

Abstract
Dr. Irving Weissman was the honored E. Donnall Thomas lecturer at the Tandem BMT Meetings, held on February 10, 2007, at Keystone, Colorado. Dr. Weissman has been a major player, and has provided us with enormous insight into many areas of biology, dating back to his high school days in Montana. He led an enormously productive career at Stanford University where he has taught us many lessons involving our understanding of lymphocyte homing, stem cell biology, both of the hematopoietic system and other types of stem cells, and also now, about cancer stem cells. Dr. Weissman has made enormous contributions to this burgeoning field that has provided us new insights and new opportunities for treatment strategies. In addition to a very productive laboratory career, he is also currently the director of both the Stem Cell Institute, as well as the Cancer Center at Stanford University. The following text is a modified transcribed version of the presentation made by Dr. Weissman.
Keywords: Hematopoietic stem cell, Hematopoiesis, Autoimmunity, Myelogenous leukemia, Aging, Leukemia stem cells
Introduction
In this presentation, I am going to talk about normal stem cells, hematopoietic stem cells, cancer stem cells, and a little bit about brain stem cells, if I have time. The point here is that we have known since Till and McCulloch [1] that hematopoietic stem cells must exist, and that the principle difference between stem cells and their progenitors is that stem cells self-renew (Figure 1). The general method that we have used to isolate stem cells is to find surface markers on stem cells, as positive selectors, and surface markers on nonstem cells, as negative selectors, so that at the end of the day you can get hematopoietic stem cells free of other unwanted cells. Of course, if you have cancer cells in the autologous transplant, you would rather get rid of them so you could have a cancer-free transplant. If you want to do allogeneic transplants, not for malignant diseases, but for other conditions where you want the donor cells to engraft without graft-versus-host disease (GVHD), again I would argue that you would like to have pure stem cells. The population of cells in the mouse that we found many years ago, that had the activity of clonal precursors of all lineages of blood cells and at which at the single cell level could give rise to long-term engraftment and growth of about 30,000 stem cells from a single cell for the life of the host, and the next host, and the next host, had this set (Sca 1, Thy1.1, ckit) of positive markers on the surface, and negative for any lineage markers (lin−).
Figure 1.
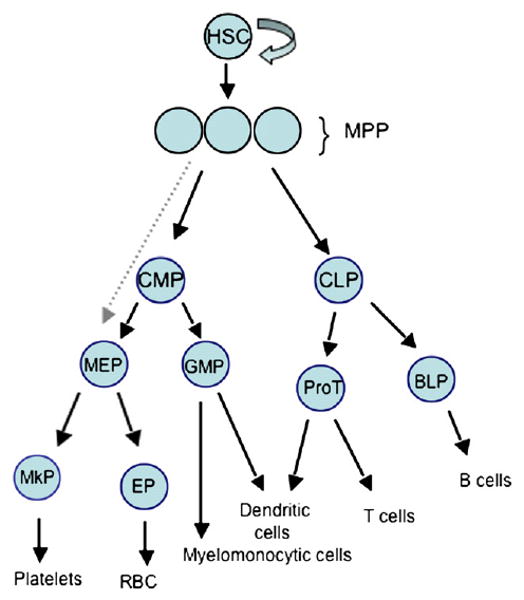
Schematic of Hematopoiesis. HSC are the long-term self-renewing cells that give rise to all mature blood cells through a hierarchy of developmental intermediates including multipotent progenitors (MPP), which have limited self-renewal and provide transient but multilineage reconstitution, and increasingly lineage restricted progenitors. CMP, common myeloid progenitor, CLP, common lymphoid progenitor, BLP, B lymphocyte protenitor, ProT, T cell progenitor, GMP, granulocyte/macrophage progenitor, MEP, megakaryocyte/erythroid progenitor, MCP, mast cell progenitor, MkP, megakaryocyte progenitor, EP, erythroid progenitor.
Now, as time has gone on, we have gotten better and better, and we know that this population has other markers on the surface by gene expression analysis. We and Sean Morrison's group [2,3], found other markers that were on (eg, CD150 and ESAM1), and markers that were off, and so one can now daily, if you have a multicolor cell sorter, get those kinds of populations of cells. And, we were also able in the 1980s to develop mice that had human fetal bone, thymus, liver, and spleen, and so we could irradiate and reconstitute those mice, and using that as 1 of the assays, we were able to isolate the human hematopoietic stem cell. It should be noted that it is not just CD34+, CD38−, nor just CD34+. One of the important points, at least if you want to study the biology of the stem cell, is to get the CD34+CD38−Thy+lin− cells. You can use ckit as a marker, although it is not the most useful marker because it is downregulated during mobilization, but then the lin− panel is very important, as well. Years ago, at a company I cofounded, called SyStemix, the population of cells in human mobilized blood that were CD34+Thy+ were used in many transplants in humans, and this same population was depleted of tumor cells.
