bars: A (for A–F), G (for G–J), 10 μm.
Neuronal loss underlies the region-specific brain atrophy in TDP-43-proteinopathies and correlates directly with clinical symptoms (Kwong et al., 2007). To determine whether expression of WT or mutant TDP43-EGFP affects neuronal survival, we cotransfected primary cortical neurons at 6 d in vitro with TDP43-EGFP constructs and mCherry, a fluorescent protein that acts as a survival marker. The transfected cells were monitored at regular intervals for 10 d by automated fluorescence microscopy (Arrasate and Finkbeiner, 2005). This system permits us to analyze and follow large numbers of live neurons over an extended period and to quantitatively relate observations at each interval to a prespecified outcome (in this case, cell death). Neuronal death was marked by morphologic changes (cell fragmentation, blebbing, or rounding) and loss of mCherry fluorescence (Fig. 3A), which correlate well with traditional measures of cell death, including annexin V staining and loss of cell membrane integrity (Arrasate et al., 2004). Control neurons were cotransfected with mCherry and EGFP alone or EGFP fused to a neurotoxic protein (exon 1 of huntingtin containing 97 consecutive glutamines, designated Htt(97Q)-EGFP).
Figure 3.
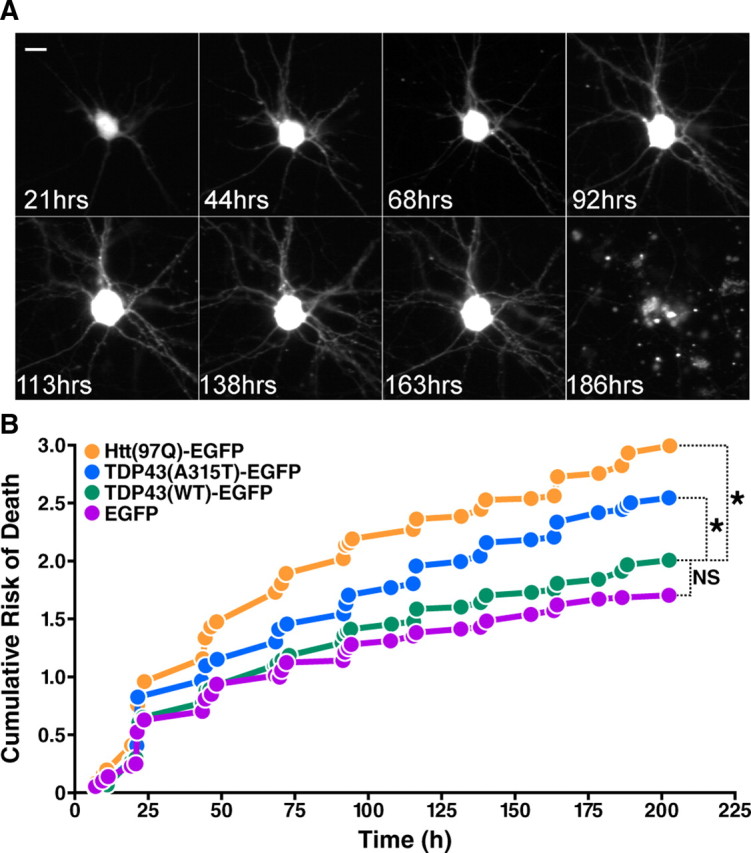
Mutant, but not WT, TDP43-EGFP is toxic when expressed in primary cortical neurons. A, A typical neuron transfected with TDP43-EGFP and mCherry and followed by automated microscopy at 12–24 h intervals until neuronal death was observed at 186 h. Scale bar, 10 μm. B, Kaplan-Meier survival analysis was used to create cumulative risk of death functions for each population of transfected neurons. In these plots, the y-axis represents a quantitative measure of the accumulated risk of cell death over time. *p < 0.0001 (log-rank test). NS, not significant (p > 0.05 by the log-rank test). The cumulative risk of death, displayed on the y-axis, represents the probability that cells in a cohort will die over the time interval for each experiment. Cumulative risk of death curves were compiled from five separate experiments, including a total of 639 neurons expressing EGFP alone, 665 neurons expressing Htt(97Q)-EGFP, 998 neurons expressing TDP43(WT)-EGFP, and 1106 neurons expressing TDP43(A315T)-EGFP.
The ability to measure the individual lifetimes of large numbers of neurons allows the use of survival analysis and related powerful statistical methods to study factors that affect cell fate. Using nonparametric Kaplan-Meier analysis, we generated survival functions for each population of neurons and plotted hazard curves depicting the cumulative risk of death (Fig. 3B). Cox proportional hazards analysis was then used to generate a hazard ratio (HR)—representing an estimate of the relative risk of death—for each cohort of neurons. Cultured neurons have a finite lifespan in vitro and therefore display a non-zero baseline risk of death. Expression of EGFP alone in these cells confers no additional risk of death above this baseline level (Arrasate and Finkbeiner, 2005). As expected, the risk of death was significantly higher in neurons expressing Htt(97Q)-EGFP than in those expressing EGFP alone (HR 1.473, p < 0.0001) (Fig. 3B) but not in neurons transfected with WT TDP43-EGFP. The rate of cell death was also significantly greater in cells expressing TDP43(A315T)-EGFP (HR 1.225, p < 0.0001) (Fig. 3B), indicating that the A315T mutation causes toxicity in transfected neurons. To determine whether this toxic effect was unique to the A315T mutation, we generated two additional TDP43-EGFP constructs carrying point mutations (N390S and G290A) associated with the development of ALS (Kabashi et al., 2008; Van Deerlin et al., 2008). When expressed in rat cortical neurons, both TDP43(N390S)-EGFP and TDP43(G290A)-EGFP significantly increased the risk of cell death over that of TDP43(WT)-EGFP (p < 0.0001 for both; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Thus, three separate mutations in TDP-43, all of which are associated with the development of ALS, caused substantial toxicity in rat cortical neurons.
Despite the simplicity of the system, expression of TDP43-EGFP in primary cortical neurons recapitulates several cardinal features of TDP-43-proteinopathies, including nuclear localization of the protein, the formation of ubiquitin-immunopositive, detergent-insoluble IBs, and neuronal toxicity related to pathogenic mutations in TDP-43. We conclude that this model is a useful tool to investigate mechanisms by which mutant TDP-43 induces neurodegeneration.
Cytoplasmic TDP-43 predicts neuronal death
Next, we used automated fluorescence microscopy to focus upon one of the TDP-43 mutations associated with ALS (A315T) and to investigate the mechanisms by which this mutation induces cell death. The ability to track changes in individual neurons over time and to relate the temporal sequence of these events to cellular survival permit us to determine the relationship of each variable to cell fate. We concentrated on molecular changes, such as aggregate formation and protein redistribution, which may account for the toxicity of the A315T mutation in TDP-43.
