

Abstract
Repeated administration of opioids not only leads to tolerance and dependence, but also results in nociceptive enhancement called opioid-induced hyperalgesia (OIH). Nociceptive mediators involved in OIH generation remain poorly understood. In the present study, we tested the hypothesis that Ca2+/calmodulin-depent protein kinase II (CaMKIIα) is critical for OIH. Opioid-induced hyperalgesia was produced by repeated morphine administration or pellet implantation in mice. Correlating with the development of tactile allodynia and thermal hyperalgesia, spinal CaMKIIα activity was significantly increased in OIH. KN93, a CaMKII inhibitor, dose- and time-dependently reversed OIH and CaMKII activation without impairing locomotor coordination. To elucidate the specific CaMKII isoform involved, we targeted CaMKIIα by using small interfering RNA and demonstrated that knockdown of spinal CaMKIIα attenuated OIH. Furthermore, morphine failed to induce OIH in CaMKIIαT286A point mutant mice, although wild-type littermate mice developed robust OIH after repeated treatments with morphine. These data implicate, for the first time, an essential role of CaMKIIα as a cellular mechanism leading to and maintaining opioid-induced hyperalgesia.
Introduction
Opioids are commonly prescribed to treat chronic pain. However, repeated administration of opioids not only leads to tolerance and dependence, but also results in opioid-induced hyperalgesia (OIH), defined as a lowered pain threshold caused by opioid exposure (Vanderah et al., 2001; Mao, 2002; Ossipov et al., 2005; Chu et al., 2008). Although hyperalgesia associated with opioid use has been reported clinically for decades, the underlying mechanism of OIH remains unclear. Opioid-induced hyperalgesia is considered a unique phenomenon that can be distinguished from opioid withdrawal-induced hyperalgesia (Li et al., 2001a; Angst and Clark, 2006). When opioids are used for the treatment of underlying painful conditions, OIH may differ from the original pain condition in its quality and affected location (Chu et al., 2008). Clinically, OIH and opioid tolerance can both result in similar increased requirement of opioid dosage; however, OIH causes a downward shift of the opioid dose–response curve whereas tolerance leads to a rightward shift of the curve (Chu et al., 2008).
A growing number of reports have reproducibly demonstrated OIH in rodent models (Kayan et al., 1971; Gardell et al., 2002; Liang et al., 2006). Although a short onset (within 2–3 h) of mechanical allodynia and thermal hyperalgesia has been reported after acute administration of opioids, such as fentanyl and heroin (Célèrier et al., 1999, 2000), chronic morphine administration with repeated intermittent injections or subcutaneous pellet implantation produced prolonged and robust OIH (Li et al., 2001a; Liang et al., 2008).
Ca2+/calmodulin-dependent protein kinase II (CaMKII) is a multifunctional serine/threonine protein kinase that is colocalized with the μ-opioid receptor in the superficial laminae of the spinal dorsal horn and in the small to medium diameter primary afferent neurons in the dorsal root ganglion (Brüggemann et al., 2000; Carlton, 2002). Activation of CaMKII requires elevated intracellular Ca2+ and activated calmodulin. Supraspinal or spinal inhibition of CaMKII has been shown to prevent or reverse morphine tolerance and dependence (Tang et al., 2006a). Moreover, spinal CaMKIIα was activated in capsaicin-induced inflammation (Fang et al., 2002), complete Freund's adjuvant (CFA)-induced inflammatory pain (Luo et al., 2008), and spinal nerve ligation (SNL)-induced neuropathic pain states (Chen et al., 2009). CFA- or SNL-induced thermal hyperalgesia and mechanical allodynia can be dose-dependently reversed by CaMKII inhibitors (Luo et al., 2008; Chen et al., 2009). A role for CaMKII in OIH has not been reported. In this study, we tested the hypothesis that CaMKIIα is a critical cellular signaling mechanism leading to opioid-induced hyperalgesia. We first used KN93, the most commonly used CaMKII inhibitor, and its kinase-inactive chemical analog KN92 in the study (Sumi et al., 1991). To overcome potential nonspecific actions of KN93 and to investigate the specific CaMKII isoform involved, we also used small interfering RNA (siRNA) to knock down CaMKIIα. Furthermore, the hypothesis was tested in mice lacking the CaMKIIα autophosphorylation site.
Materials and Methods
Materials.
Morphine sulfate (MS), morphine, and placebo pellets were obtained from the National Institute on Drug Abuse (Rockville, MD). 2-[N-(2-Hydroxyethyl)-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine) (KN93) and 2-[N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine(KN92) were purchased from Calbiochem. Other chemicals were purchased from Sigma.
Animals.
Male ICR mice (20–25 g, Harlan Laboratories) were kept under standard conditions with a 14/10 h light/dark cycle (5:00 A.M. to 7:00 P.M.) and allowed food and water ad libitum before experimental procedures. For CaMKIIαT286A mutant mice, breeders were generously provided by Dr. Alcino Silva, University of California Los Angeles (Giese et al., 1998). These mice were back-crossed with C57BL/6J mice for 10 generations. Heterozygous breeding was used to generate male homozygous mutant mice and littermate wild-type control mice for this study. Both genotypes were viable and showed normal growth and reproduction. Genotyping of litters was performed by PCR using a set of primers (5′-CTGTACCAGCAGATCAAAGC-3′, 5′-ATCACTAGCACCATGTGGTC-3′). The PCR products for wild-type and mutant alleles are 200 and 290 bp, respectively. Each experimental mouse was genotyped twice using DNA from two separate extractions from the tail tissue samples. The investigator who performed the biochemical and behavioral tests was blind to mouse genotypes. All breeding and experiments were performed in accordance with the policies and recommendations of the International Association for the Study of Pain (IASP) and the NIH guidelines after approval by the University of Illinois Institutional Animal Care and Use Committee.
OIH induced by repeated subcutaneous administration.
To induce OIH, mice were treated subcutaneously according to a previously published schedule (Liang et al., 2006). Mice received 20 mg/kg morphine sulfate (twice per day, s.c.) for 3 consecutive days and two more injections of 40 mg/kg morphine sulfate on day 4. Control mice received equal volume and number of saline injections (subcutaneous). Mechanical and thermal sensitivities were tested before and after morphine treatment.
OIH induced by morphine pellet implantation.
Separate groups of 5 male ICR mice were implanted subcutaneously with morphine pellets or placebo pellets (1 pellet/mouse, each morphine pellet contains 75 mg of morphine base; a placebo pellet is contains no morphine). This treatment not only induces opioid dependence and tolerance (Tang et al., 2006a), but also OIH (Li et al., 2001b). To determine the presence of OIH, mechanical and thermal sensitivities were tested before and after morphine treatment as described below.
Drug and siRNA administration.
KN93 and KN92 were administered intrathecally by percutaneous puncture through the L5-L6 intervertebral space, as described previously (Hylden and Wilcox, 1980; Chen et al., 2009). A lateral tail flick was considered as success of the intrathecal injection. To inhibit CaMKIIα, CaMKIIα was targeted by small interfering RNA (siRNA). Four days after morphine pellet implantation, mice were treated with CaMKIIα siRNA (5′-CACCACCAUUGAGGACGAAdTdT-3′, 3′-dTdTGUGGUGGUAACUCCUGCUU-5′) (Zayzafoon et al., 2005) or Stealth RNAi negative control (Invitrogen) (2 μg, i.t., twice per day for 3 consecutive days). These oligos were mixed with the transfection reagent i-Fect (Neuromics), in a ratio of 1:5 (w/v) (Luo et al., 2005). Mechanical and thermal sensitivity tests were performed daily.
Mechanical sensitivity.
Mechanical sensitivity was assayed using calibrated von Frey filaments (Stoelting) according to the “up-down” paradigm (Luo et al., 2008; Chen et al., 2009). Briefly, mice were placed on wire mesh platforms in individual Plexiglas containers. After 30 min of acclimation, each von Frey filament was pressed upward to the mid-plantar surface for 5 s or until a withdrawal response occurred. Withdrawal of the hindpaw was scored as a response. The up-down algorithm was used to determine 50% probability of paw withdrawal threshold (Luo et al., 2008).
Thermal sensitivity.
