

Abstract
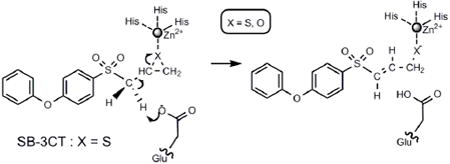
SB-3CT is a 2-[(arylsulfonyl)methyl]thiirane that achieves potent inhibition, by a thiirane-opening mechanism, of the MMP2 and MMP9 zinc metalloproteases. The deprotonation mechanism for thiirane opening of SB-3CT and for the opening of its oxirane analog, both relevant to the inhibition of MMP2, were investigated computationally using acetate anion as the Brønsted base and in methanol and acetonitrile as solvents. The activation barriers for the reaction show a significant stereoelectronic effect. The lowest energy paths have the breaking C–H bond gauche to both sulfone oxygens, and with this C-H bond anti to the breaking C–S bond of the thiirane. The calculated primary isotope effect agrees with experimental data.
Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases that regulate functions of the extracellular matrix. Gelatinases A (MMP2) and B (MMP9) are zinc metalloproteases that digest type IV collagens.1 Excessive activities of these two enzymes are implicated in tumor metastasis and angiogenesis. Since the unregulated activities of these two enzymes have been implicated in many diseases, they are targets for selective inhibitor design. Both the R and S enantiomers of SB-3CT selectively inhibit MMP2 with high activity, and MMP9 with somewhat lower activity. 2
The key event in the inhibition of MMP2 by SB-3CT is enzyme-catalyzed ring-opening of the thiirane, giving a stable zinc-thiolate species.2-5 In the previously proposed mechanism for MMP inhibition, nucleophilic addition of the carboxylate of the active site glutamate to a thiirane carbon results in ring opening and covalent binding to the enzyme (Scheme 1a). This mechanism is precedented with oxirane inhibitors of carboxypeptidase A, a structurally different but mechanistically related protease.6 Recent experiments from the Mobashery laboratory have revealed a different mechanism for SB-3CT (Scheme 1b).7 In this new mechanism, the carboxylate of glutamate-404 abstracts a hydrogen from the methylene group juxtaposed between the sulfone and the thiirane. This deprotonation initiates ring opening, and also produces a thiolate capable of coordination to the zinc at the active site. This latter mechanism is supported by the observation of a primary deuterium kinetic isotope effect for the methylene group. 7
Scheme 1.
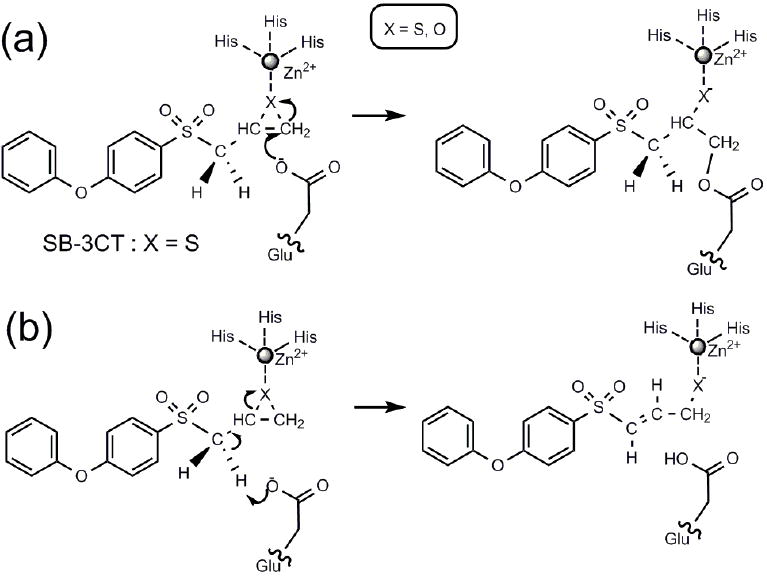
Mechanisms for MMP2 inhibition by SB-3CT: (a) previously proposed mechanism, (b) current mechanism.