Transfected neurons were scored for several variables, including fluorescence intensity, IB formation, and subcellular localization of TDP43-EGFP (nuclear or cytoplasmic). Since the fluorescence intensity of EGFP in live neurons is directly related to the level of proteins to which it is fused (Arrasate and Finkbeiner, 2005), we used the normalized fluorescence intensity in each neuron as a measure of the level of TDP43-EGFP expression.
To examine the role of IB formation in TDP43-mediated disease, we compared the survival of neurons that formed IBs and those that did not. Because we were interested in IB formation per se, we prospectively followed neurons with diffuse TDP43-EGFP to identify those that subsequently formed IBs. To avoid selection bias against neurons with shorter survival times, we included only neurons that were alive 24 h after transfection. Cells that form an IB after 24 h should have a higher risk of death if IB formation is toxic and a lower risk if it is a coping mechanism. Alternatively, if IB formation is unrelated to cellular survival, the risk of death should be similar in both populations.
Previously, we reported that the expression level of aggregation-prone proteins is an important predictor of IB formation (Arrasate et al., 2004). Indeed, IB formation was significantly and positively affected by expression level in neurons transfected with WT or mutant TDP43-EGFP (Fig. 4A). Moreover, the level of expression predicted the timing of IB formation, such that neurons with the highest expression levels formed IBs within 24 h after transfection.
Figure 4.

IB formation is unrelated to cellular survival in neurons expressing WT and mutant TDP43-EGFP. A, Neurons with diffuse TDP43-EGFP had lower expression levels than neurons that formed IBs before or >24 h after transfection. B, C, Cohorts of neurons with IBs (n = 52) and those with diffuse TDP43-EGFP (n = 201) displaying similar expression levels (B) were analyzed by Kaplan-Meier survival analysis, and curves depicting the cumulative risk of death (C) were plotted for each population. D, Cumulative risk of death curves for neurons with IBs and those with diffuse TDP43-EGFP expressing WT (n = 149) or mutant TDP43-EGFP (n = 104). There is no significant difference in the risk of death between neurons with and without IBs, expressing either WT or mutant TDP43-EGFP. *p < 0.01; **p < 0.001; NS, not significant (p > 0.05). p values were determined by Tukey's multiple-comparison test (A, B) and log-rank test (C, D). Results represent data combined from three independent experiments.
To control for the effects of expression level on IB formation and cellular survival, we analyzed cohorts of neurons with equivalent expression levels 24 h after transfection. The normalized expression level was similar for neurons expressing WT and mutant TDP43-EGFP, with and without IBs (Fig. 4B). For a given expression level, IBs formed more readily in neurons expressing TDP43(A315T)-EGFP than in those expressing TDP43(WT)-EGFP (21% vs 10%, p < 0.015, χ2 test). Nevertheless, Kaplan-Meier and Cox proportional hazards analyses of these cohorts failed to demonstrate an effect of IB formation on cell survival (Fig. 4C,D, Table 1). These results indicate that IB formation is not required for mutant TDP43-EGFP toxicity and suggest that diffuse or soluble forms of the protein have a neurotoxic effect that is independent of IB formation.
Table 1.
Cox proportional hazards analysis of the effects of cellular phenotypic variables on neuronal survival
| Variable | Univariate HR (95% CI) | p | Multivariate HR (95% CI) | p |
|---|---|---|---|---|
| TDP43(WT)-EGFP (n = 579) | ||||
| Cytoplasmic localization | 1.739 (1.248–2.423) | 0.0011 | 1.724 (1.235–2.406) | 0.0014 |
| IB formation | 0.840 (0.450–1.567) | NS | 0.897 (0.480–1.675) | NS |
| Fluorescence intensity(a.u.)a | 6.201 (3.703–10.384) | <0.0001 | 2.608 (1.392–4.885) | 0.0028 |
| TDP43(A315T)-EGFP (n = 650) | ||||
| Cytoplasmic localization | 1.772 (1.319–2.380) | 0.0001 | 1.747 (1.297–2.354) | 0.0002 |
| IB formation | 1.143 (0.696–1.880) | NS | 0.903 (0.533–1.528) | NS |
| Fluorescence intensity(a.u.)a | 3.587 (2.476–5.195) | <0.0001 | 1.608 (0.992–2.604) | 0.0537 |
CI, Confidence interval; n, number of neurons; NS, not significant (p > 0.05); a.u., arbitrary units.
aHR is reported in relation to SD of TDP43-EGFP fluorescence intensity.
Since the redistribution of TDP-43 from the nucleus to the cytoplasm is a prominent characteristic of TDP-43-associated diseases (Mackenzie and Rademakers, 2007), we next asked whether the subcellular localization of TDP-43 predicts cellular survival. Neurons with purely nuclear TDP43-EGFP or those with cytoplasmic and nuclear TDP43-EGFP were identified by comparing the distribution of TDP43-EGFP to that of mCherry, a fluorescent protein that is coexpressed in the cytoplasm of every transfected neuron (Fig. 5A–C). The subcellular localization of TDP43-EGFP in these cells was confirmed by immunocytochemical staining of fixed neurons for nuclear markers (Fig. 2A–C). By Cox proportional hazards analysis, the presence of cytoplasmic TDP43-EGFP 24 h after transfection was directly and significantly associated with the risk of cell death in neurons expressing WT or mutant TDP43-EGFP (HR 2.877, p < 0.0001 by log-rank test), even in multivariate analyses controlling for expression level (Table 1).
Figure 5.