Thermal sensitivity was measured using a plantar tester (model 7372, UGO BASILE) as described previously (Hargreaves et al., 1988; Wang et al., 2001; Chen et al., 2009). In these experiments, mice were placed in a clear plastic enclosure with a glass floor. After a 30 min period of habituation, radiant heat stimulation was applied to the middle portion of the planter surface of the hindpaw, and paw withdrawal latencies were measured. The heat intensity was adjusted to produce a baseline response around 10 s. To prevent tissue damage, a cutoff time of 20 s was applied.
Rotarod test.
To exclude the possibility that KN93 may cause locomotor impairment, a rotarod test was performed as described previously (Prestori et al., 2008). Mice were trained to remain on a fixed speed (4 rpm) rotarod for 60 s (model series 8; IITC Life Science). On the following day, mice were retrained and those that failed to stay on the rotarod for 60 s were not used in further studies. Baseline was tested 30 min later by placing the mice on an accelerating rotarod (4–40 rpm over 300 s). The latency to fall off the rotarod was recorded. Mice were then treated intrathecally with either KN93 (45 nmol), KN92 (45 nmol), or saline and retested 0.5, 1, 2, 4, and 8 h later. The cutoff was set at 300 s.
Immunoblotting.
Mice were killed and lumbar spinal cords were quickly dissected and frozen on dry ice for Western blotting analysis, as previously described (Tang et al., 2006a; Chen et al., 2009). Briefly, tissues were homogenized in ice-cold radioimmunoprecipitation assay buffer and centrifuged. Aliquots of supernatant samples were used to determine protein content by a modified Bradford method (Pierce). Samples (60 μg of total protein) were separated by 12% SDS-PAGE and electrotransferred onto PVDF membrane. The membrane was probed with a rabbit anti-(T286)pCaMKIIα antibody (1:1000, Santa Cruz Biotechnology) or a mouse anti-CaMKIIα antibody (1:1000, Santa Cruz Biotechnology) at room temperature for 3 h, followed by incubation with HRP-conjugated donkey anti-rabbit (for pCaMKIIα) or anti-mouse (for CaMKIIα) secondary antibody (1:1000, GE Healthcare). An enhanced chemiluminescence detection system (ECL, GE Healthcare) was applied for development. The membrane was then stripped and reprobed with a mouse anti-β-actin antibody (1:10,000, Santa Cruz Biotechnology) followed by a HRP-conjugated anti-mouse secondary antibody (1:1000, GE Healthcare) and developed as above. ECL signals were detected using a ChemiDoc system and analyzed using the Quantity One program (Bio-Rad). CaMKII immunoreactivity was expressed as the ratio of the optical densities of pCaMKII or CaMKIIα to those of β-actin.
Immunohistochemistry.
Immunohistochemical staining of spinal CaMKII was carried out according to the method previously described (Wang et al., 2001). Mice were deeply anesthetized with ketamine (100 mg/kg) and xylazine (5 mg/kg, i.p.). The vascular system was perfused with 60 ml of 4°C PBS, pH 7.4, and followed by 50 ml of 2% paraformaldehyde solution. The lumbar spinal cords were removed and postfixed overnight in 4% paraformaldehyde at 4°C, and cryoprotected in 30% sucrose for 24 h at 4°C. Spinal cord sections were cut on a cryostat at 20 μm thickness and washed 2 times with cold PBS. Sections were preincubated with 0.3% H2O2 and blocked with 10% goat serum (Invitrogen) and 0.3% Triton X-100 in PBS for 20 min. Floated sections were incubated with the primary antibody for pCaMKIIαThr286 (1:1000) overnight at room temperature, followed by another incubation with biotinylated goat anti-rabbit IgG secondary antibody (1:200, Vector Laboratories) at room temperature for 2 h. Incubation in the absence of the first antibody or in the presence of pCaMKIIαThr286 blocking peptide (Santa Cruz Biotechnology) was used to control for antibody specificity. The sections were developed using Elite Vectastain ABC kit (Vector Laboratories). Diaminobenzidine (DAB) stained sections were imaged by inverted microscopy (Olympus) and quantified using the MetaMorph Imaging Software (Universal Imaging). For each condition, 5 sections and 6 areas from each section were analyzed and averaged.
Statistical analysis.
All data are presented as mean ± SEM. Comparisons between groups were analyzed using Student's t test (two groups) or a two-way repeated measure ANOVA followed by post hoc analyses using Dunnett's t test (multiple groups). Statistical significance was established at 95% confidence limit.
Results
Chronic morphine exposure-induced mechanical allodynia and thermal hyperalgesia in mice
Repeated subcutaneous morphine administration and subcutaneous morphine pellet implantation are two commonly used OIH models in mice. Four days of subcutaneous morphine administration by intermittent injections significantly increased mechanical and thermal sensitivities compared with saline-treated mice (Fig. 1A,B). The mechanical allodynia and thermal hyperalgesia were detectable on day 5 and lasted for approximately 2 weeks before recovery (p < 0.001 compared with saline control, n = 5).
Figure 1.
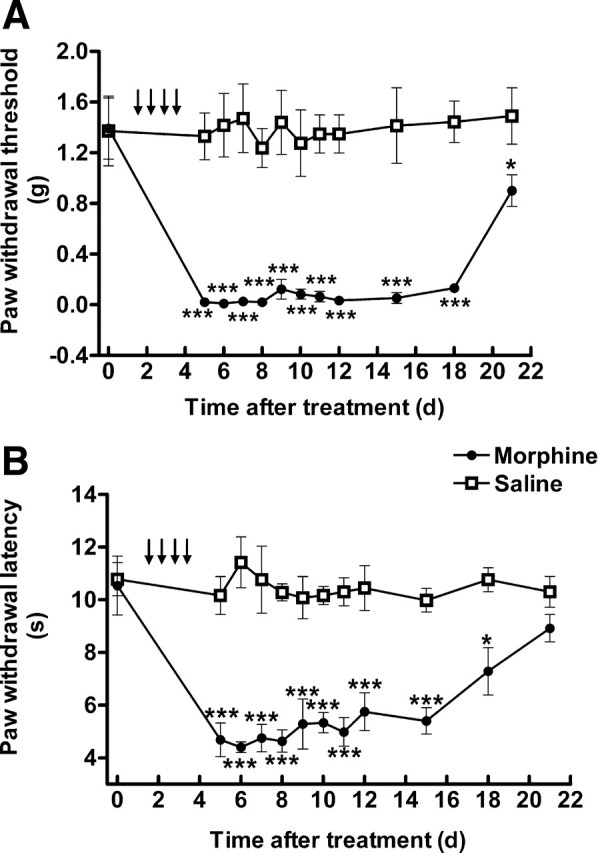
Repeated intermittent morphine administration-induced mechanical allodynia (A) and thermal hyperalgesia (B). Mice received saline or morphine sulfate (day 1–3: 20 mg/kg; day 4: 40 mg/kg; twice daily, s.c.). The paw withdrawal threshold to von Frey filament probing and withdrawal latency to radiant heat were determined. Data are expressed as mean ± SEM *p < 0.05, **p < 0.01, ***p < 0.001, compared with the saline-treated group; n = 5 for each group.
Continuous morphine exposure using pellet implantation also induced OIH. Mice were implanted subcutaneously with morphine pellets (75 mg/pellet) or placebo pellet and mechanical and thermal sensitivities were measured daily for 15 d. Morphine implantation initially produced antinociception in both mechanical (p < 0.001, compared with the placebo group, n = 5) and thermal sensitivity tests (p < 0.05, compared with the placebo group, n = 5). This was followed by a decrease in paw withdrawal threshold and latency. Mechanical allodynia was developed on day 6 and lasted for 7 d (p < 0.001, compared with placebo group, n = 5) (Fig. 2A,B). Thermal hyperalgesia was also observed from day 5 to day 9 (p < 0.001, compared with placebo group, n = 5) after morphine implantation. Comparing the two OIH models, repeated intermittent morphine administration led to longer lasting and more robust mechanical allodynia and thermal hyperalgesia in ICR mice. Therefore, this model was used for the CaMKII intervention studies.
Figure 2.