Eli Hanania performed an experiment for SyStemix in the mid-1990s, where he added authentic breast cancer cells or non-Hodgkin lymphoma (NHL) or myeloma cells to mobilized blood so that 7 cells per thousand nucleated cells were tumor cells. He then ran the population through the cell sorter, and the combination of flow sorting and multiple antibodies purged the graft 250,000-fold if it was breast cancer, 220,000-fold if it was NHL, and about 145,000-fold if it was myeloma. In contrast, the same sorter with any single antibody alone or by any other single antibody method would not purge the graft more than 100- or 1000-fold. Rob Negrin et al. [4] led the clinical trial at Stanford started in 1996, and showed a dose-response in myeloablated stage IV breast cancer patients, accruing 22 patients by 1998, 15 at Stanford, and 7 at Detroit. The dose-response characteristics pretty much mirrored what we had in mice. That is, that syngeneic or autologous transplants at a dose of 2 × 105 stem cells per kilogram body weight would give you 10-13 days for white cells and a prolonged time for platelets but anything above 8 × 105 per kilo you averaged about 10 days for 500 white cells and 20,000 platelets. Those patients were followed for at least 4 years, and displayed about 45% disease-free survival (DFS) at 3 years and 37% at 4 years time.
Hematopoietic Stem Cell Transplantation (HSCT)
Rapid and sustained engraftment can occur in humans with a stem cell transplant in an autologous setting. You can deliver a cancer-free graft and then decide whether that is important to what you are doing. In mice, Judy Shizuru and I showed in several experiments that even in the most aggressive allogeneic setting of C57BL/6 transplantation into BALB/c mice, with no MHC match, something you would never do, that at the dose that a syngeneic mouse gives you 22 days to engraftment, it takes an allogeneic HSC transplant 10 times that dose, a thousand cells [5]. However, by the time you get to 10,000 cells in the allogeneic, it is equivalent to 5000 syngeneic or autologous cells; both engraft platelets and neutrophils by 10 days. So, these results suggest, if mice and humans turn out to be the same, that you could accomplish allogeneic engraftment. Now Judy Shizuru and I have lowered that dose considerably by pretreating the host with antibodies that are even more lymphoablative, so they eliminate T cells and natural killer cells. And you have to eliminate both of those if you have a haploidentical or a fully allogeneic donor in this setting.
In these pure HSC allotransplants, there was no GVHD or residual host-versus-graft response, because there were no T cells in the transplant in these mice. Shizuru and I went on to show, as did Kim Gandy with me, that when you transplant C57BL/6 into BALB/c mice you can cotransplant the same day or a year later, a heart or an islet, and they are permanently tolerant [6,7]. We thought it would just be a matter of a year or 2 before everybody would try to use this for organ graft tolerance. Of course, that did not happen right away, but I have been known to be a little enthusiastic. When I was 16, in Great Falls, Montana, I transplanted male bone marrow into female mice in Ernst Eichwald's lab, and showed that I could induce permanent transplant tolerance. The mice were given a sublethal dose of irradiation, and third-party grafts were rejected. So, that day in 1957, I went home and told my mom, “it's going to be just a matter of a couple of years before everybody is going to get organ transplants this simple way.” The new setting of this approach is that you can now do it under entirely nonlethal conditions, where you lymphoablate and only myelo-suppress the host, and you can get engraftment, long-term chimerism, and graft acceptance without any GVHD.
HSCT and Autoimmune Disease
Now, Shizuru went on with me and George Beilhack to look at type I diabetes, which, as you know, is not a disease of the islets, but a disease of the immune system [8]. And, just as you would like to treat sickle cell, beta thalassemia, or severe combined immunodeficiency with stem cells that do not give GVHD, but do engraft, type I diabetes is another disease that may be correctable by giving hematopoietic stem cells from a donor that is resistant to the developing autoimmune disease. Nonobese diabetic (NOD) mice develop type I diabetes at 5-6 months of age by an autoimmune attack, and it is a multigenic disorder. If we give them myeloablative conditioning, which is also lymphoablative, and give back their bone marrow, which has T cells in it, much like the Richard Burt studies to reset the clock, I am told, the approach buys you a couple of months. If you give back the NOD, autologous stem cells alone buys you another couple of months, but the genetic predilection of the disease always comes back and the animals eventually develop diabetes. However, if you give third-party strain, AKR, which is distantly related but much more relevant, transplant MHC H2g7 matched HSC (these mice have at least 3 independent genes that are resistance genes to diabetes), it is curative in the polydypsic polyuric stage of the disease. All you have to fix at that time, in these mice, is the autoimmune part of the disease and the islets regenerate. The islets are still having infiltrative B cells, but not T cells, and that does not lead to a progressive disease. It is important to note that you can cure mice of progression in this autoimmune disease in both the myeloablative and the nonmyeloablative settings in mice.
Shizuru went on to show, with me, that you can cotransplant already diabetic mice at 6 months or so (the most difficult thing about these experiments is providing insulin every day), and cure them of their autoimmune disease with stem cells, and of their islet deficiency with islets from the same donor. If we had a method of growing or getting islets more efficiently this would be a good therapeutic chance. The transplant can be done with total lymphoid irradiation (TLI) and antithymocyte globulin (ATG), a setting that allows MHC matched HSC to engraft. Eventually, we think that we would be able to treat diabetic patients. It would have to be in the nonmyeloablative setting to compete with insulin as a treatment for these patients.