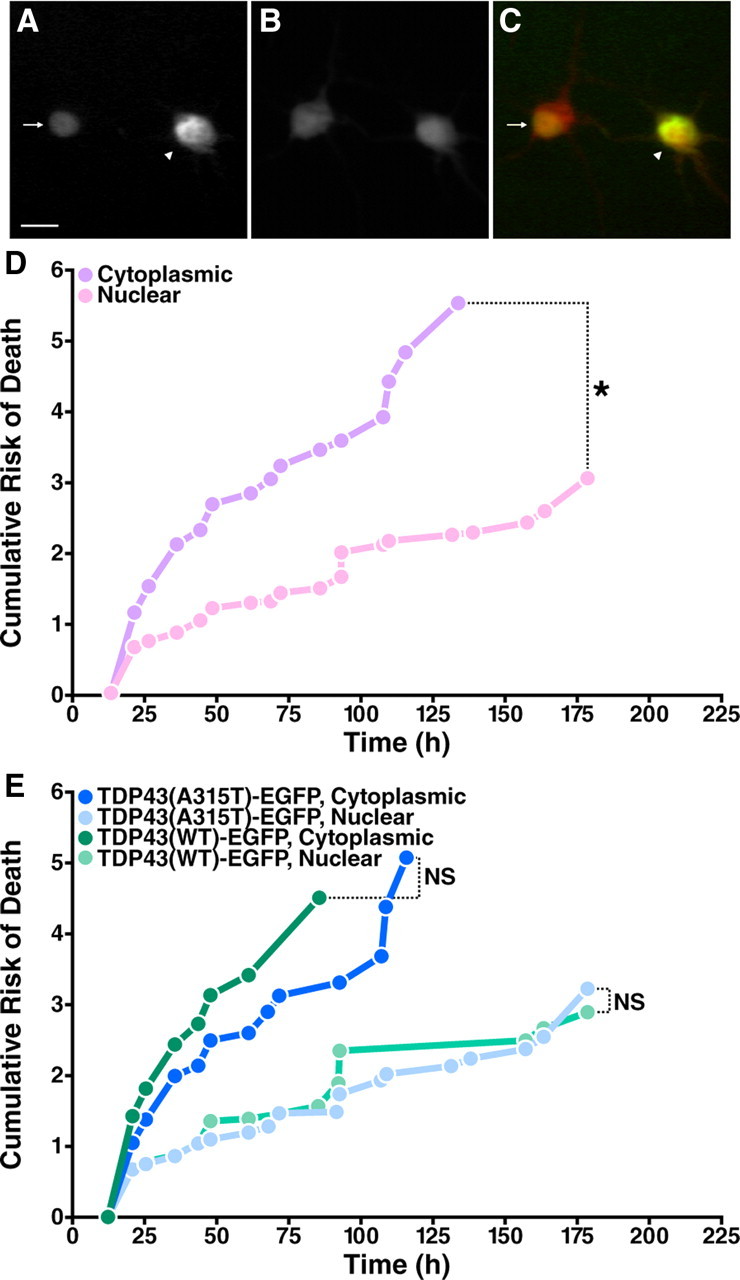
Cytoplasmic TDP43-EGFP significantly increases the risk of death in transfected neurons. A–C, Cells displaying purely nuclear (arrow) or both nuclear and cytoplasmic (arrowhead) TDP43-EGFP were identified by fluorescence microscopy. A, EGFP fluorescence. B, mCherry fluorescence. C, Merged image with EGFP fluorescence in green, mCherry fluorescence in red, and overlap in yellow. Scale bar, 10 μm. D, Kaplan-Meier survival analysis of neurons with matched expression levels and either nuclear localization of TDP43-EGFP (nuclear, n = 153) or nuclear and cytoplasmic protein (cytoplasmic, n = 258), demonstrating the toxicity of cytoplasmic TDP43-EGFP. E, The toxic effect of cytoplasmic mislocalization was similar for both WT (n = 170) and mutant TDP43-EGFP (n = 241). *p < 0.0001 (log-rank test). NS, not significant (p > 0.05). Cumulative risk of death curves were constructed with data from three separate experiments.
To fully isolate the effects of protein mislocalization on cell fate, we analyzed neurons with matched expression levels of TDP43(WT)-EGFP or TDP43(A315T)-EGFP (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material) and compared the risk of death in neurons with cytoplasmic (and nuclear) TDP43-EGFP to those with solely nuclear TDP43-EGFP (Fig. 5D). Cytoplasmic TDP43-EGFP increased the risk of death two- to threefold, regardless of whether the neurons expressed WT or mutant TDP43-EGFP (Fig. 5E, Table 1). Surprisingly, neurons with 2purely nuclear protein—either TDP43 (WT)-EGFP or TDP43(A315T)-EGFP—had similar survival times, suggesting that the toxicity is caused by cytoplasmic TDP43-EGFP itself and does not require the A315T mutation.
The initial identification of several disease-associated proteins emerged from genetic studies demonstrating a link between inherited neurodegenerative disorders and mutations of specific genes. In some of these disorders, including Parkinson's disease and Alzheimer's disease, duplication of the WT gene, leading to overexpression of the gene product, recapitulates the disorder (Salehi et al., 2006; Theodore et al., 2008). Could overexpression of TDP-43 likewise be sufficient to induce neurodegeneration in our model? To answer this question, we took advantage of the fact that transient transfection normally results in a population of transfected neurons with a range of expression levels, allowing us to examine the cellular response to different “doses” of TDP-43. High-level expression of both WT and mutant TDP43-EGFP resulted in the mislocalization of the protein from the nucleus to the cytoplasm (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material). Moreover, elevated expression levels of both TDP43(WT)-EGFP and TDP43(A315T)-EGFP were associated with an increased risk of cell death, consistent with dose-dependent neurotoxic effects (Table 1). The expression level of EGFP alone did not affect cellular survival. Thus, overexpression of TDP43-EGFP results in protein mislocalization and induces neurodegeneration independently of the A315T mutation.
The A315T mutation increases the proportion of cytoplasmic TDP-43
Because the cytoplasmic distribution of TDP43-EGFP was clearly linked to neuronal toxicity, we hypothesized that the A315T mutation stimulates cell death indirectly by enhancing mislocalization of the protein. To test this hypothesis, we counted neurons exhibiting a purely nuclear distribution of TDP43-EGFP and those with nuclear and cytoplasmic protein. Cytoplasmic mislocalization was significantly more frequent in neurons expressing TDP43(A315T)-EGFP than in those expressing TDP43(WT)-EGFP (39.15% in comparison to 27.10%, respectively; p < 0.0001, χ2 test), consistent with our hypothesis. The effect of the A315T mutation on protein localization was also evaluated by subcellular fractionation (Fig. 6A), showing that cells expressing TDP43(A315T)-EGFP displayed approximately twice as much cytoplasmic protein as those expressing TDP43(WT)-EGFP, but no significant difference in the amount of nuclear TDP43-EGFP was detected. These results extend and confirm the observations made by fluorescence microscopy.
Figure 6.