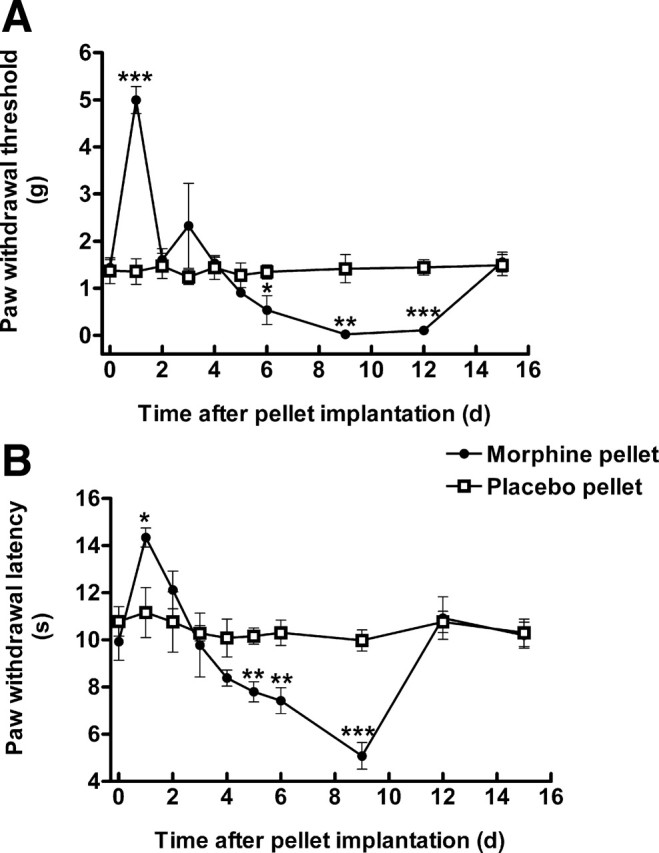
Morphine pellet implantation-induced mechanical allodynia (A) and thermal hyperalgesia (B). Mice were implanted subcutaneously with morphine or placebo pellets. The paw withdrawal threshold to von Frey filament probing and withdrawal latency to radiant heat were measured daily. Morphine initially produced antinociception followed by a steep decrease in mechanical threshold (A) and thermal withdrawal latency (B), indicative of the development of OIH. Data are expressed as mean ± SEM *p < 0.05, **p < 0.01, ***p < 0.001, compared with the placebo pellet group; n = 5 for each group.
CaMKII inhibition by KN93 reversed morphine-induced hyperalgesia
In order to investigate the possible role of CaMKII in OIH, we used KN93, a CaMKII inhibitor, in the initial study. KN92, a kinase inactive analog of KN93, was used as a negative control. Both mechanical allodynia and thermal hyperalgesia were significantly developed on day 5. At that point, mice were treated with KN93 (15–45 nmol, i.t.) or KN92 (45 nmol, i.t.) and mechanical and thermal sensitivities were monitored for 24 h. Opioid-induced hyperalgesia was found to be attenuated by KN93 in a dose- and time-dependent manner. At the highest dose, KN93 (45 nmol) completely reversed allodynia and hyperalgesia. Its anti-allodynic/anti-hyperalgesic effect appeared 30 min after KN93 administration and peaked at 2 h (Fig. 3). The anti-allodynic/anti-hyperalgesic action of KN93 diminished at 8 h postadministration. At lower doses, KN93 (30 nmol, i.t.) partially suppressed allodynia and hyperalgesia, while KN93 at 15 nmol did not affect either thermal or mechanical sensitivity at any time point tested. At the peak-effect time (2 h), ED50 values were estimated to be 26.1 ± 3.4 nmol (anti-allodynic) and 17.8 ± 3.0 nmol (anti-hyperalgesic). In contrast, KN92 (45 nmol, i.t.) did not alter the pain threshold at any time point tested, which strongly supported a CaMKII-mediated effort exhibited by KN93. To rule out a potential negative effect of KN93 on locomotor coordination that can be a confounding factor in data interpretation, we further tested the effect of KN93 on locomotor activity in a rotarod test. Administration of KN93 (45 nmol, i.t.) or KN92 (45 nmol, i.t.) to naive mice did not produce significant changes to the locomotor coordination in the rotarod test (p > 0.05, compared with saline-treated group, n = 5) (Fig. 4). These data are in agreement with our previous observation that KN93 (45 nmol, i.t.) did not change mechanical and thermal sensitivities in naive mice (Luo et al., 2008).
Figure 3.

Reversal of morphine-induced mechanical allodynia (A) and thermal hyperalgesia (B) by KN93. OIH was induced by intermittent morphine injections. On day 5, mice received KN93 (15–45 nmol, i.t.), KN92 (45 nmol, i.t.), or saline (intrathecally) at time 0. Mechanical allodynia and thermal hyperalgesia were tested at the different time points as indicated. KN93, but not KN92, reversed the established morphine-induced mechanical allodynia and thermal hyperalgesia in a dose- and time-dependent manner. Data are expressed as mean ± SEM *p < 0.05, ***p < 0.001, compared with the saline-treated group; †p < 0.05, †††p < 0.001, compared with the morphine-treated group; n = 8 for each group.
Figure 4.

Effect of KN93 on locomotor activity. Mice were trained to remain on a fixed speed (4 rpm) rotarod for 60 s. On the following day, each mouse was retrained to ensure that it could stay on the rotarod for at least 60 s. Baseline was tested by placing the mice on an accelerating rotarod (4–40 rpm over 300 s). The latency to fall from the rotarod was recorded. Mice were then administered with either KN93 (45 nmol), KN92 (45 nmol) or saline intrathecally and retested 0.5, 1, 2, 4 and 8 h later. Neither KN93 nor KN92 significantly impaired locomotor activity. Data are expressed as mean ± SEM p > 0.05, compared with the saline-treated group; n = 5 for each group.
To correlate behavioral effects with biochemical changes, CaMKIIα activity was determined by analyzing the degree of autophosphorylation (pCaMKIIα) (Fang et al., 2002; Xu and Huang, 2004). Compared with the saline control, repeated morphine administration significantly increased spinal pCaMKIIα (Fig. 5A; p < 0.05, n = 4), without significantly altering its expression (Fig. 5B; p > 0.05, n = 4). KN93, at the highest dose used (45 nmol, 1 h), significantly reversed the morphine-induced activation of CaMKIIα (p < 0.05, compared with morphine group, n = 4). In contrast, KN92 (45 nmol) did not change morphine-induced pCaMKIIα. Activation of spinal CaMKIIα was also examined using immunohistochemical approach. After chronic morphine exposure, increased pCaMKIIα immunoreactivity was found mostly in the superficial laminae of the spinal dorsal horn compared with the saline-treated mice (Fig. 6I). Some pCaMKIIα immunoreactivity was also found in the neuropils which has been reported by others as well (Liang et al., 2004; Fujiyoshi et al., 2007; Song et al., 2009). However, the enhanced pCaMKIIα immunostaining was significantly suppressed by KN93 (45 nmol, i.t.) (Fig. 6II). These data suggested that chronic morphine-induced mechanical allodynia and thermal hyperalgesia, and their reversal by KN93, are biochemically correlated with morphine-induced CaMKIIα activation and subsequent inactivation in the presence of KN93, but not KN92.
Figure 5.