The mechanism of the Brønsted base-mediated ring opening of sulfonylmethyl-substituted oxiranes and thiiranes is known from the study of the base-mediated solvolysis of these systems undertaken by Piras and Stirling.8 For both the sulfonylmethyl-substituted oxirane and thiirane with ethoxide as the base, they observed a mechanism analogous to Scheme 1b. Cleavage of the carbon-hydrogen bond in the TS was established by a primary deuterium kinetic isotope effect (kH/kD = 3.2 with ethoxide as the base).8 A primary deuterium-kinetic-isotope effect is also observed for the methylene group in SB-3CT in the inhibition of MMP2 (kH/kD = 5.0), strongly suggesting a mechanistic parallel between the solvolysis and enzyme-catalyzed reactions.7
In the present work, model molecules 1 and 2 (Scheme 2) were used to study the energetics and stereoelectronic effects of the deprotonation and ring opening of SB-3CT (Scheme 1b). Calculations were carried out with the development version of the GAUSSIAN series of programs.9 B3LYP density functional theory10-12 was chosen since it performs better than other levels of theory when compared to CBS-QB313,14 calculations on these systems (see Supporting Information for details). The 6-31+G(d) basis set was used for geometry optimization followed by single point calculations with the 6-311+G(d,p) basis set. All calculations were carried out in solution using the integral-equation-formalism of the polarizable continuum model (IEF-PCM),15 with CH3OH (ε = 32.63) and CH3CN (ε = 36.64) as solvents. These dielectric constants are comparable to the estimated dielectric constant of the active site cavity of related zinc dependent enzymes.16
Scheme 2.
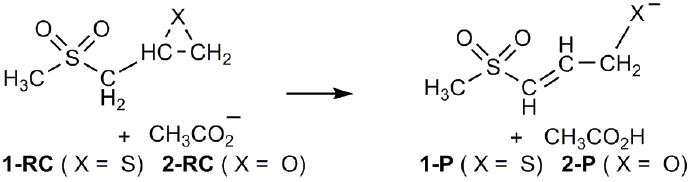
Ring opening of 2-(methylsulfonylmethyl)-thiirane and -oxirane by acetate.
For mechanism (a) in methanol, the barriers and reaction enthalpies are 14.8 and −1.9 kcal/mol for the thiirane versus 18.5 and −0.6 kcal/mol for the oxirane. For mechanism (b) (see Table 1) the barriers are 3.5 lower for 1 and and 1.3 kcal/mol lower for 2, in accord with the observation that the ring opening of SB-3CT with acetate in methanol occurs by mechanism (b).7 Switching to acetonitrile as solvent further lowers the barrier for 1 by approximately 1 kcal/mol, but has negligible effect for 2. Replacing the methyl group with phenyl reduces the barriers for 1 for mechanism (b) yet further, by approximately 2.5 kcal/mol. In MMP2, the energetics are altered further by interaction with the active site zinc ion, making both reactions rather exothermic. These energetics will be explored in a separate paper since a full QM/MM treatment of the inhibitor in the active site of MMP2 is required. Moreover, because of constraints imposed by the active site, the enzymatic reaction may involve conformations other than the lowest energy reactant complex and transition state.
Table 1.
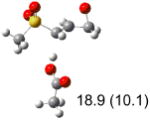
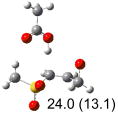
Calculated transition states, reaction barriers and reaction enthalpies in methanol solution for different conformers of the model inhibitorsa,b
| thiirane | oxirane | |
|---|---|---|
| (a) | 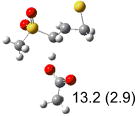 |
 |
| (b) | 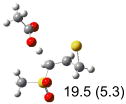 |
 |
| (c) | 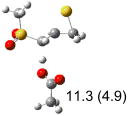 |
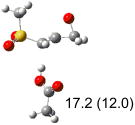 |
| (d) | 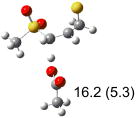 |
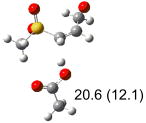 |
| (e) | 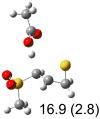 |
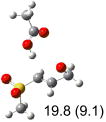 |
| (f) | 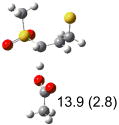 |
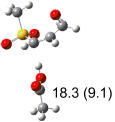 |
IEF-PCM/B3LYP/6-311+G(d,p) energies with optimized geometries and zero point energies obtained with the 6-31+G(d) basis set. The reaction enthalpies are listed in the parenthesis. The color scheme is the same as in Figure 1.