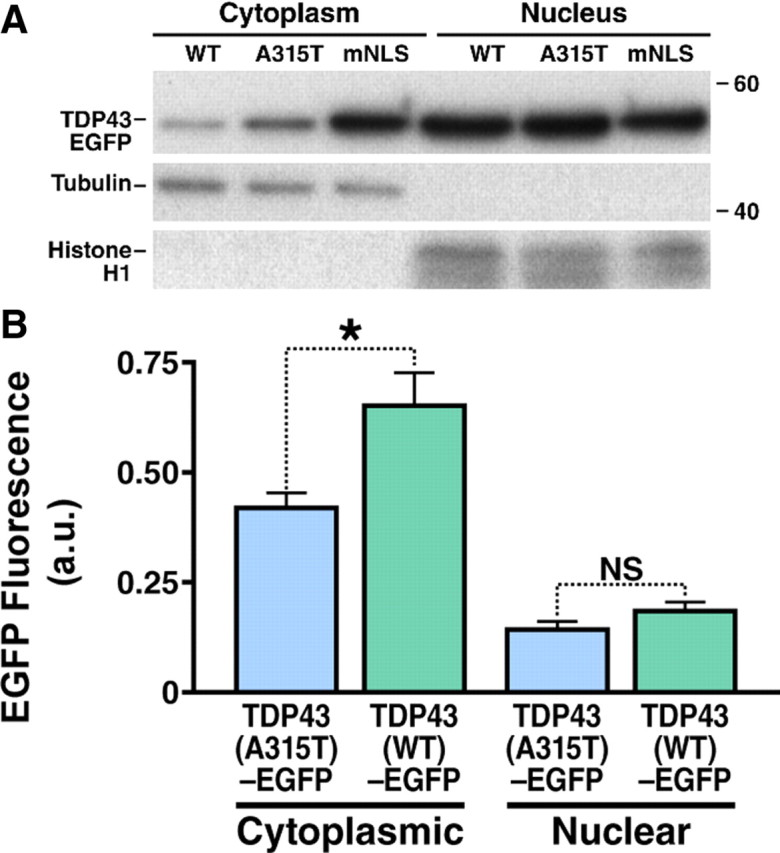
Both the A315T mutation and disruption of the nuclear localization signal increase cytoplasmic mislocalization of TDP43-EGFP. A, HEK293 cells were transfected with either TDP43(WT)-EGFP, TDP43(A315T)-EGFP, and TDP43(mNLS), and cell lysate from each group was separated into cytosolic and nuclear fractions. TDP43-EGFP can be found within the nuclear fractions (marked by the presence of histone H1) of each group. After normalization, the amount of TDP43-EGFP within the cytoplasmic fraction (marked by α-tubulin) increases by a factor of 1.79 and 3.86 in cells transfected with TDP43(A315T)-EGFP and TDP43(mNLS)-EGFP, respectively, compared with those expressing TDP43(WT)-EGFP. A band migrating at 43 kDa and corresponding to endogenous TDP-43 was noted in the nuclear fraction of each lysate (data not shown). B, The mean expression level of neurons with cytoplasmic TDP43-EGFP was significantly higher in cells transfected with TDP43(WT)-EGFP than in those transfected with TDP43(A315T)-EGFP, indicating that cytoplasmic mislocalization of mutant TDP43-EGFP compared with wild-type cannot be due to relative overexpression alone. *p = 0.0008 (two-tailed t test); NS, not significant (p > 0.05). Results represent pooled data from three different experiments.
Since overexpression of TDP43-EGFP also increased mislocalization and cell death, we wondered whether the neurotoxic effects of the A315T mutation were related to selective high-level expression of the mutant protein. In fact, the mean expression level of TDP43(A315T)-EGFP in transfected neurons was lower than that of TDP43(WT)-EGFP [0.2271 ± 0.014 vs 0.2892 ± 0.025 arbitrary units (mean ± SEM); p = 0.0252, two-tailed t test]. Furthermore, cytoplasmic mislocalization of TDP43-EGFP was observed at a lower mean expression level in neurons transfected with TDP43(A315T)-EGFP (Fig. 6B), arguing against artifactual mislocalization of mutant TDP43-EGFP from overexpression. These findings suggest that the A315T mutation specifically induces the mislocalization of TDP43-EGFP from the nucleus to the cytoplasm in transfected neurons.
If the A315T mutation increases the proportion of cytoplasmic TDP43-EGFP, the ratio of nuclear to cytoplasmic protein (NCR) should be lower in cells expressing mutant TDP43-EGFP. To investigate this possibility, we measured the amounts of TDP43-EGFP in the nucleus and cytoplasm of individual cells and calculated the NCR for each neuron. Indeed, the mean NCR was significantly lower in neurons expressing TDP43(A315T)-EGFP (p = 0.003, Kolmogorov–Smirnov test) (Fig. 7A), confirming that the A315T mutation enhances cytoplasmic mislocalization. By Cox proportional hazards analysis, the NCR was a significant predictor of cellular survival (Table 2) and thus is a sensitive determinant of outcome in neurons expressing TDP43-EGFP.
Figure 7.
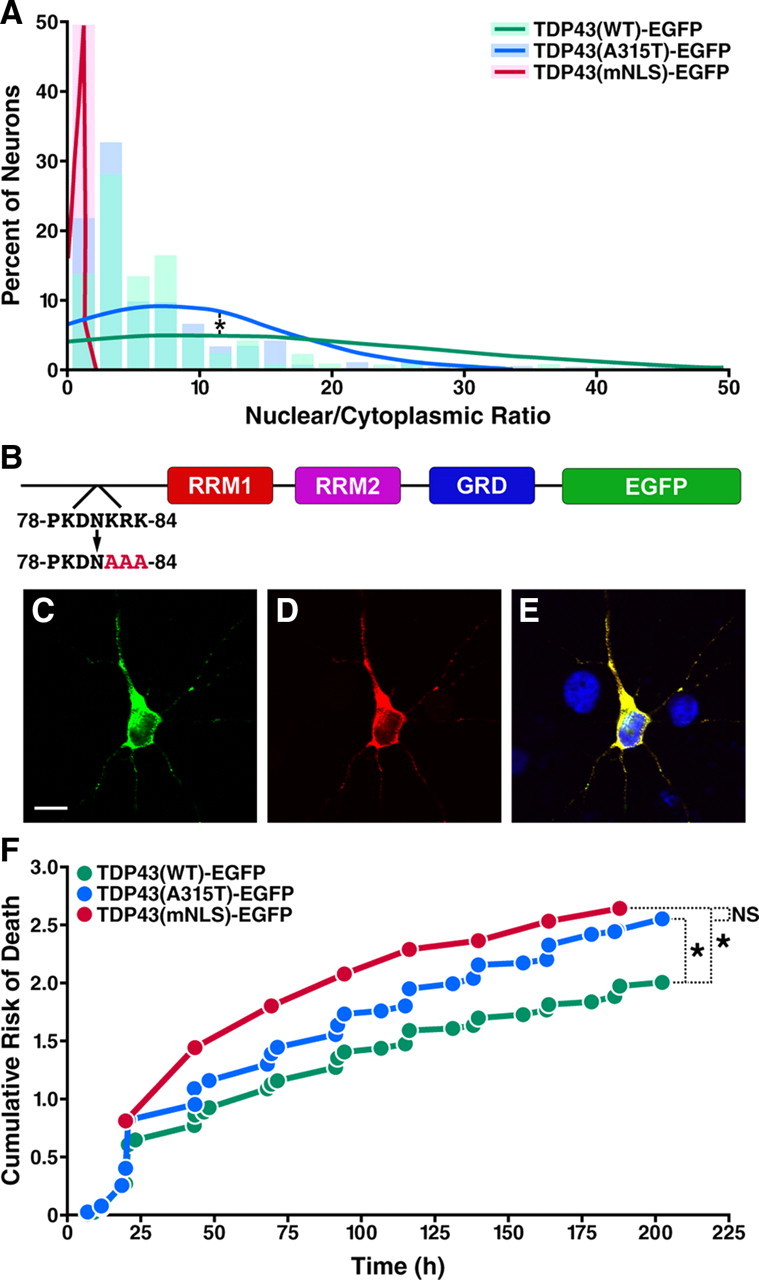
Mutant TDP43-EGFP is cytoplasmically mislocalized, and cytoplasmic TDP43-EGFP is sufficient to produce neurodegeneration. A, The mean NCR of TPD43-EGFP, calculated from the fluorescence intensity in the nucleus and cytoplasm of transfected cells, was lower in cells expressing mutant TDP-43-EGFP (blue, n = 261) than in those expressing WT protein (green, n = 214), consistent with the mislocalization of TDP43(A315T)-EGFP to the cytoplasm (*p = 0.003, Kolmogorov–Smirnov test). Solid lines mark the normal distribution curves for each population, and shaded regions represent the proportions of neurons with the indicated NCR values plotted as a histogram. Virtually all neurons expressing TDP43(mNLS)-EGFP (red, n = 107) had an NCR <1, confirming the predominantly cytoplasmic localization of the protein. NCR distribution curves and histograms were generated from expression data pooled from three separate experiments. B, Schematic of TDP43-EGFP highlighting the NLS encompassing residues 78–84. TDP43(mNLS)-EGFP was created by substituting nonpolar alanine residues (AAA) for the basic residues (KRK) at positions 82–84 of TDP43(WT)-EGFP. C–E, Confocal fluorescence microscopy of neurons expressing