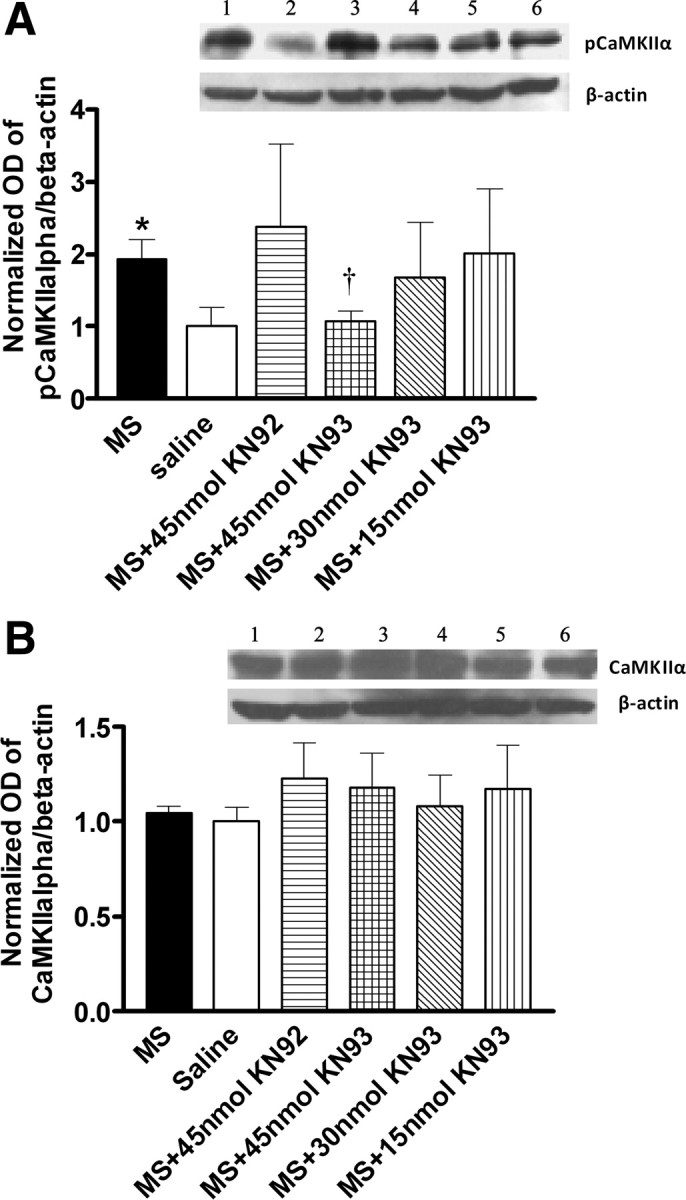
Suppression of morphine-induced CaMKIIα activation by KN93. Morphine or saline-treated mice were administered intrathecally with KN93 (15–45 nmol), KN92 (45 nmol), or saline on day 5. One hour later, mice were killed and the lumbar spinal cords were taken for the analysis of CaMKIIα activation using the immunoblotting method, by determining the degree of CaMKIIα autophosphorylation (pCaMKIIα). Morphine enhanced pCaMKIIα (A), without altering CaMKIIα expression (B). KN93, but not its inactive analog, KN92, reversed morphine enhanced CaMKIIα activation. Data are expressed as mean ± SEM, *p < 0.05, compared with the saline-treated group; †p < 0.05, compared with the morphine-treated group; n = 4 for each group.
Figure 6.
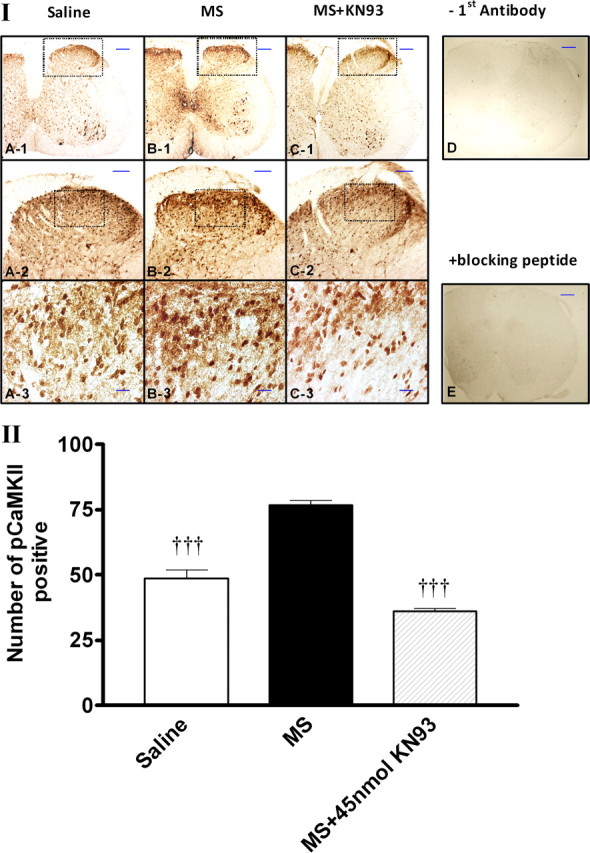
Immunohistochemical staining of pCaMKIIα expression after the treatment with KN93. I, Morphine- or saline-treated mice were administered (intrathecally) with KN93 (15–45 nmol), KN92 (45 nmol), or saline on day 5. Mice were killed 1 h after the treatment with saline or KN93 (45 nmol, i.t.). The lumbar spinal section was dissected out and immunostained with pCaMKIIα antibody. II, Quantitative analysis of pCaMKIIα immunoreactivity was performed by counting the number of positively stained cells using the MetaMorph Imaging Software. No pCaMKIIα immunoreactivity was detected if the first antibody was omitted (D) or if the first antibody was incubated in the presence of pCaMKIIαT286 blocking peptide (Santa Cruz Biotechnology). Data are expressed as mean ± SEM, †††p < 0.001, compared with the morphine-treated group; n = 3 for each group. Scale bars are 200 μm (A-1, B-1, C-1, D, E), 100 μm (A-2, B-2, C-2), or 20 μm (A-3, B-3, C-3).
Small interfering RNA(siRNA)-mediated knockdown of CaMKIIα and reversal of morphine- induced hyperalgesia
To further investigate the specific isoform of CaMKII involved in OIH, siRNA targeting CaMKIIα (Zayzafoon et al., 2005) was applied to knock down the expression of CaMKIIα in the spinal cord. Mice received repeated morphine administration in phase 1 (Fig. 7) and, as expected, developed mechanical allodynia and thermal hyperalgesia 18 h after the final morphine injection. In phase 2, mice received intrathecal administration of CaMKIIα siRNA or scrambled siRNA (2 μg/injection, twice per day for 3 consecutive days) and the sensitivities to mechanical and thermal stimuli were measured daily. Treatment with CaMKIIα siRNA gradually attenuated morphine-induced mechanical allodynia (Fig. 7A) and thermal hyperalgesia (Fig. 7B). After 3 d of treatment, OIH was completely reversed (Fig. 7, p < 0.001, compared with morphine-treated group, n = 5). Correlating with the behavioral effect, pCaMKIIα immunostaining in the spinal dorsal horn was significantly suppressed (Fig. 8). In contrast, scrambled siRNA did not alter OIH (Fig. 7) or morphine-induced pCaMKIIα immunoreactivity (Fig. 8).
Figure 7.
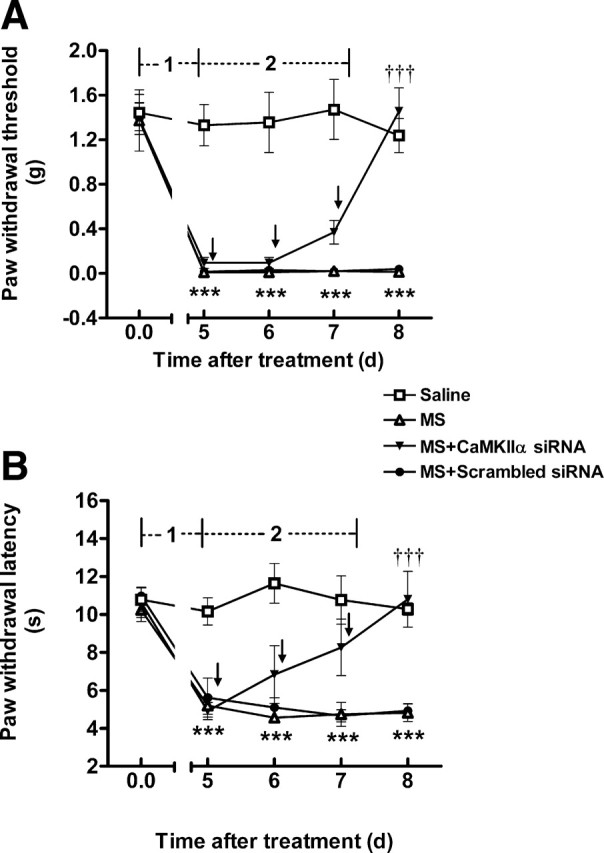
Reversal of OIH by siRNA-mediated CaMKIIα knockdown. In phase 1, mice received repeated saline or morphine administration. Mechanical and thermal sensitivities were measured on day 5. In phase 2, after OIH had fully developed, mice were treated with CaMKIIα siRNA or scrambled siRNA (2 μg, twice per day for 3 d). Mechanical and thermal sensitivities were tested daily. Established OIH was reversed by CaMKIIα siRNA. Data are expressed as mean ± SEM ***p < 0.001, compared with the saline-treated group; †††p < 0.001, compared with the morphine-treated group; n = 5 for each group. Arrows indicated saline, CaMKIIα, or scrambled siRNA administration.
Figure 8.