Using the most stable conformer for the reactant complex of the R and S enantiomers of 1 for reactants, and infinitely separated acetic acid and ring opened structures for products.
To explore the conformations of the inhibitor in solution, we considered rotation about the CC bond connecting the methylene to the three-membered ring, and the CS bond to the sulfone (see Scheme 3a). Each bond is a three-fold rotor and is expected to have a low barrier to rotation. Except for three conformations where the sulfone and the three-membered ring bump into each other, there are six low-energy conformations within 0.8 kcal/mol of the lowest energy minimum (Supporting Info., Fig. S2).
Scheme 3.
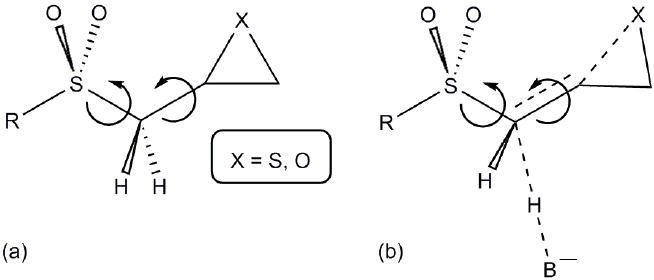
Stereochemistry of deprotonation
The ring opening paths starting from the lowest energy conformers are illustrated in Figure 1 for both 1 and 2. In 1-TS, the thiirane ring is partially opened with the CS bond elongated to 2.04 Å. The deprotonation is well advanced with the migrating proton 1.56 Å from the donor carbon, and 1.13 Å from the acceptor oxygen. Similarly in 2-TS, the epoxide ring is partially opened with the CO bond elongated to 1.79 Å; the deprotonation is somewhat closer to completion.
Figure 1.
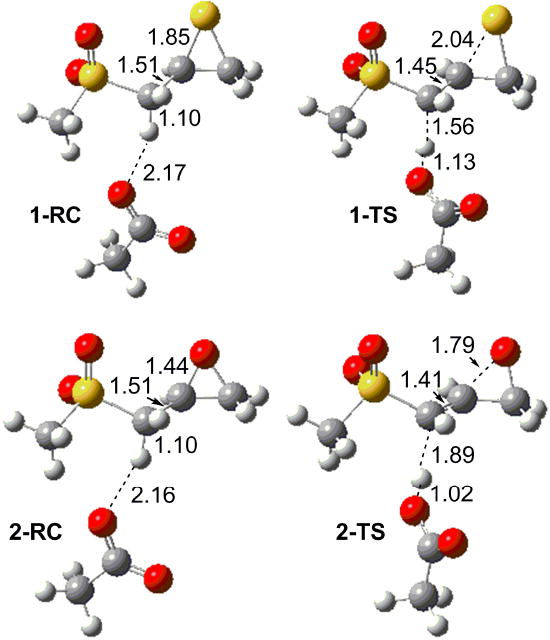
Reactant complexes and transition states for model sulfone thiirane and oxirane reactions with acetate as base. Key bond lengths are in Ångstroms. Atoms are colored according to atom types (H, white C, gray; O, red; S, yellow). The transition state structures 1-TS and 2-TS correspond to row (a) in Table 1.
The departing proton may be either syn or anti to the CX bond that is being broken in the thiirane or oxirane. In both 1-TS and 2-TS in Figure 1, the CC bonds adjacent to three membered ring are shorter than reactant complexes 1-RC and 2-RC. Because of the partial double bond character of this CC bond, it is a two-fold rotor with a substantial barrier (see Scheme 3b). Therefore, the transition state has less conformational flexibility because of the partially formed CC double bond. Rigid scans show that the two-fold barriers for CC bond rotation in the TS are about 3 to 4 times higher than in the reactants.