TDP43(mNLS)-EGFP, demonstrating cytoplasmic localization of the fusion protein. C, EGFP fluorescence. Scale bar, 10 μm. D, TDP43 immunostaining. E, Merged image with Hoechst nuclear staining in blue and overlap in yellow. F, Kaplan-Meier survival analysis and cumulative risk of death curves for neurons expressing TDP43(WT)-EGFP (n = 998), TDP43(A315T)-EGFP (n = 1106), and TDP43(mNLS)-EGFP (n = 732). Both TDP43(A315T)-EGFP and TDP43(mNLS)-EGFP were highly toxic in transfected cells (*p < 0.0001, log-rank test), but there was no statistically significant difference between the effects of TDP43(A315T)-EGFP and TDP43(mNLS)-EGFP on cellular survival (NS, not significant; p > 0.05, log rank test). Cumulative risk of death plots included data from five independent experiments.
Table 2.
Cox proportional hazards analysis of the effects of expression level and subcellular distribution of TDP43 on neuronal survival
| Variable | HRa | 95% CI | p |
|---|---|---|---|
| TDP43(WT)-EGFP (n = 238) | |||
| NCR | 0.982 | 0.969–0.996 | 0.0122 |
| Cytoplasmic TDP43-EGFP (fluorescence intensity, a.u.) | 1.660 | 1.189–2.317 | 0.0029 |
| Nuclear TDP43-EGFP (fluorescence intensity, a.u.) | 0.972 | 0.885–1.068 | NS |
| TDP43(A315T)-EGFP (n = 272) | |||
| NCR | 0.981 | 0.965–0.998 | 0.0278 |
| Cytoplasmic TDP43-EGFP (fluorescence intensity, a.u.) | 1.346 | 1.080–1.678 | 0.0082 |
| Nuclear TDP43-EGFP (fluorescence intensity, a.u.) | 0.972 | 0.872–1.084 | NS |
CI, Confidence interval; n, number of neurons; NS, not significant (p > 0.05); a.u., arbitrary unit.
aHR is reported in relation to SD of the NCR or TDP43-EGFP fluorescence intensity.
What is the relationship between the subcellular mislocalization of TDP-43 and neurodegeneration? Too much TDP-43 in the cytoplasm may be toxic to neurons, or cellular survival may depend instead upon the presence of adequate amounts of nuclear TDP-43. Alternatively, the relative distribution of TDP-43 between the nucleus and the cytoplasm may be essential for cellular survival. Our system can determine whether cell fate is best predicted by any one of these variables. Cox proportional hazards analysis showed that the amount of cytoplasmic TDP-43 per cell was a strong and significant predictor of cell death in neurons expressing WT or mutant TDP43-EGFP (Table 2). In contrast, the amount of nuclear TDP-43 per cell did not correlate with neuronal survival. Moreover, as shown in Figure 6A, expression of TDP43(A315T)-EGFP is associated with a twofold increase in the amount of cytoplasmic protein without substantially affecting the amount of nuclear TDP43-EGFP. These results strongly suggest that cytoplasmic TDP43-EGFP exhibits a toxic gain-of-function that leads to cell death regardless of the amount of protein in the nucleus.
If the A315T mutation stimulates cell death through mislocalization of TDP-43, then targeting the protein to the cytoplasm should recapitulate the toxicity of TDP43(A315T)-EGFP. To test this hypothesis, we largely restricted the distribution of WT TDP43-EGFP to the cytoplasm by mutating several amino acids of the predicted NLS of the protein (Fig. 7B). Employing a similar strategy, Winton et al. (2008) showed that disruption of the NLS leads to cytoplasmic retention of TDP-43. When expressed in primary cortical neurons, TDP43(mNLS)-EGFP was primarily localized to the cytoplasm, as expected (Fig. 7C–E), and the NCR was <1 (Fig. 7A), consistent with the protein's nearly complete cytoplasmic localization. The cytoplasmic distribution of TDP43(mNLS)-EGFP was confirmed by quantitative fluorescence microscopy of live neurons (data not shown), and biochemical fractionation of transfected cells (Fig. 6A). Mutating WT TDP43-EGFP in this way to mimic the cytoplasmic mislocalization of TDP43(A315T)-EGFP significantly increased its toxicity, as shown by Kaplan-Meier analysis (HR 1.286, p < 0.0001) (Fig. 7F). The mean expression level of TDP43(mNLS)-EGFP in transfected neurons was comparable to that of the WT protein (data not shown), excluding the possibility that the toxic effect of TDP43(mNLS)-EGFP arises from overexpression.