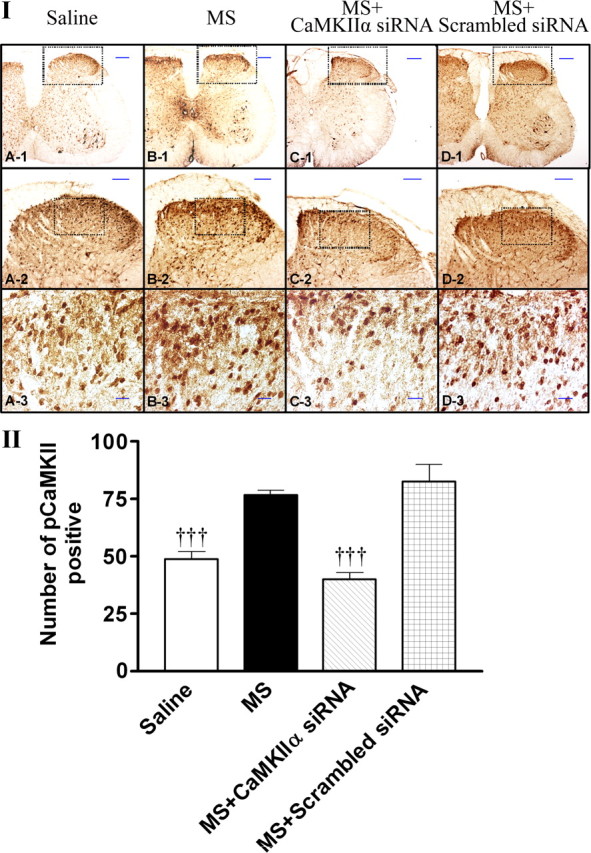
Suppression of morphine-induced CaMKIIα activation by CaMKIIα siRNA. I, Naive mice (A) or mice with OIH were treated with saline (B), CaMKIIα siRNA (2 μg, twice daily for 3 d) (C), or scrambled siRNA (2 μg, twice daily for 3 d). One day after the last injection of siRNA, the spinal lumbar region was immunostained with pCaMKIIα antibody. II, Quantitative analysis of pCaMKIIα immunoactivity was performed by counting the number of positively stained cells using MetaMorph Imaging Software. Data are expressed as mean ± SEM, †††p < 0.001, compared with the morphine-treated group; n = 3 for each group. Scale bars are 200 μm (A-1, B-1, C-1, D-1), 100 μm (A-2, B-2, C-2, D-2), or 20 μm (A-3, B-3, C-3, D-3).
Absence of morphine-induced hyperalgesia in CaMKIIαT286A point mutation mice
To investigate whether functional CaMKIIα is also required for the development of OIH, we used CaMKIIαT286A mice in the study. These mice were engineered to carry a T286A point mutation, lacking the ability to undergo CaMKIIα autophosphorylation and further activation (Giese et al., 1998). We investigated whether inactivation of CaMKIIα by T286A point mutation affected morphine-induced hyperalgesia. Separate group of 6 male CaMKIIαT286A mutant and littermate wild-type mice received repeated injections of morphine every 12 h for 4 d using the same treatment schedule as described above. Baseline pain thresholds were not significantly different between wild-type and mutant mice (Fig. 9). The wild-type mice exhibited mechanical allodynia and thermal hyperalgesia on day 5, indicative of the presence of OIH (p < 0.01, compared with baseline, n = 6). However, mechanical allodynia and thermal hyperalgesia were not detected in morphine-treated CaMKIIαT286A mutant mice. These data indicated that CaMKIIα is required for the initiation of OIH (Fig. 9).
Figure 9.
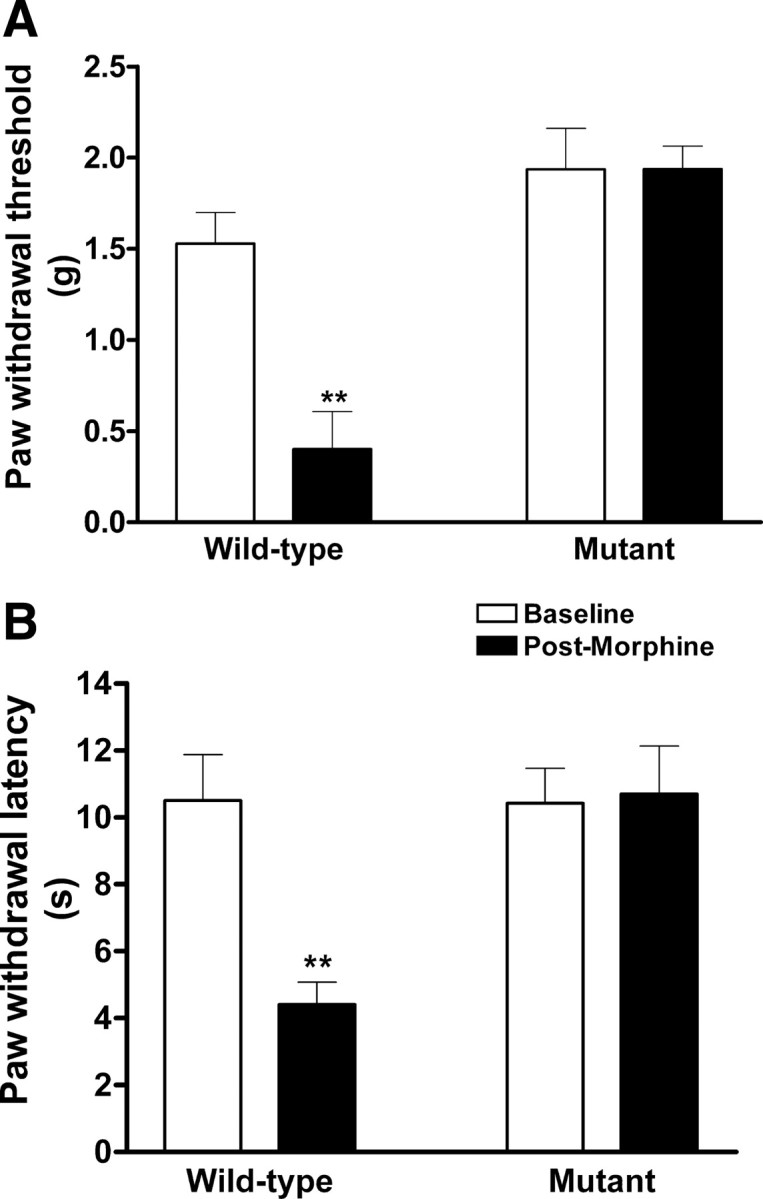
Morphine failed to induce hyperalgesia in CaMKIIαT286A mutant mice. Male CaMKIIαT286A mutant and littermate wild-type mice were subcutaneously administered saline or morphine sulfate (day 1–3: 20 mg/kg/d; day 4: 40 mg/kg, twice daily). Baseline pain thresholds were not significantly different between wild-type and mutant mice. Mechanical and thermal sensitivities were tested on day 5. Morphine treatment successfully induced hyperalgesia in CaMKIIαT286A+/+ mice (**p < 0.01, compared with the baseline). However, CaMKIIαT286A−/− mice failed to develop OIH (p > 0.05, compared with the baseline). Data are expressed as mean ± SEM; n = 6 for each group.
Discussion
In this study, we tested the hypothesis that CaMKIIα is a required component for the development and maintenance of OIH. We found that acute CaMKII inhibition by KN93 or siRNA-mediated knockdown of CaMKIIα reversed opioid-induced mechanical allodynia and thermal hyperalgesia. OIH was absent in CaMKIIαT286A mutant mice. Comparing two mouse models of OIH, the intermittent injection method offered longer lasting and more robust OIH compared with the pellet model, and was chosen as the model for CaMKII intervention studies. CaMKII inhibition by KN93 reversed OIH, correlating with decreased CaMKIIα activation by KN93 in the spinal cord. In contrast, the kinase-inactive control compound KN92 did not affect CaMKIIα activity or OIH. In the previous studies, we found that KN92 did not inhibit other isoforms of CaMKII, either (Tang et al., 2006a; Luo et al., 2008; Chen et al., 2009). The methoxybenzenesulfonamide KN93 is the most commonly used CaMKII inhibitor. It does not inhibit protein kinase C, protein kinase A, or even other CaM-dependent enzymes such as MLCK or CaM-dependent phosphodiesterase at very high concentrations (Sumi et al., 1991; Niki et al., 1993). Due to its wide applications, however, several nonspecific actions have been reported for the compound. For example, both KN93 and KN92 blocked voltage-dependent potassium channels in vascular myocytes (Ledoux et al., 1999; Rezazadeh et al., 2006) and L-type calcium channels in SH-SY5Y cells (Gao et al., 2006). That is why we have applied the negative control, KN92, at a high dose in all studies involving KN93. The isoquinolinesulfonamide derivative, KN62, another CaMKII inhibitor, has been found to act as an antagonist of the P2X7 receptor (Chessell et al., 1998; Baraldi et al., 2003), although such a CaMKII-independent effect has not been reported for KN93 or KN92.