Now, does it stop there? No. In experiments recently published by Julie Christensen at Cellerant [9], she used the NZB × NZW (NZBW) model for lupus, in which as the mice age, they develop autoantibodies, which include anti-DNA and antihistone antibodies, and get glomerulonephritis, probably from circulating immune complexes. In this myeloablative trial with animals that were 73-79 days of age, Christensen et al. gave them a lethal split dose of irradiation, and then transplanted them. The irradiation had some deleterious effect in some of those animals, and syngeneic transplantation did not, in this case, offer an advantage. But haploidentical allogeneic stem cell (DBA × C57BL)F1 mice, which share the H2d haplotype but also have an unshared H2b haplotype, were transplanted into an autoimmune [NZB(d) × NZW(w)]F1 hybrid strain. So, to make it simpler, it is a d × b into d × w transplant, like a haploidentical situation within a family. These allogeneic transplanted recipients had a later onset of disease, and many of them did not progress. When you look at their proteinuria, from <100 mg per deciliter up to very high proteinurias (>500 mg/dL), the syngeneic recipients did not have many animals in the low proteinuria class, whereas the haploallogeneic recipients had a lot with <100, <8% with >500 mg/dL, and at the time the age-matched control group all had high proteinuria. What was true for proteinuria was also true for circulating immune complexes. Now, can you do this in a nonmyeloablative setting? To answer that question, mice were given sublethal doses of irradiation after treatment with 2 doses of anti-T cell globulin, at doses known to deplete all classes of T cells, and also anti-asialoGM 1 antibody, which depletes NK cells. Mice were treated at about 280 days of age, when they already had extensive disease. Conditioning alone was sufficiently immunosuppressive that it delayed the progression of disease, but the allotransplant led to a cure of the disease in many of the animals. And, again their proteinuria at the high level was lowest in the allotransplanted versus age-matched or the conditioned-alone animals.
These were the 2 practical applications that we have looked at in mouse preclinical testing. I believe that it will be possible to have a protocol to go into allogeneic transplants in humans. Obviously, autoimmune patients are about the hardest to do. So, you would have to have proof of principle in patients who needed to have a healthy hematopoietic system, like in sickle cell, beta thalassemia, severe combined immunodeficiency, or some other indication.
Normal Stem Cells
I now want to discuss the origins of the blood-forming system, because it gets at the cell types that form it and it involves what has become conventional wisdom. The very first site of blood formation in the mouse, and in the human, is in the yolk sac blood islands. Mesodermal precursors migrate out into the visceral entoderm and form islands of blood inside and blood vessel outside. They connect up to each other and then they form the vitelline vasculature that connects up with the dorsal aorta. The first site of appearance of hematopoiesis within the embryo starts in the yolk sac, goes to the dorsal aorta, and although nobody quotes it, we showed in 1975-1978 that you could transplant yolk sac blood islands from previtelline vasculature connection embryos into the yolk sac cavity of others and they were chimeric for life, including hematopoietic stem cells and T cells. The establishment of that primitive hematopoietic system is the precursor of the establishment of the definitive hematopoietic system. Since then, a number of people, especially Gordon Keller, [10,11] have shown that a cell called the hemangioblast, that he had purified to a single cell level from ES cells, can give rise to blood and blood vessel cells. So it is now conventional wisdom that the hemangioblast is the necessary precursor of hematopoietic and endothelial cells. Backing that belief were the experiments where a VEGF receptor called Flk-1 is knocked out in mice. Flk-1 is on the hemangioblast and flk-1 knockout mice develop neither blood nor blood vessels. However, a gene knockout does not tell you the lineage relationship between cells, and the presence of a hemangioblast in an embryonic stem cell culture does not mean it is necessarily the cell type that gives rise to blood and blood vessels. Therefore, Hiroo Ueno and I [9] asked whether yolk sac blood islands, when they arise, are from a single cell, and if they are from a single cell, are they from a hemangioblast. And the method we used to test it was to make embryonic stem cells that had different colors.
We made, by gene knock-in to the rosa locus, a blue fluorescent protein encoding gene, green fluorescent protein, and red ES cells, each expressing only the color they should [12]. We injected those into the blastocysts of uncolored mice, implanting them in the uterus, and we thus generated tetra-chimeric mice. Using about 20 uncolored ES cells and exactly 5 each of the 3 different colored ES cells, 4 days later, the embryo had developed 3 germ layers and in the visceral endoderm we could detect the first blood island forming. The cells migrating up are polyclonal in mice where there are clonal organizations of epithelial cells. Without going through all the data in detail, I will just say there were no clonal blood islands from a common hemangioblast. None. There were no biclonal yolk sac blood islands with a single endothelial precursor and a single hematopoietic precursor forming it. About 20% of the blood islands could be explained by hemangioblasts participating in a polyclonal seeding, but 80% had no relationship between blood and blood vessel in this polyclonal blood island at the time they first formed. We included a flk-1 marking system so that if a cell turned on flk-1, the cell changed its color, and again only 20% of the blood cells in the organism went through a flk-1-positive intermediate.
We can conclude then that the beginnings of the blood-forming system in the yolk sac are polyclonal, and although some cells could be derived from hemangioblasts, the vast majority are not. Now, those chimeric animals are useful to look at other organs. First, throughout the endoderm, from the tongue to the rectum, what you can see is that both the filiform papillae in the tongue and the villae in the small intestine are derived from a single cell. They are all blue, all green, all red, or all uncolored. The second point is that if you look at an adult chimeric mouse, cut a section through the crypts where the stem cells are supposed to be, one can see dozens of villi that are formed from a single gut stem cell. This observation does not correlate with the current conventional wisdom that the 4th cell up around is a stem cell and it regenerates the villous. It is not proof that it is not true, but it is not consistent with it. The fact that you only have single colored crypts whenever you look at an animal means that you are not replacing it, turning it over, with external stem cells. So, it is a stem cell system. I will not go into all the other tissues, but I will just say that in the liver you have a clonal organization all coming out from the portal vein, and the colors stay consistent going out from the portal vein; whether those are stem or progenitors, we do not know.