The rotation about the CS bond to the sulfone is also more restricted in the transition state than in the reactant. The barrier for CS rotation in the TS is about 2 to 3 times higher than in the reactant. A carbanion next to a sulfone prefers to be gauche to both oxygens.17-22 Thus, proton abstraction in 1 and 2 should also be favored gauche to both oxygens (i.e. anti to the R group shown in Scheme 3b). Ring opening could lead to products with either E or Z configurations in the TSs.
To study these stereoelectronic effects, the transition states for abstraction of the pro-S hydrogen from the R and S isomers were calculated for the six lowest energy conformers of 1 and 2 (Table 1). Rows (a), (b) and (c) show the S isomer, and the remaining three rows show the R isomer. Rows (a), (c), and (e) give products in the E configuration, and the others give the Z configuration. By symmetry, these TSs are equivalent in energy to abstraction of the pro-R hydrogen from the respective S and R isomers.
In row (a) of Table 1 (same as the TSs in Figure 1) the breaking CH bond is gauche to only one sulfone oxygen, and anti to the opening CX bond in thiirane and oxirane (E product). When the breaking CH bond is syn to the ring opening bond (row (b)), the reaction barriers are significantly higher (Z product). Steric interactions may also contribute to higher reaction barriers in row (b). The lowest barrier for the S isomers of 1 in solution has the departing proton anti to the breaking CS bond and gauche to both sulfone oxygens (row (c)). Since TSs in both row (a) and (c) have the departing proton anti to the breaking CS bond, and lead to products with the E configuration, the lower barrier in row (c) for thiirane must be due to the stereoelectronic effect from the sulfone group.17-22
The above stereoelectronic effect is also evident in the R isomers of 1. The TS with the lowest barrier has the departing proton anti to the breaking CS bond and gauche to both sulfone oxygens (row (f), Z product). The TS with the breaking CH bond anti to the ring opening bond (row (d), Z product) has a barrier close to the TS with the syn orientation (row (e), E product) because of the steric repulsion between sulfone and thiirane groups in the Z configuration. Similarly, when comparing row (a) with (d), and row (b) with (e), the barriers leading to Z products (row (d) and (b)) are higher due to steric interaction between sulfone and thiirane ring.
The trends for the oxirane reactions are similar to those for thiirane. The anti orientation is preferred over syn, and TSs leading to E products are favored over those giving Z products. The stereoelectronic effect of the orientation between the sulfone group and the breaking CH bond on the barriers (row (a) vs (c) and row (d) vs (f)) is comparable to the thiirane case.
In summary, the thiirane reactions show a significant stereoelectronic effect with respect to the relative orientation between the sulfone group and the breaking CH bond. TSs with the breaking CH bond gauche to both sulfone oxygens have lower barriers than other cases. For both thiirane and oxirane, the TSs with the breaking CH bond anti to the CS bond are more favorable than those with a syn orientation. In addition, steric interaction between the sulfone and the three membered ring can increase the barriers leading to Z products. In all of the orientations, the thiirane reactions with acetate have lower barriers than the oxirane reactions and are much less endothermic.
The primary kinetic isotope effect (KIE) for hydrogen abstraction and ring opening for thiirane using the model system was computed in methanol using the reaction pathway in row (c) of Table 1. Both changes in the zero point energy and quantum tunneling corrections using the Wigner model23 (factor 1.15 in this case) were taken into account. The computed KIE, kH/kD = 5.0, agrees with the experimentally observed KIE for the inhibition of MMP2 by SB-3CT (kH/kD = 5.0).7 Similar calculations for other pathways also lead to good agreement (for example, kH/kD = 4.7 for the TS of 1 in row (a)).
In this study, the ring opening of 1 and 2 were investigated as models for the inhibition of MMP2 by SB-3CT. Calculations reveal strong stereoelectronic effects for both thiirane and oxirane. These calculations provide insight into the deprotonation mechanism for the concerted ring opening of the adjacent thiirane or oxirane ring.