To ascertain a CaMKII-mediated mechanism and to investigate the specific isoform involved, we further targeted CaMKIIα by genetic approaches. Knockdown of CaMKIIα by siRNA and resultant suppression of OIH suggested that CaMKIIα was the relevant isoform. These studies were controlled by scrambled siRNA to exclude off-target interference. Some siRNA molecules have been reported to cause nonspecific interferon response (Bridge et al., 2003; Sledz et al., 2003; Hornung et al., 2005), leading to nonspecific inhibition of protein synthesis and nonspecific degradation of endogenous mRNA. In a separate study, we tested the effect of CaMKIIα siRNA on the expression of five key genes: OAS1, OAS2, MX1, ISGF3γ and IFITM1 that are involved in the interferon response (Veals et al., 1992; Deblandre et al., 1995; Hovnanian et al., 1998; Kochs et al., 2002) and found that the sequence did not produce an interferon response. Moreover, lack of morphine-induced hyperalgesia in CaMKIIαT286A−/− mice further indicated that CaMKIIα is required for the initiation of OIH. These data, for the first time, implicated a critical role of CaMKIIα in OIH.
In addition to CaMKIIα, several other mechanisms underlying OIH have been suggested, such as the β2-adrenergic receptor (Liang et al., 2006), spinal cyclooxygenase (COX) (Dunbar et al., 2000), as well as local cytokine production (Liang et al., 2008). It has been suggested that both peripheral and central sensitization are important for the development of OIH (Chu et al., 2008). Blockade of the N-methyl-d-aspartate (NMDA) receptor by MK801 or ketamine has been shown to reverse (Li et al., 2001a) or prevent (Rivat et al., 2002) OIH. A vast amount of information is available for the interaction of CaMKIIα and the NMDA receptor, although these data are mostly from learning and memory studies. Both CaMKIIα and the NMDA receptor are required for the generation of long-term potentiation (LTP) in hippocampal neurons (Mayford et al., 1996). Inhibition of CaMKII or deletion of the CaMKII gene impairs spatial learning (Silva et al., 1992; Wolfman et al., 1994; Silva and Josselyn, 2002). It has been demonstrated that Ca2+ influx via the activation of NMDA receptors results in CaMKIIα activation and autophosphorylation of CaMKIIα at position Thr286 (Fukunaga et al., 1992; Strack et al., 2000). Cytosolic free Ca2+ was increased after treatment with opioids (Smart et al., 1997; Spencer et al., 1997; Quillan et al., 2002). We and others have demonstrated that CaMKIIα activity is increased after chronic treatment with opioids (Tang et al., 2006a). On the other hand, CaMKIIα can phosphorylate and activate the NMDA receptor, leading to the influx of Ca2+ through the channels (Kitamura et al., 1993). Therefore, CaMKII and the NMDA receptor may interact in a feedforward manner in OIH. Such a mechanism may explain the persistent nature of OIH, especially if one considers that desensitization can occur to a protein kinase after prolonged activation.
The μ-opioid receptor is another potential substrate of CaMKIIα. The receptor is expressed in the primary afferent neurons and is colocalized with CaMKII in pain-processing regions such as dorsal root ganglion sensory neurons and superficial layers of the spinal dorsal horn (Brüggemann et al., 2000; Carlton, 2002). The interaction between CaMKIIα and the μ-opioid receptor was further supported by cellular studies. The μ-opioid receptor contains consensus CaMKIIα phosphorylation sites. Desensitization to the μ-opioid receptor was enhanced when a constitutively active CaMKIIα was expressed in Xenopus oocytes (Mestek et al., 1995; Koch et al., 1997), which was abolished if the receptor was mutated to delete the putative CaMKIIα phosphorylation sites (Koch et al., 1997). In addition to the NMDA receptor and the μ-opioid receptor, there are likely many other potential substrates for CaMKIIα and it will be imperative to identify these targets in future studies to better understand and ultimately treat OIH. Such searches may benefit from increasingly available proteomics tools.
Both CaMKII and PKC are important kinases mediating Ca2+ signaling. Ca2+ influx through ion channels such as the NMDA receptor results in the activation of CaMKII and PKC. Enhanced activation of PKC has been reported in OIH (Célèrier et al., 2004). Opioid-induced hyperalgesia is absent in mice lacking the gene coding for PKCγ (Célèrier et al., 2004). It will be very interesting to study the possible crosstalk between CaMKII and PKC in OIH. The NR2A subunit of the NMDA receptor serves as the CaMKIIα docking site that binds to autophosphorylated CaMKIIα, a process that is known to be regulated by PKC (Gardoni et al., 2001; Leonard et al., 2002). Recently, we have identified the neuronal PKC substrate, neurogranin, as a mechanism that synchronizes the action of CaMKII and PKC in opioid tolerance and dependence (Shukla et al., 2006; Tang et al., 2006b, 2007).
In summary, our data strongly implicate CaMKIIα as a cellular mechanism leading to and maintaining opioid-induced hyperalgesia. Accumulating evidence highlights an important role for CaMKII in morphine tolerance and dependence (Tang et al., 2006a), CFA-induced inflammatory pain (Luo et al., 2008), and spinal nerve injury-induced neuropathic pain (Chen et al., 2009). These findings add to the growing literature that OIH, opioid tolerance, opioid dependence, and neuropathic pain may share common mechanisms at a certain level, although they are distinct phenomena (Mayer et al., 1999; Horvath and DeLeo, 2009). Targeting CaMKII may be useful for the prevention and treatment of OIH.
Footnotes
This work was supported in part by National Institutes of Health Grants HL098141, AT003647, and DA005050. Y.C. was supported by a University of Illinois Fellow scholarship.