Hematopoiesis
I hinted to you that there were several migrations to form the blood-forming system. A migration from the mesenchyme to the yolk sac, from the yolk sac to the dorsal aorta, and from the dorsal aorta to the fetal liver. The liver gives out during fetal life and they go to the bone marrow. So, migration is critical for each of the steps of movement of hematopoietic stem cells during the developmental process, and migration continues at a pace none of us really suspected, unless our interpretation of this experiment is wrong. Doug Wright and I did several experiments following up 2 beautiful findings: 1 by Joan Goodman and George Hodson in 1962, that the hematopoietic capacity for restoration of the blood was about a thousandth of that of the bone marrow, but it was there [13]. Then, of course, the BMT field contributed to the knowledge that you can mobilize stem cells from marrow to blood with cytoxan plus granulocyte-colony stimulating factor (G-CSF) [14-16].
Now, the amazing thing, if you think about it, is that the first bone marrow transplant worked. Cells that were resident in the bone marrow and placed, not put it in the bone marrow, but into the vascular system, found their way to the radiated hematopoietic bone marrow and engrafted there. I would argue from that observation that there had to be a system already in place for cells that were in the blood, Goodman's very tiny number, to go to the bone marrow. To make a long story short, we found that there are steady-state 100 hematopoietic stem and multipotent cells in the blood of the mouse. And, that when we isolated those stem cells and put them into the blood stream their residence time in the blood stream was between 1 and 5 minutes. We tracked them with luciferase, thanks to a lot of work by Rob Negrin and Chris Contag, and we found they homed to bone marrow, liver, and spleen—the hematopoietic foci in a mouse [17]. They did not stop in the lung; the only cell that I have ever injected that did not get stuck in lung capillaries. Well, given even a 5-minute residence time to maintain 100 hematopoietic stem cells in the blood you have to flux 30,000 hematopoietic stem/progenitor cells through the blood every day.
So, now it is 1 of the biggest recirculating systems we can imagine. With that knowledge we went back and looked at those experiments where people said brain cells could give rise to stem cells that could give rise to brain and blood, muscle stem cells could give rise to muscle and blood. And, it was always separate hematopoietic stem cells and muscle stem cells, or hematopoietic stem cells and brain stem cells. The presence of all those circulating hematopoietic stem cells was very curious, and there is a lot of things that come from this. We know by parabiosis that the ones that cross in the blood establish themselves as grafts in the bones. So, they participate in hematopoiesis. What happens when they become malignant? Well, you have a malignancy of a cell that is spreading throughout the bone marrow cavity. You have never seen a myelogenous leukemia that is in 1 bone only, so that you could just cut off the arm or irradiate it and cure the disease. And, I would say you have not seen it because you are looking at the normal property of the precursors that become leukemic. Using migration, integrin α4β1 and SDF 1 find their way back to the hematopoietic site. It has a lot to do with niches and what we think niches are, because a lot of the cells in that traffic will be near blood vessels and others will be near other sites. It also has a lot to do with trying to figure out what the steps are in forming those niches. How about the next step down? We know long-term stem cells give rise to short-term stem cells, then they have a decision to lymphoid or myeloid. You might have hoped to work it out in the microanatomy of the bone marrow, but if the cells on average leave 1 site and then enter at another site, they may find another niche that is a myeloid or a lymphoid niche. It is curious that HSCs and inflammatory T cells both express integrin α4β1 [18,19] and home to SDF1 [20]. Perhaps a subset of HSC in circulation enter VCAM1+ vessels in inflamed tissues, there to respond to local signals for direct bursts of myelomonocytic cells.
In an experiment by Yuan Cao, Amy Wagers, Chris Contag, and myself [17], a single visually observed hematopoietic stem cell from a luciferase-positive animal was injected with host cells, and in different hosts hematopoietic foci that go from the spleen to the vertebrae, those that stay in the vertebrae, or those that reach the skull and develop several independent foci, resulted, consistent with the recircularory property of these cells. Thinking about this observation, he began to examine how many empty niches there are in the bone marrow, because if they are going out they must leave a niche and when they come back they fill the niche. So, Deepta Battacharya injected a large number of hematopoietic stem cells, up to 5000 of them, into a syngeneic animal that differed either only by the Ly5 allele or a green fluorescent protein (GFP) in the donor, not the host [21]. None of those grafts worked, because of rejection—even the tiniest differences were seen by an intact host and led to rejection of the hematopoietic stem cells. So, he then moved on to severe combined immunodeficient recipient mice, and he found that a single injection of 250 hematopoietic stem cells fills about a 0.5% of the niches, and cures the animal by 6 weeks of its immunodeficiency. Although these mice only produced about 0.5% of the myeloid cells from the donor, donor cells sustain the production of both T and B cells, because there is no competition for B cell and T cell niches; all aspects of their immune system are normal. When he did the experiment he actually went from 0 cells to 5000 cells and found that he saturated the open 0.5% of niches with the single bolus injection of 250 cells. However, if he gave 250 cells a day apart, the next day the niches are open again and so he gets a 0.5% from each. So, there may be something there about slow opening of niches and maybe slow infusion of stem cells, but I will not go that far yet.