If the toxicity of the A315T mutation depends on cytoplasmic mislocalization of TDP43-EGFP, then preventing nuclear export of the protein by disrupting the predicted nuclear export signal (NES) of TDP-43 should attenuate the toxic effect of the A315T mutation. Indeed, we found that mutation of the NES significantly blunted the toxicity of the A315T mutation, such that expression of TDP43(mNES-A315T)-EGFP had no significant effect upon cellular survival compared with TDP43(WT)-EGFP (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). These results confirm that cytoplasmic mislocalization of TDP43-EGFP is essential for A315T-specific cellular toxicity.
As demonstrated by immunocytochemistry and subcellular fractionation, neurons transfected with TDP43(mNLS)-EGFP display significantly more protein within the cytoplasm than those expressing TDP43(A315T)-EGFP. Nevertheless, the toxicity of TDP43(mNLS)-EGFP was not significantly different from that of mutant TDP43(A315T)-EGFP (HR 1.011, p = 0.7705 by log-rank test), suggesting that the toxic effect may be saturated by relatively low amounts of TDP43-EGFP in the cytoplasm. In support of this conclusion, we found that the toxic effect of cytoplasmic TDP-43 is similar for neurons expressing WT, A315T, or mNLS TDP43-EGFP, and that the toxicity of cytoplasmic TDP-43 in each case reaches a maximum at relatively modest levels of cytoplasmic protein (data not shown). Moreover, if the A315T mutation induces toxicity through cytoplasmic mislocalization of TDP-43, then a double-mutant of TDP-43 incorporating both the mNLS and A315T mutations should not display any additional toxic effect beyond that of either mutation by itself. In fact, the cumulative risk of cell death was no different in neurons expressing TDP43(mNLS)-EGFP or TDP43(mNLS- A315T)-EGFP (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). These results confirm that the toxicity of the A315T mutation is closely related to cytoplasmic mislocalization of TDP-43, and that the mechanism underlying the toxic effect involves an abnormal accumulation of TDP-43 within the cytoplasm.
Discussion
In this study, we sought to understand the role of TDP-43 mutations in the pathogenesis of FTLD and ALS. We established a primary neuron-based model that recapitulates key features of TDP-43-proteinopathies (e.g., nuclear localization of WT TDP-43 in healthy neurons, mislocalization of mutant TDP-43 to the cytoplasm in affected neurons, the formation of ubiquitinated, detergent-insoluble IBs, and neurodegeneration) and is highly amenable to experimental manipulation. We expect it will enable the rigorous evaluation of mechanistic hypotheses regarding TDP-43-induced neurodegeneration and yield physiologically relevant information. As shown by automated fluorescence microscopy, several mutations in TDP-43 associated with the development of ALS caused significant neuronal toxicity in this model. Cellular survival was independent of IB formation, suggesting that diffuse forms of TDP-43 are toxic. One of these mutations, A315T, increased the mislocalization of TDP43-EGFP to the cytoplasm, and the amount of cytoplasmic TDP43-EGFP was a powerful predictor of cell death. Furthermore, restricting TDP43-EGFP to the cytoplasm by mutating the NLS was toxic to neurons, while preventing cytoplasmic mislocalization of the protein by mutating the NES abrogated the toxic effects of the A315T mutation. These observations suggest that the A315T mutation increases cell death by enhancing cytoplasmic mislocalization of TDP-43.
Although IB formation was initially presumed to be toxic, a growing body of evidence indicates that IB formation can, in certain situations, function as a coping response by neurons expressing aggregation-prone proteins (Arrasate et al., 2004; Ko et al., 2008; Lesné et al., 2008; Berlau et al., 2009; Mitra et al., 2009). We found that IB formation in cells expressing TDP43-EGFP was not necessary for mutation-specific cell death, suggesting that similar to at least some other proteins associated with neurodegenerative disorders, TDP-43 forms aggregates that are relatively inert. However, we were unable to detect a beneficial effect of TDP43-EGFP IB formation upon cellular survival. This could be due to inadequate sensitivity or resolution of our system. Alternatively, it is possible that IB formation could cause neuronal dysfunction without affecting cellular survival; since our analysis focused solely on cell death, this effect would go undetected.
Overexpression of WT or mutant TDP43-EGFP was toxic to primary neurons and resulted in cytoplasmic mislocalization of the protein, consistent with findings in yeast expressing high levels of human TDP-43 (Johnson et al., 2008). Analysis of neurons with equivalent expression levels showed that cytoplasmic TDP43-EGFP per se is a strong predictor of cell death, independent of expression level. However, overexpression of either WT or mutant TDP43-EGFP caused neurodegeneration, suggesting that modifiers of TDP-43 expression in vivo could significantly affect the risk of developing sporadic ALS or FTLD.
In contrast to prior investigations of TDP-43 in cultured cells (Ayala et al., 2008; Winton et al., 2008), we detected cytoplasmic mislocalization of the protein in a significant number of neurons and found that mutant TDP-43 expression was toxic to transfected cells. Whereas we used primary cortical neurons for our experiments, previous studies were conducted in immortalized cell lines. Rodent primary neurons accurately localize a wide variety of fluorescently labeled transfected proteins (Saudou et al., 1998; Arrasate et al., 2004; Bradley et al., 2006; Pintchovski et al., 2009), confirming that these cells can faithfully recapitulate subcellular protein distribution. The prominent mislocalization of TDP-43 in primary neurons, but not in immortalized cell lines, suggests that neuron-specific mechanisms govern its metabolism and distribution. These mechanisms may explain why TDP-43 mutations linked to familial ALS and FTLD are toxic to neurons but not to other cell types.
In human brain sections and cultured neurons, cytoplasmic redistribution of TDP-43 is closely linked to nuclear depletion of the protein (Arai et al., 2006; Neumann et al., 2006). The neurodegeneration in TDP-43-proteinopathies could thus reflect two possibly overlapping mechanisms: loss of an essential function of TDP-43 in the nucleus or gain of a toxic function unique to cytoplasmic TDP-43. In our system, mutant TDP43-EGFP stimulated cell death in primary WT rat neurons expressing endogenous TDP-43. This toxic gain-of-function is consistent with the autosomal-dominant inheritance of nearly all TDP-43 mutations associated with familial ALS, including A315T (Kabashi et al., 2008; Kühnlein et al., 2008; Sreedharan et al., 2008; Van Deerlin et al., 2008). Alternatively, mutant TDP-43 could act in a dominant-negative manner by sequestering endogenous TDP-43 within the nucleus or otherwise disrupting its function. However, the amount of cytoplasmic TDP43-EGFP was a strong and independent predictor of cell death, and there was no clear relation between the amount of nuclear TDP43-EGFP and neuronal survival. Moreover, localizing TDP43-EGFP to the cytoplasm was detrimental to transfected neurons, and the cell death induced by mutant TDP43-EGFP was intimately associated with cytoplasmic mislocalization of the protein. These results provide no evidence that nuclear depletion of TDP-43 is important for the toxicity of the mutant protein but do indicate that mislocalization of TDP-43 to the cytoplasm is sufficient to induce neurodegeneration. However, the fact that these experiments were conducted in the presence of normal levels of endogenous TDP-43 limits our ability to fully exclude the possibility that loss of nuclear TDP-43 is itself toxic.