References
- Angst MS, Clark JD. Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology. 2006;104:570–587. doi: 10.1097/00000542-200603000-00025. [DOI] [PubMed] [Google Scholar]
- Baraldi PG, del Carmen Nuñez M, Morelli A, Falzoni S, Di Virgilio F, Romagnoli R. Synthesis and biological activity of N-arylpiperazine-modified analogues of KN-62, a potent antagonist of the purinergic P2X7 receptor. J Med Chem. 2003;46:1318–1329. doi: 10.1021/jm021049d. [DOI] [PubMed] [Google Scholar]
- Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R. Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet. 2003;34:263–264. doi: 10.1038/ng1173. [DOI] [PubMed] [Google Scholar]
- Brüggemann I, Schulz S, Wiborny D, Höllt V. Colocalization of the mu-opioid receptor and calcium/calmodulin-dependent kinase II in distinct pain-processing brain regions. Brain Res Mol Brain Res. 2000;85:239–250. doi: 10.1016/s0169-328x(00)00265-5. [DOI] [PubMed] [Google Scholar]
- Carlton SM. Localization of CaMKIIalpha in rat primary sensory neurons: increase in inflammation. Brain Res. 2002;947:252–259. doi: 10.1016/s0006-8993(02)02932-3. [DOI] [PubMed] [Google Scholar]
- Célèrier E, Laulin J, Larcher A, Le Moal M, Simonnet G. Evidence for opiate-activated NMDA processes masking opiate analgesia in rats. Brain Res. 1999;847:18–25. doi: 10.1016/s0006-8993(99)01998-8. [DOI] [PubMed] [Google Scholar]
- Célèrier E, Rivat C, Jun Y, Laulin JP, Larcher A, Reynier P, Simonnet G. Long-lasting hyperalgesia induced by fentanyl in rats: preventive effect of ketamine. Anesthesiology. 2000;92:465–472. doi: 10.1097/00000542-200002000-00029. [DOI] [PubMed] [Google Scholar]
- Célèrier E, Simonnet G, Maldonado R. Prevention of fentanyl-induced delayed pronociceptive effects in mice lacking the protein kinase Cgamma gene. Neuropharmacology. 2004;46:264–272. doi: 10.1016/j.neuropharm.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Chen Y, Luo F, Yang C, Kirkmire CM, Wang ZJ. Acute inhibition of Ca2+/calmodulin-dependent protein kinase II reverses experimental neuropathic pain in mice. J Pharmacol Exp Ther. 2009;330:650–659. doi: 10.1124/jpet.109.152165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chessell IP, Michel AD, Humphrey PP. Effects of antagonists at the human recombinant P2X7 receptor. Br J Pharmacol. 1998;124:1314–1320. doi: 10.1038/sj.bjp.0701958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu LF, Angst MS, Clark D. Opioid-induced hyperalgesia in humans: molecular mechanisms and clinical considerations. Clin J Pain. 2008;24:479–496. doi: 10.1097/AJP.0b013e31816b2f43. [DOI] [PubMed] [Google Scholar]
- Deblandre GA, Marinx OP, Evans SS, Majjaj S, Leo O, Caput D, Huez GA, Wathelet MG. Expression cloning of an interferon-inducible 17-kDa membrane protein implicated in the control of cell growth. J Biol Chem. 1995;270:23860–23866. doi: 10.1074/jbc.270.40.23860. [DOI] [PubMed] [Google Scholar]
- Dunbar SA, Karamov IG, Buerkle H. The effect of spinal ibuprofen on opioid withdrawal in the rat. Anesth Analg. 2000;91:417–422. doi: 10.1097/00000539-200008000-00035. [DOI] [PubMed] [Google Scholar]
- Fang L, Wu J, Lin Q, Willis WD. Calcium-calmodulin-dependent protein kinase II contributes to spinal cord central sensitization. J Neurosci. 2002;22:4196–4204. doi: 10.1523/JNEUROSCI.22-10-04196.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiyoshi K, Yamada M, Nakamura M, Yamane J, Katoh H, Kitamura K, Kawai K, Okada S, Momoshima S, Toyama Y, Okano H. In vivo tracing of neural tracts in the intact and injured spinal cord of marmosets by diffusion tensor tractography. J Neurosci. 2007;27:11991–11998. doi: 10.1523/JNEUROSCI.3354-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga K, Soderling TR, Miyamoto E. Activation of Ca2+/calmodulin-dependent protein kinase II and protein kinase C by glutamate in cultured rat hippocampal neurons. J Biol Chem. 1992;267:22527–22533. [PubMed] [Google Scholar]
- Gao L, Blair LA, Marshall J. CaMKII-independent effects of KN93 and its inactive analog KN92: reversible inhibition of L-type calcium channels. Biochem Biophys Res Commun. 2006;345:1606–1610. doi: 10.1016/j.bbrc.2006.05.066. [DOI] [PubMed] [Google Scholar]
- Gardell LR, Wang R, Burgess SE, Ossipov MH, Vanderah TW, Malan TP, Jr, Lai J, Porreca F. Sustained morphine exposure induces a spinal dynorphin-dependent enhancement of excitatory transmitter release from primary afferent fibers. J Neurosci. 2002;22:6747–6755. doi: 10.1523/JNEUROSCI.22-15-06747.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni F, Bellone C, Cattabeni F, Di Luca M. Protein kinase C activation modulates alpha-calmodulin kinase II binding to NR2A subunit of N-methyl-d-aspartate receptor complex. J Biol Chem. 2001;276:7609–7613. doi: 10.1074/jbc.M009922200. [DOI] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, Noronha A, Manoharan M, Akira S, de Fougerolles A, Endres S, Hartmann G. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. 2005;11:263–270. doi: 10.1038/nm1191. [DOI] [PubMed] [Google Scholar]
- Horvath RJ, DeLeo JA. Morphine enhances microglial migration through modulation of P2X4 receptor signaling. J Neurosci. 2009;29:998–1005. doi: 10.1523/JNEUROSCI.4595-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovnanian A, Rebouillat D, Mattei MG, Levy ER, Marié I, Monaco AP, Hovanessian AG. The human 2′,5′-oligoadenylate synthetase locus is composed of three distinct genes clustered on chromosome 12q24.2 encoding the 100-, 69-, and 40-kDa forms. Genomics. 1998;52:267–277. doi: 10.1006/geno.1998.5443. [DOI] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- Kayan S, Woods LA, Mitchell CL. Morphine-induced hyperalgesia in rats tested on the hot plate. J Pharmacol Exp Ther. 1971;177:509–513. [PubMed] [Google Scholar]
- Kitamura Y, Miyazaki A, Yamanaka Y, Nomura Y. Stimulatory effects of protein kinase C and calmodulin kinase II on N-methyl-d-aspartate receptor/channels in the postsynaptic density of rat brain. J Neurochem. 1993;61:100–109. doi: 10.1111/j.1471-4159.1993.tb03542.x. [DOI] [PubMed] [Google Scholar]
- Koch T, Kroslak T, Mayer P, Raulf E, Höllt V. Site mutation in the rat mu-opioid receptor demonstrates the involvement of calcium/calmodulin-dependent protein kinase II in agonist-mediated desensitization. J Neurochem. 1997;69:1767–1770. doi: 10.1046/j.1471-4159.1997.69041767.x. [DOI] [PubMed] [Google Scholar]
- Kochs G, Haener M, Aebi U, Haller O. Self-assembly of human MxA GTPase into highly ordered dynamin-like oligomers. J Biol Chem. 2002;277:14172–14176. doi: 10.1074/jbc.M200244200. [DOI] [PubMed] [Google Scholar]
- Ledoux J, Chartier D, Leblanc N. Inhibitors of calmodulin-dependent protein kinase are nonspecific blockers of voltage-dependent K+ channels in vascular myocytes. J Pharmacol Exp Ther. 1999;290:1165–1174. [PubMed] [Google Scholar]
- Leonard AS, Bayer KU, Merrill MA, Lim IA, Shea MA, Schulman H, Hell JW. Regulation of calcium/calmodulin-dependent protein kinase II docking to N-methyl-d-aspartate receptors by calcium/calmodulin and alpha-actinin. J Biol Chem. 2002;277:48441–48448. doi: 10.1074/jbc.M205164200. [DOI] [PubMed] [Google Scholar]
- Li X, Angst MS, Clark JD. Opioid-induced hyperalgesia and incisional pain. Anesth Analg. 2001a;93:204–209. doi: 10.1097/00000539-200107000-00040. [DOI] [PubMed] [Google Scholar]
- Li X, Angst MS, Clark JD. A murine model of opioid-induced hyperalgesia. Brain Res Mol Brain Res. 2001b;86:56–62. doi: 10.1016/s0169-328x(00)00260-6. [DOI] [PubMed] [Google Scholar]
- Liang D, Li X, Clark JD. Increased expression of Ca2+/calmodulin-dependent protein kinase II alpha during chronic morphine exposure. Neuroscience. 2004;123:769–775. doi: 10.1016/j.neuroscience.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Liang D, Shi X, Qiao Y, Angst MS, Yeomans DC, Clark JD. Chronic morphine administration enhances nociceptive sensitivity and local cytokine production after incision. Mol Pain. 2008;4:7. doi: 10.1186/1744-8069-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang DY, Liao G, Wang J, Usuka J, Guo Y, Peltz G, Clark JD. A genetic analysis of opioid-induced hyperalgesia in mice. Anesthesiology. 2006;104:1054–1062. doi: 10.1097/00000542-200605000-00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo F, Yang C, Chen Y, Shukla P, Tang L, Wang LX, Wang ZJ. Reversal of chronic inflammatory pain by acute inhibition of Ca2+/calmodulin-dependent protein kinase II. J Pharmacol Exp Ther. 2008;325:267–275. doi: 10.1124/jpet.107.132167. [DOI] [PubMed] [Google Scholar]