So far, I've told you that hematopoietic stem cells self-renew, we have prospectively isolated them, and with a number of people in the lab and a number of people in other labs have found that there are least 2 downstream multipotent cells that hardly self-renew. One, the short-term stem cells is present at 6 weeks; the next 1 down, a multipotent cell, is there for <2 weeks. At the single cell level these multipotent cells give robust engraftment and multilineage engraftment, but they do not self-renew. They never go back even a single step no matter how many cells you inject. So, this is a lineage. And then, outside of that, they go either to the lymphoid or the myeloid progenitor, erythrocyte progenitors go another way. We can prospectively isolate each of those populations and they do what their name says they do, make myeloid cells, lymphocytes, or erythrocytes, and that they do it with very high efficiency. The only cell that permanently self-renews in the hematopoietic system is the long-term hematopoietic stem cell, which keeps migrating in and out of its niches.
Aging and Myelopoiesis
Next, we wanted to look at what are the preconditions that lead to leukemia and at what stage of development do the leukemias break out. Is it a known stage or is it a mixture … a mixed phenotype? And, does a cell that never self-renewed now gain self-renewal capacity to be a leukemia stem cell? Some of the experiments on the way turned out to be very interesting. First, Sean Morrison and I, and then again recently Derrick Rossi and I, looked at the aging of stem cells, and here is what happens to them [22-24]. From 2-month to 2-year-old mice, stem cells actually increase their number dramatically—maybe 20-fold. A 2-year-old mouse, like a very old human, does not make many new lymphocytes, but still makes lots of myeloid cells. Usually, there is an anemia of aging, of red cells, but you can have too many platelets. Certainly that is the phenotype of the aged mouse. When Derrick, David Bryder, and I transplanted equal transplantable units of purified hematopoietic stem cells, young and old, into the same young mouse, the cells from old mice gave rise to 20-fold the number of stem cells, decreased numbers of lymphocytes, and lots of myeloid cells, whereas in the same microenvironment, the young cells gave rise to lots of myeloid, lots of lymphoid, and not many more stem cells. So, every property that we were looking at was intrinsic to the transplanted hematopoietic stem cells. Our early experiments with Uchida [25] showed that in adult mice no cells other than HSC give rise to HSC, and probably from early fetal life on there are only the original clones of HSC and their progeny that sustain hematopoiesis. These clones change properties as they go through their self-renewing divisions. The changes they go through are not random. They may look random, but they always lose lymphopoiesis and always gain myelopoiesis in aging mice.
Considering that the shift toward myelopoiesis is intrinsic, we studied the gene expression profile at the level of the purified long-term stem cells, and we found that the transcription factors for myelopoiesis were up and others for lymphoid development were down [23]. There is a correlation of the fate of the cells and the transcription factors they are expressing at the level of the hematopoietic stem cell. Thus, very distant fates are proscribed already at the level of the stem cell. We published something like that in 1990 for fetal life as well [26]. Looking at the names of some of those myeloid genes that are overexpressed, out of the top 32, 17 have been involved in translocations, duplications, or inversions in human acute myelogenous or promyelocytic leukemia. That led to a speculation that these translocations were probably at loci that are transcriptionally active rather than transcriptionally silent. The fact that some genes on different chromosomes that are linked together in transcriptional foci [27] will lead us to test whether among the transcriptionally active genes if there is random translocation or not.
Now, there is accelerated aging in a number of progeria syndromes. Derrick picked 3 to study that involved errors in repair of DNA damage, because he figured that accumulated DNA damage might play a role in aging [24]. That is a popular hypothesis. So, we had defective nucleotide excision repair mice, XPD, we had mice that lacked the RNA component of their telomerase, so they cannot extend their telomers efficiently. We had mice that lacked Ku 80, a protein that binds to blunt end DNA double-strand breaks, and then through H2 AX and other proteins is involved in signaling the repair process. The big shock is that these animals as they age did not selectively lose their hematopoietic stem cells. However, of the long-term hematopoietic stem cells, only 1% to 4% go into cell cycle per day, whereas for the MPPs, about 15% to 20% of their multipotent progeny go into cycle every day. Therefore, a lot of those cells could have been hanging around without going into cell cycle. When we put them into protocols to test their activity and when we transplanted them, the age-matched stem cells from those 3 diseases—the Tert, the XPD, and the Ku 80—transplanted self-renewing capacity not nearly as well. Furthermore, as they got older, it got worse, and if you put them directly from young versus old in a proliferation assay, they did not proliferate as well, and this further decreased as they got older. In addition, they undergo apoptosis more then their counterparts from young mice when they go into their first cell divisions, and they do not self-renew. Thus, they have lost in vitro self-renewal capacity.
Next, we wanted to know if there was accumulating DNA damage in these cells. We looked for double-strand breaks and other DNA damage using the finding that right after a DNA double-strand break, a rare nucleosome component, H2AX, which is 1 in 6 of the H2As, so it is in 1 of 3 of the nucleosomes, becomes phosphorylated, creating a megalocus of H2AXs and triggering the repair process. We X-rayed the cells, and immediately after we stained for H2 AX and found increasing numbers of foci with increasing doses of irradiation. In old mice, there is the equivalent of somewhere between 50 and 100 rads of irradiation worth of breaks that LT HSCs have accumulated. These foci are largely gone in the multipotent progenitors. The explanation for this is that you are accumulating double-strand breaks as you age. For H2 AX foci, nothing is going to happen unless the cells are called into cell division. We speculated that exposed cells either activate the repair process, or go from G0 to G1 and die in the S phase because they have a double-strand break, clearing out by death all of those that were there and that would explain this kind of data.