How might mutant TDP-43 be involved in the development of familial ALS? The A315T mutation induced cytoplasmic mislocalization of TDP43-EGFP in rat cortical neurons. Because it is located in the putative protein-protein interaction domain of TDP-43, the A315T mutation could increase the proportion of cytoplasmic protein by enhancing the association of TDP-43 with cytoplasmic binding partners, which would impair its nuclear import, and facilitate its nuclear export, or by disrupting its affinity for nuclear binding partners. Restricting TDP-43 to the cytoplasm may disproportionately expose the protein to endogenous proteases, in the process generating high cytoplasmic concentrations of C-terminal TDP-43 fragments, which are enriched in cytoplasmic inclusions found in affected neurons (Igaz et al., 2008).
Increasing the amount of cytoplasmic full-length or C-terminal TDP-43 may result in a toxic gain-of-function or a pathologic augmentation of the protein's normal function. For instance, cytoplasmic mislocalization of full-length or C-terminal TDP-43 could lead to the sequestration of essential mRNA transcripts, abnormal cytoplasmic-nuclear RNA transport, or local translation of mRNA. The demonstration that mutations in another RNA-binding protein, FUS/TLS (fused-in-sarcoma/translocated-in-liposarcoma) are involved in familial ALS provides additional evidence that disruption of RNA processing could be involved in neurodegeneration (Kwiatkowski et al., 2009; Lagier-Tourenne and Cleveland, 2009). Several neuromuscular disorders other than ALS, including myotonic dystrophy and fragile X syndrome, are characterized by aberrant RNA processing within the nucleus as well as the cytoplasm (Ranum and Cooper, 2006). TDP-43 mislocalization could induce motor neuron death through a similar mechanism. Consistent with this hypothesis, deficiencies in low-molecular-weight neurofilament mRNA, to which TDP-43 binds with high affinity (Strong et al., 2007), are associated with motor neuron degeneration (Wong et al., 2000). Even slight changes in neurofilament stoichiometry can lead to neurofilament aggregation and cell death in transgenic models of motor neuron disease (Wong et al., 2000; Lariviere and Julien, 2004). Thus, one possibility is that cytoplasmic full-length or C-terminal TDP-43 effectively sequesters neurofilament transcripts, leading to perturbations in neurofilament stoichiometry, aggregation, and motor neuron death.
In conclusion, we have established a primary cortical neuron model that exhibits many characteristic features of TDP-43, proteinopathies. As in other neurodegenerative diseases, overexpression of the WT protein was sufficient to induce neuronal death. Thus, the risk of developing sporadic ALS and FTLD may be directly proportional to the overall amount of TDP-43, and could be strongly affected by regulators of TDP-43 expression or degradation. Furthermore, we demonstrated that cytoplasmic retention of TDP43-EGFP was also toxic, and that expression of mutant TDP43-EGFP increased the risk of cell death by enhancing cytoplasmic mislocalization of the protein. While the amount of cytoplasmic TDP43-EGFP per cell accurately predicted cellular mortality, the amount of nuclear TDP43-EGFP was unrelated to cellular survival, suggesting that the toxicity results mainly from the downstream effects of cytoplasmic TDP-43. If the mislocalization of TDP-43 is necessary for disease pathogenesis, inhibition of protein redistribution may be an important target for future therapeutics in both FTLD and ALS. Alternatively, if cytoplasmic accumulation of TDP-43 leads to cell death by sequestering mRNA transcripts or transport machinery, small-molecule inhibitors of interactions involving TDP-43 might prevent disease progression. The cellular model presented here will be a valuable system for investigating the pathogenesis of TDP-43 proteinopathies and for evaluating novel and potentially neuroprotective strategies.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants 2R01 NS039074, from the National Institute of Neurological Disorders and Stroke, and 2P01 AG022074, from the National Institute on Aging, and by the J. David Gladstone Institutes (to S.F.), and Grant GM070967/EY014576 (to J.Y.W). The animal care facility was partly supported by an NIH Extramural Research Facilities Improvement Program Project (C06 RR018928). We thank Drs. J. Engstrom and S. Hauser and the members of the Finkbeiner laboratory, including Eva LaDow, Carol Peebles, Jason Miller, and Kelley Nelson, for all their generous support and advice. We thank Paul J. Taylor for commenting on an earlier version of this manuscript and for disclosing unpublished results. We thank G. Howard and S. Ordway for editorial assistance, and K. Nelson for administrative assistance. We thank members of the Consortium for Frontotemporal Dementia Research (memory.ucsf.edu/ftd/research/laboratory/cfr) for their critical reviews and discussions. J.Y.W. and S.J.B. brought this project to the attention of S.F. E.J.R. and J.Y.W. made the expression plasmids encoding TDP-43(WT)-EGFP and TDP43(A315T)-EGFP. S.F. invented and refined the automated imaging and analysis techniques, and S.J.B., G.S., and S.F. designed the experiments to apply these approaches to TDP-43. S.J.B. made the expression plasmids encoding TDP-43(G290A)-EGFP, TDP43(N390S)-EGFP, TDP43(mNLS)-EGFP, TDP-43(mNES)-EGFP, TDP-43(mNLS-A315T)-EGFP, and TDP-43(mNES-A315T)-EGFP. S.J.B., G.S., and E.K. isolated the primary neurons from rodents. S.J.B. conducted all primary neuron transfections, automated imaging, survival analyses, immunocytochemical staining, fluorescence intensity measurements, cell counts, subcellular fractionations, and statistical calculations and performed all fluorescence microscopy. S.J.B. designed and constructed the figures with advice from S.F. and G.S. S.J.B. and S.F. wrote and edited this manuscript.