Supplementary Material
Acknowledgments
This work was supported at Wayne State University by the National Science Foundation (CHE0512144) and at the University of Notre Dame by the National Institutes of Health (CA122417). We thank Wayne State University for generous allocations of computer time on its computational grid.
Footnotes
Supporting Information Available: Details of the computational methods, benchmark calculations in the gas phase, computational results for mechanism (a), and low energy conformers of (R)-2-[(methyl-sulfonyl)-methyl] thiirane. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Liotta LA, Tryggvason K, Garbisa S, Robey PG, Abe S. Biochemistry. 1981;20:100–104. doi: 10.1021/bi00504a017. [DOI] [PubMed] [Google Scholar]
- 2.Brown S, Bernardo MM, Li Z-H, Kotra LP, Tanaka Y, Fridman R, Mobashery S. J Am Chem Soc. 2000;122:6799–6800. [Google Scholar]
- 3.Kleifeld O, Kotra LP, Gervasi DC, Brown S, Bernardo MM, Fridman R, Mobashery S, Sagi I. J Biol Chem. 2001;276:17125–17131. doi: 10.1074/jbc.M011604200. [DOI] [PubMed] [Google Scholar]
- 4.Solomon A, Rosenblum G, Gonzales PE, Leonard JD, Mobashery S, Milla ME, Sagi I. J Biol Chem. 2004;279:31646–31654. doi: 10.1074/jbc.M401310200. [DOI] [PubMed] [Google Scholar]
- 5.Ikejiri M, Bernardo MM, Bonfil RD, Toth M, Chang M, Fridman R, Mobashery S. J Biol Chem. 2005;280:33992–34002. doi: 10.1074/jbc.M504303200. [DOI] [PubMed] [Google Scholar]
- 6.Kim D, Mobashery S. Curr Med Chem. 2001;8:959–965. doi: 10.2174/0929867013372805. [DOI] [PubMed] [Google Scholar]
- 7.Forbes C, Shi Q, Fisher JF, Lee M, Hesek D, Larrull LI, Toth M, Gossing M, Fridman R, Mobashery S. doi: 10.1111/j.1747-0285.2009.00881.x. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piras PP, Stirling CJM. J Chem Soc Perkin Trans 2. 1987:1265–1271. [Google Scholar]
- 9.Frisch MJ, et al. Revision F.02. Gaussian, Inc.; Wallingford, CT: 2007. See Supporting Information for full reference. [Google Scholar]
- 10.Lee C, Yang W, Parr RG. Phys Rev B: Condens Matter. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 11.Becke AD. Phys Rev A: Gen Phys. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 12.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 13.Montgomery JJA, Frisch MJ, Ochterski JW, Petersson GA. J Chem Phys. 1999;110:2822–2827. [Google Scholar]
- 14.Montgomery JJA, Frisch MJ, Ochterski JW, Petersson GA. J Chem Phys. 2000;112:6532–6542. [Google Scholar]
- 15.Tomasi J, Mennucci B, Cances E. THEOCHEM. 1999;464:211–226. [Google Scholar]
- 16.Sigel H, Martin RB, Tribolet R, Haring UK, Malinibalakrishnan R. Eur J Biochem. 1985;152:187–193. doi: 10.1111/j.1432-1033.1985.tb09180.x. [DOI] [PubMed] [Google Scholar]
- 17.Wolfe S, Rauk A, Csizmadia IG. J Am Chem Soc. 1967;89:5710–5712. [Google Scholar]
- 18.Wolfe S, Rauk A, Csizmadia IG. J Am Chem Soc. 1969;91:1567–1569. [Google Scholar]
- 19.Wolfe S. Acc Chem Res. 1972;5:102–111. [Google Scholar]
- 20.Wolfe S, Tel LM, Liang JH, Csizmadia IG. J Am Chem Soc. 1972;94:1361–1364. [Google Scholar]
- 21.Wolfe S, Stolow A, Lajohn LA. Tetrahedron Lett. 1983;24:4071–4074. [Google Scholar]
- 22.Wolfe S, Stolow A, Lajohn LA. Can J Chem. 1984;62:1470–1475. [Google Scholar]
- 23.Wigner EZ. Physik Chem. 1932;B19:203–216. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.