Leukemic Stem Cells
Many leukemias have translocations as an event leading to the leukemia, and I have just told you 2 things: (1) aging mice, and perhaps people, have highly transcriptionally active sites that turn out to be partners of translocation, and (2) double-strand breaks accumulate and can reside without repair. I propose that the coincidence of those 2 developments leads to nonrandom translocations. Toshihiro Miyamoto and Koichi Akashi in my lab, hematologists and bone marrow transplanters from Japan, knew that at the Hiroshima Hospital they saved viable cell suspensions of the bone marrow of patients who developed hematopoietic malignancies. We focused on those people who had early onset acute myelogenous leukemia (AML)1-ETO [28]. John Dick and his group, in a beautiful set of experiments, showed that he could transplant AML from humans into immunodeficient mice [29,30]. He and I had been working on these mice as xenotransplant recipients in concert for a long time [29,31]. John showed that the leukemia would be transferred only with CD34+38− cells. The presumption was that AML was a stem cell disease, but, as I mentioned previously, CD34,38 is not sufficient as a marker. We looked at CD34+38lo Thy1+ Lin− HSC and 34+38lo Lin−Thy1− cells that we now know to be multipotent progenitors (that normally do not self-renew beyond 12 weeks), and all of the leukemia activity was in the multipotent progenitor population. They make leukemia blasts, in vitro, and we now know that they can transplant it. However, the most important finding was that the stem cells in those patients do not transplant the disease; CD34+38−, Thy+, Lin− cells make normal mixtures of methylcellulose colonies, full distribution, but up to 40% of them have the AML1-ETO translocation [28]. Thus, the AML1-ETO translocation may be necessary for the leukemia that these people got, but it is not sufficient on its own. Patients can survive to 150 months at least with 1% of their stem cells still being AML1-ETO positive. This suggested to us that one has to have many events in the progression before one gets an AML. We then looked at a number of leukemias to test this hypothesis.
Catriona Jamieson and I looked at polycythemia vera, and our only question was whether this was a disease at the level of the stem cell [32]. Emmanuelle Passegue and I had shown in mice that a chronic myelogenous leukemia could be induced by a JunB knockout; this myeloproliferative disease was only transplantable at the level of the hematopoietic stem cell while it was still a chronic myeloproliferative disease, but when it went to blast crisis or acute leukemia, the leukemia was a downstream cell that we could identify, not the hematopoietic stem cell [33]. Other people, not us, showed that there is an activated mutated JAK2 kinase as part of the pathogenesis of polycythemia vera, this myeloproliferative disease, and we showed that purified hematopoietic stem cells from those patients show an abnormal phenotype-methylcellulose overproduction of red cells and larger colonies that are blocked with a JAK2 inhibitor. To summarize all of our experiments in a mouse model, if we add 1 event after another to try to get to AML, we have to avoid program cell death every which way the cell can do it. We used myelomonocytic expression of Bcl-2, along with a knock out of the fas death pathway. The leukemias that derive from these mice overexpress CD47, which enhances their mobility and prevents macrophages from phagocytosing them. The leukemias that develop only grow in T cell-deficient mice (Jamieson, Jaiswal, and Weissman, unpublished). If you have a functional T cell system they do not grow, implying immune recognition; these leukemias were transplantable at the GM progenitor stage of development, and had turned on beta-catenin expression. A couple of years ago Tannishta Reya, Karl Willert, Roeland Nusse, and I showed that the Wnt pathway was involved in the self-renewal of mouse hematopoietic stem cells, and found that if you block the beta-catenin pathway they cannot self-renew [34,35]. So, in the leukemias I am talking about they are myelogenous leukemias and they have activated a self-renewal component that is used in hematopoiesis only by the long-term hematopoietic stem cell.
In every example at which we have looked, the first phase of the leukemia in its chronic form is a disease at the level of the hematopoietic stem cell. Now, probably the early phase of AML1-ETO, those Hiroshima samples, probably they did not come in and complain of having a translocation only in the hematopoietic stem cells. But, then the argument goes on that a progeny cell from that has accumulated the 2 or more events and the leukemia breaks out in its acute phase at a downstream phase. So, that has been true so far of the examples I have told you. AML1-ETO has something going on in the stem cells, but the multipotent progenitor is the acute leukemia. In the mouse model, the acute leukemia is at the GM progenitor, but it starts in the hematopoietic stem cell So, Jamieson and I got a lot of samples of CML, and we looked at the chronic phase, the accelerated phase, and the myeloid blast crisis phase. She noted that in the myeloid blast crisis phase, the 1 cell population, beside the blasts, that were increased in frequency were the GM progenitors [36], which are several developmental steps downstream from the hematopoietic stem cell. We know that CML starts with a multipotent hematopoietic cell; Phil Fialkow and colleagues [35] showed by allelic X chromosomal markers that the CML clone includes myeloerythroid cells and B cells (but not T cells). Of course, the canonical bcr-abl translocation is in these progeny. Catriona and I first showed that chronic phase CML HSC indeed harbors the translocation. These and normal HSC contained in their nuclei the unphosphorylated [active] form of beta-catenin. In the normal or chronic phase GM progenitor, you hardly see it, but it is blazing in the myeloid blast crisis GM progenitor [37]. Along with Laurie Ailles, we introduced by lentivirus, a reporter with LEF-1/TCF (the nuclear partners of beta catenin) canonical sequences followed by the GFP gene, to test whether the presence of nuclear catenin resulted in the reporter being turned on. In this experiment, GMP in myeloid blast crisis patients, but not from normal donors, turn on the green protein. The final point is that normal GM progenitors do not replate in the methylcellulose assay. If you let them make the colonies and then replate them, they make GM colonies but do not renew GMP. The myeloid blast crisis cells replate just as well as hematopoietic stem cells, and we could block the replating by introducing into those myeloid blast crisis, leukemia stem cells, the pathway inhibitor Axin (expressed at real high levels). Catriona has gone on to show that one can transplant these leukemias. The GMP from normal mice do not transplant the disease, but the GM progenitors from myeloid blast crisis transplant blast crisis serially from 1 mouse to the next; the blast cells do not transplant the disease. Therefore, it is not the hematopoietic stem cell precursors, nor the blast cell progeny, but the GM progenitor that is the target and it has turned on, in addition to bcr-abl, the beta-catenin pathway. It is also CD47+ and has increased levels of bcl-2, so, there were many events that probably occurred.