References
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Finkbeiner S. Automated microscope system for determining factors that predict neuronal fate. Proc Natl Acad Sci U S A. 2005;102:3840–3845. doi: 10.1073/pnas.0409777102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. 2008;121:3778–3785. doi: 10.1242/jcs.038950. [DOI] [PubMed] [Google Scholar]
- Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, Legallic S, Salachas F, Hannequin D, Decousus M, Lacomblez L, Guedj E, Golfier V, Camu W, Dubois B, Campion D, Meininger V, Brice A French Clinical and Genetic Research Network on Frontotemporal Lobar Degeneration/Frontotemporal Lobar Degeneration with Motoneuron Disease. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 2009;65:470–473. doi: 10.1002/ana.21612. [DOI] [PubMed] [Google Scholar]
- Berlau DJ, Corrada MM, Head E, Kawas CH. APOE epsilon2 is associated with intact cognition but increased Alzheimer pathology in the oldest old. Neurology. 2009;72:829–834. doi: 10.1212/01.wnl.0000343853.00346.a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley J, Carter SR, Rao VR, Wang J, Finkbeiner S. Splice variants of the NR1 subunit differentially induce NMDA receptor-dependent gene expression. J Neurosci. 2006;26:1065–1076. doi: 10.1523/JNEUROSCI.3347-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008;13:867–878. doi: 10.2741/2727. [DOI] [PubMed] [Google Scholar]
- Elvira G, Wasiak S, Blandford V, Tong XK, Serrano A, Fan X, del Rayo Sánchez-Carbente M, Servant F, Bell AW, Boismenu D, Lacaille JC, McPherson PS, DesGroseillers L, Sossin WS. Characterization of an RNA granule from developing brain. Mol Cell Proteomics. 2006;5:635–651. doi: 10.1074/mcp.M500255-MCP200. [DOI] [PubMed] [Google Scholar]
- Graff-Radford NR, Woodruff BK. Frontotemporal dementia. Semin Neurol. 2007;27:48–57. doi: 10.1055/s-2006-956755. [DOI] [PubMed] [Google Scholar]
- Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, Clark CM, Elman LB, Miller BL, Grossman M, McCluskey LF, Trojanowski JQ, Lee VM. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol. 2008;173:182–194. doi: 10.2353/ajpath.2008.080003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquier A, Bellouze S, Blanchard S, Bohl D, Haase G. Astrocytic protection of spinal motor neurons but not cortical neurons against loss of Als2/alsin function. Hum Mol Genet. 2009;18:2127–2139. doi: 10.1093/hmg/ddp136. [DOI] [PubMed] [Google Scholar]
- Johnson BS, McCaffery JM, Lindquist S, Gitler AD. A yeast TDP-43 proteinopathy model: exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 2008;105:6439–6444. doi: 10.1073/pnas.0802082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman D. Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci U S A. 1999;96:11404–11409. doi: 10.1073/pnas.96.20.11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S. Amyotrophic lateral sclerosis models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:97–114. doi: 10.1007/s00401-007-0308-4. [DOI] [PubMed] [Google Scholar]
- Ko LW, Ko HH, Lin WL, Kulathingal JG, Yen SH. Aggregates assembled from overexpression of wild-type alpha-synuclein are not toxic to human neuronal cells. J Neuropathol Exp Neurol. 2008;67:1084–1096. doi: 10.1097/NEN.0b013e31818c3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühnlein P, Sperfeld AD, Vanmassenhove B, Van Deerlin V, Lee VM, Trojanowski JQ, Kretzschmar HA, Ludolph AC, Neumann M. Two German kindreds with familial amyotrophic lateral sclerosis due to TARDBP mutations. Arch Neurol. 2008;65:1185–1189. doi: 10.1001/archneur.65.9.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Kwong LK, Neumann M, Sampathu DM, Lee VM, Trojanowski JQ. TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol. 2007;114:63–70. doi: 10.1007/s00401-007-0226-5. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136:1001–1004. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lariviere RC, Julien JP. Functions of intermediate filaments in neuronal development and disease. J Neurobiol. 2004;58:131–148. doi: 10.1002/neu.10270. [DOI] [PubMed] [Google Scholar]
- Lesné S, Kotilinek L, Ashe KH. Plaque-bearing mice with reduced levels of oligomeric amyloid-beta assemblies have intact memory function. Neuroscience. 2008;151:745–749. doi: 10.1016/j.neuroscience.2007.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Rademakers R. The molecular genetics and neuropathology of frontotemporal lobar degeneration: recent developments. Neurogenetics. 2007;8:237–248. doi: 10.1007/s10048-007-0102-4. [DOI] [PubMed] [Google Scholar]
- Mitra S, Tsvetkov AS, Finkbeiner S. Single neuron ubiquitin-proteasome dynamics accompanying inclusion body formation in Huntington disease. J Biol Chem. 2009;284:4398–4403. doi: 10.1074/jbc.M806269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Ou SH, Wu F, Harrich D, García-Martínez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69:3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintchovski SA, Peebles CL, Kim HJ, Verdin E, Finkbeiner S. The serum response factor and a putative novel transcription factor regulate expression of the immediate-early gene Arc/Arg3.1 in neurons. J Neurosci. 2009;29:1525–1537. doi: 10.1523/JNEUROSCI.5575-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranum LP, Cooper TA. RNA-mediated neuromuscular disorders. Annu Rev Neurosci. 2006;29:259–277. doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, Takimoto-Kimura R, Kleschevnikov AM, Sambamurti K, Chung PP, Xia W, Villar A, Campbell WA, Kulnane LS, Nixon RA, Lamb BT, Epstein CJ, Stokin GB, Goldstein LS, Mobley WC. Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51:29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong MJ, Volkening K, Hammond R, Yang W, Strong W, Leystra-Lantz C, Shoesmith C. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci. 2007;35:320–327. doi: 10.1016/j.mcn.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Theodore S, Cao S, McLean PJ, Standaert DG. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J Neuropathol Exp Neurol. 2008;67:1149–1158. doi: 10.1097/NEN.0b013e31818e5e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, Steinbart E, McCluskey L, Grossman M, Neumann M, Wu IL, Yang WS, Kalb R, Galasko DR, Montine TJ, Trojanowski JQ, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Wu LS, Chang HY, Shen CK. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem. 2008;105:797–806. doi: 10.1111/j.1471-4159.2007.05190.x. [DOI] [PubMed] [Google Scholar]
- Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem. 2008;283:13302–13309. doi: 10.1074/jbc.M800342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong NK, He BP, Strong MJ. Characterization of neuronal intermediate filament protein expression in cervical spinal motor neurons in sporadic amyotrophic lateral sclerosis (ALS) J Neuropathol Exp Neurol. 2000;59:972–982. doi: 10.1093/jnen/59.11.972. [DOI] [PubMed] [Google Scholar]
- Yokoseki A, Shiga A, Tan CF, Tagawa A, Kaneko H, Koyama A, Eguchi H, Tsujino A, Ikeuchi T, Kakita A, Okamoto K, Nishizawa M, Takahashi H, Onodera O. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol. 2008;63:538–542. doi: 10.1002/ana.21392. [DOI] [PubMed] [Google Scholar]