- Luo MC, Zhang DQ, Ma SW, Huang YY, Shuster SJ, Porreca F, Lai J. An efficient intrathecal delivery of small interfering RNA to the spinal cord and peripheral neurons. Mol Pain. 2005;1:29. doi: 10.1186/1744-8069-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J. Opioid-induced abnormal pain sensitivity: implications in clinical opioid therapy. Pain. 2002;100:213–217. doi: 10.1016/S0304-3959(02)00422-0. [DOI] [PubMed] [Google Scholar]
- Mayer DJ, Mao J, Holt J, Price DD. Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc Natl Acad Sci U S A. 1999;96:7731–7736. doi: 10.1073/pnas.96.14.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- Mestek A, Hurley JH, Bye LS, Campbell AD, Chen Y, Tian M, Liu J, Schulman H, Yu L. The human mu opioid receptor: modulation of functional desensitization by calcium/calmodulin-dependent protein kinase and protein kinase C. J Neurosci. 1995;15:2396–2406. doi: 10.1523/JNEUROSCI.15-03-02396.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niki I, Okazaki K, Saitoh M, Niki A, Niki H, Tamagawa T, Iguchi A, Hidaka H. Presence and possible involvement of Ca/calmodulin-dependent protein kinases in insulin release from the rat pancreatic beta cell. Biochem Biophys Res Commun. 1993;191:255–261. doi: 10.1006/bbrc.1993.1210. [DOI] [PubMed] [Google Scholar]
- Ong ST, Li F, Du J, Tan YW, Wang S. Hybrid cytomegalovirus enhancer-h1 promoter-based plasmid and baculovirus vectors mediate effective RNA interference. Hum Gene Ther. 2005;16:1404–1412. doi: 10.1089/hum.2005.16.1404. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, King T, Vanderah TW, Porreca F. Underlying mechanisms of pronociceptive consequences of prolonged morphine exposure. Biopolymers. 2005;80:319–324. doi: 10.1002/bip.20254. [DOI] [PubMed] [Google Scholar]
- Prestori F, Rossi P, Bearzatto B, Lainé J, Necchi D, Diwakar S, Schiffmann SN, Axelrad H, D'Angelo E. Altered neuron excitability and synaptic plasticity in the cerebellar granular layer of juvenile prion protein knock-out mice with impaired motor control. J Neurosci. 2008;28:7091–7103. doi: 10.1523/JNEUROSCI.0409-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quillan JM, Carlson KW, Song C, Wang D, Sadée W. Differential effects of mu-opioid receptor ligands on Ca(2+) signaling. J Pharmacol Exp Ther. 2002;302:1002–1012. doi: 10.1124/jpet.302.3.1002. [DOI] [PubMed] [Google Scholar]
- Rezazadeh S, Claydon TW, Fedida D. KN-93 (2-[N-(2-Hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinn amyl)-N-methylbenzylamine), a calcium/calmodulin-dependent protein kinase II inhibitor, is a direct extracellular blocker of voltage-gated potassium channels. J Pharmacol Exp Ther. 2006;317:292–299. doi: 10.1124/jpet.105.097618. [DOI] [PubMed] [Google Scholar]
- Rivat C, Laulin JP, Corcuff JB, Célèrier E, Pain L, Simonnet G. Fentanyl enhancement of carrageenan-induced long-lasting hyperalgesia in rats: prevention by the N-methyl-d-aspartate receptor antagonist ketamine. Anesthesiology. 2002;96:381–391. doi: 10.1097/00000542-200202000-00025. [DOI] [PubMed] [Google Scholar]
- Samakoglu S, Lisowski L, Budak-Alpdogan T, Usachenko Y, Acuto S, Di Marzo R, Maggio A, Zhu P, Tisdale JF, Rivière I, Sadelain M. A genetic strategy to treat sickle cell anemia by coregulating globin transgene expression and RNA interference. Nat Biotechnol. 2006;24:89–94. doi: 10.1038/nbt1176. [DOI] [PubMed] [Google Scholar]
- Shukla PK, Tang L, Wang ZJ. Phosphorylation of neurogranin, protein kinase C, and Ca2+/calmodulin dependent protein kinase II in opioid tolerance and dependence. Neurosci Lett. 2006;404:266–269. doi: 10.1016/j.neulet.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Josselyn SA. The molecules of forgetfulness. Nature. 2002;418:929–930. doi: 10.1038/418929a. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:206–211. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- Sledz CA, Holko M, de Veer MJ, Silverman RH, Williams BR. Activation of the interferon system by short-interfering RNAs. Nat Cell Biol. 2003;5:834–839. doi: 10.1038/ncb1038. [DOI] [PubMed] [Google Scholar]
- Smart D, Hirst RA, Hirota K, Grandy DK, Lambert DG. The effects of recombinant rat mu-opioid receptor activation in CHO cells on phospholipase C, [Ca2+]i and adenylyl cyclase. Br J Pharmacol. 1997;120:1165–1171. doi: 10.1038/sj.bjp.0701012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MS, Seo HS, Yang M, Kim JS, Kim SH, Kim JC, Wang H, Sim KB, Kim H, Shin T, Moon C. Activation of Ca2+/calmodulin-dependent protein kinase II alpha in the spinal cords of rats with clip compression injury. Brain Res. 2009;1271:114–120. doi: 10.1016/j.brainres.2009.03.032. [DOI] [PubMed] [Google Scholar]
- Spencer RJ, Jin W, Thayer SA, Chakrabarti S, Law PY, Loh HH. Mobilization of Ca2+ from intracellular stores in transfected neuro2a cells by activation of multiple opioid receptor subtypes. Biochem Pharmacol. 1997;54:809–818. doi: 10.1016/s0006-2952(97)00243-8. [DOI] [PubMed] [Google Scholar]
- Strack S, McNeill RB, Colbran RJ. Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 2000;275:23798–23806. doi: 10.1074/jbc.M001471200. [DOI] [PubMed] [Google Scholar]
- Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, Hidaka H. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12 h cells. Biochem Biophys Res Commun. 1991;181:968–975. doi: 10.1016/0006-291x(91)92031-e. [DOI] [PubMed] [Google Scholar]
- Tang L, Shukla PK, Wang LX, Wang ZJ. Reversal of morphine antinociceptive tolerance and dependence by the acute supraspinal inhibition of Ca(2+)/calmodulin-dependent protein kinase II. J Pharmacol Exp Ther. 2006a;317:901–909. doi: 10.1124/jpet.105.097733. [DOI] [PubMed] [Google Scholar]
- Tang L, Shukla PK, Wang ZJ. Attenuation of opioid tolerance by antisense oligodeoxynucleotides targeting neurogranin. Eur J Pharmacol. 2006b;542:106–107. doi: 10.1016/j.ejphar.2006.05.024. [DOI] [PubMed] [Google Scholar]
- Tang L, Shukla PK, Wang ZJ. Disruption of acute opioid dependence by antisense oligodeoxynucleotides targeting neurogranin. Brain Res. 2007;1143:78–82. doi: 10.1016/j.brainres.2007.01.058. [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Suenaga NM, Ossipov MH, Malan TP, Jr, Lai J, Porreca F. Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. J Neurosci. 2001;21:279–286. doi: 10.1523/JNEUROSCI.21-01-00279.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veals SA, Schindler C, Leonard D, Fu XY, Aebersold R, Darnell JE, Jr, Levy DE. Subunit of an alpha-interferon-responsive transcription factor is related to interferon regulatory factor and Myb families of DNA-binding proteins. Mol Cell Biol. 1992;12:3315–3324. doi: 10.1128/mcb.12.8.3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gardell LR, Ossipov MH, Vanderah TW, Brennan MB, Hochgeschwender U, Hruby VJ, Malan TP, Jr, Lai J, Porreca F. Pronociceptive actions of dynorphin maintain chronic neuropathic pain. J Neurosci. 2001;21:1779–1786. doi: 10.1523/JNEUROSCI.21-05-01779.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfman C, Fin C, Dias M, Bianchin M, Da Silva RC, Schmitz PK, Medina JH, Izquierdo I. Intrahippocampal or intraamygdala infusion of KN62, a specific inhibitor of calcium/calmodulin-dependent protein kinase II, causes retrograde amnesia in the rat. Behav Neural Biol. 1994;61:203–205. doi: 10.1016/s0163-1047(05)80001-9. [DOI] [PubMed] [Google Scholar]
- Xu GY, Huang LY. Ca2+/calmodulin-dependent protein kinase II potentiates ATP responses by promoting trafficking of P2X receptors. Proc Natl Acad Sci U S A. 2004;101:11868–11873. doi: 10.1073/pnas.0401490101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zayzafoon M, Fulzele K, McDonald JM. Calmodulin and calmodulin-dependent kinase IIalpha regulate osteoblast differentiation by controlling c-fos expression. J Biol Chem. 2005;280:7049–7059. doi: 10.1074/jbc.M412680200. [DOI] [PubMed] [Google Scholar]