Let me summarize this in a speculative model of what is happening (Figure 2). Long-term hematopoietic stem cells go to the myeloid series in the quantal steps of differentiation shown on the top line. The only self-renewing cell is the long-term HSC. Let us imagine that if 1 of these cells is bcr-abl, it is a clonal cell; that we know from our studies, Jamieson and I, that it becomes the vast majority of the hematopoietic stem cells in those patients. It does not increase the frequency of stem cells, but rather they have out-competed the cells that do not have bcr-abl [38]. So long as that clone can exist, we propose that another event could happen. Let us say that in a bcr-abl HSC subclone you turn on Tert, and then in a further bcr-abl+, tert+ HSC subclone, you turn on Bcl-2, and then further along, CD47, or all the events you can imagine until you have a cell that is able to live long, keeps proliferating long, has dominated the pool, and something else happens. And the something else that happens here, we propose, is that you do not shut off the beta-catenin system as you go from the long-term stem cell to the short, and on down. We know that this system is on intrinsically in those cells. They do not need Wnt to drive it, which may be an important point when you plan therapies, eventually. We know it transplants the disease, and we propose that if we can isolate in pure form the leukemia stem cells, which are only 6% of the cells in the bone marrow, that they and only they have the genetic epigenetic changes that are shown speculatively in the figure as events 1, 2, 3, 4, 5, 6, 7, etc.
Figure 2.
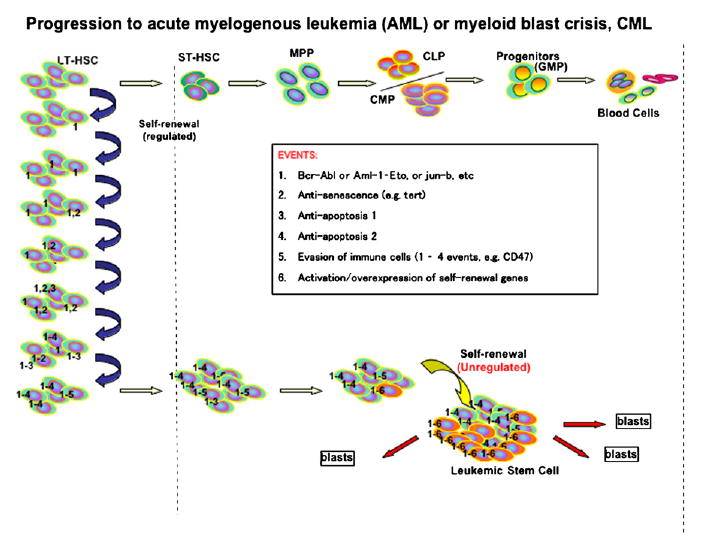
Progression to acute myelogenous leukemia (AML) or myeloid blast crisis (CML).
Conclusion
Saying that stem cell biology is important for us to understand, of course, is something that hematopoietic stem cell transplanters have been doing all along— trying to regenerate the system, the hematopoietic system. BMT was developed by oncologists, so it is mainly used for oncologic indications. We hope that we can move human allogeneic HSC into the therapy of nonmalignant diseases, because pure HSC cannot cause any level of GVHD: these cells should be useful in blocking autoimmune disease and repairing other genetic diseases of the blood-forming system. Once you know that hematopoietic stem cells can induce permanent transplantation tolerance, which we have shown over and over again, then you should be able some day to cotransplant hematopoietic stem cells and liver stem cells, if they exist, or lung, and so on, and change the way that you treat these diseases. When we get to the tumor field, I propose that cancer stem cells (CSCs) are using the same self-renewal genes that the normal stem cells use, but regulation of self-renewal is out of control; and these CSCs have in addition to the hit of the self-renewal gene, so many other genes that prevent surveillance over those cells internal or extrinsic. We know that they have ABC transporters and so they are tougher to get drugs into. In 2001, Mike Clarke, Tannishta Reya, Sean Morrison, and I [39] noted that perhaps the CSCs were more resistant to drugs and radiation than the majority of cells found in a tumor. Because remember, all those drugs were found because they shrunk tumors by maybe 90% to 95%, which did not mean that in the 90% to 95% were 90% to 95% of the tumor stem cells. It just meant it shrunk the tumor. The promise of applying “stem cell thinking and approaches” to the cancer field is that the isolation of such cells will allow new insights, new drugs, and new immune modalities designed to kill CSCs.
References
- 1.Till JE, McCulloch EA. A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat Res. 1961;14:213–22. [PubMed] [Google Scholar]
- 2.Forsberg EC, Prohaska SS, Katzman S, Heffner GC, Stuart JM, Weissman IL. Differential Expression of Novel Potential Regulators in Hematopoietic Stem Cells. PLoS Genet. 2005;1(3):e28. doi: 10.1371/journal.pgen.0010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 4.Negrin RS, Atkinson K, Leemhuis T, Hanania E, Juttner C, Tierney K, Hu WW, Johnston LJ, Shizurn JA, Stockerl-Goldstein KE, Blume KG, Weissman IL, Bower S, Baynes R, Dansey R, Karanes C, Peters W, Klein J. Transplantation of highly purified CD34+Thy-1+ hematopoietic stem cells in patients with metastatic breast cancer. Biol Blood Marrow Transplant. 2000;6(3):262–271. doi: 10.1016/s1083-8791(00)70008-5. [DOI] [PubMed] [Google Scholar]
- 5.Shizuru JA, Jerabek L, Edwards CT, Weissman IL. Transplantation of purified hematopoietic stem cells: requirements for overcoming the barriers of allogeneic engraftment. Biol Blood Marrow Transplant. 1996;2(1):3–14. [PubMed] [Google Scholar]
- 6.Gandy KL, Weissman IL. Tolerance of allogeneic heart grafts in mice simultaneously reconstituted with purified allogeneic hematopoietic stem cells. Transplantation. 1998;65(3):295–304. doi: 10.1097/00007890-199802150-00001. [DOI] [PubMed] [Google Scholar]
- 7.Shizuru JA, Weissman IL, Kernoff R, Masek M, Scheffold YC. Purified hematopoietic stem cell grafts induce tolerance to alloantigens and can mediate positive and negative T cell selection. Proc Natl Acad Sci U S A. 2000;97(17):9555–9560. doi: 10.1073/pnas.170279297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beilhack GF, Scheffold YC, Weissman IL, Taylor C, Jerabek L, Burge MJ, Masek MA, Shizuru JA. Purified allogeneic hematopoietic stem cell transplantation blocks diabetes pathogenesis in NOD mice. Diabetes. 2003;52(1):59–68. doi: 10.2337/diabetes.52.1.59. [DOI] [PubMed] [Google Scholar]
- 9.Smith-Berdan S, Gille D, Weissman IL, Christensen JL. Reversal of autoimmune disease in lupus-prone New Zealand black/New Zealand white mice by nonmyeloablative transplantation of purified allogeneic hematopoietic stem cells. Blood. 2007;110(4):1370–1378. doi: 10.1182/blood-2007-03-081497. [DOI] [PubMed] [Google Scholar]
- 10.Kennedy M, Firpo M, Choi K, Wall C, Robertson S, Kabrun N, Keller G. A common precursor for primitive erythropoiesis and definitive haematopoiesis. Nature. 1997;386(6624):488–493. doi: 10.1038/386488a0. [DOI] [PubMed] [Google Scholar]
- 11.Huber TL, Kouskoff V, Fehling HJ, Palis J, Keller G. Haemangioblast commitment is initiated in the primitive streak of the mouse embryo. Nature. 2004;432(7017):625–630. doi: 10.1038/nature03122. [DOI] [PubMed] [Google Scholar]
- 12.Ueno H, Weissman IL. Clonal analysis of mouse development reveals a polyclonal origin for yolk sac blood islands. Dev Cell. 2006;11(4):519–533. doi: 10.1016/j.devcel.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 13.Goodman JW, Hodgson GS. Evidence for stem cells in the peripheral blood of mice. Blood. 1962;19:702–714. [PubMed] [Google Scholar]
- 14.Richman CM, Weiner RS, Yankee RA. Increase in circulating stem cells following chemotherapy in man. Blood. 1976;47(6):1031–1039. [PubMed] [Google Scholar]
- 15.Socinski MA, Cannistra SA, Elias A, Antman KH, Schnipper L, Griffin JD. Granulocyte-macrophage colony stimulating factor expands the circulating haemopoietic progenitor cell compartment in man. Lancet. 1988;1(8596):1194–1198. doi: 10.1016/s0140-6736(88)92012-0. [DOI] [PubMed] [Google Scholar]
- 16.Duhrsen U, Villeval JL, Boyd J, Kannourakis G, Morstyn G, Metcalf D. Effects of recombinant human granulocyte colony-stimulating factor on hematopoietic progenitor cells in cancer patients. Blood. 1988;72(6):2074–2081. [PubMed] [Google Scholar]
- 17.Cao YA, Wagers AJ, Beilhack A, Dusich J, Bachmann MH, Negrin RS, Weissman IL, Contag CH. Shifting foci of hematopoiesis during reconstitution from single stem cells. Proc Natl Acad Sci U S A. 2004;101(1):221–226. doi: 10.1073/pnas.2637010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagers AJ, Allsopp RC, Weissman IL. Changes in integrin expression are associated with altered homing properties of Lin(-/lo)Thy1.1(lo)Sca-1(+)c-kit(+) hematopoietic stem cells following mobilization by cyclophosphamide/granulocyte colony-stimulating factor. Exp Hematol. 2002;30(2):176–185. doi: 10.1016/s0301-472x(01)00777-9. [DOI] [PubMed] [Google Scholar]
- 19.Yang XD, Michie SA, Mebius RE, Tisch R, Weissman I, McDevitt HO. The role of cell adhesion molecules in the development of IDDM: implications for pathogenesis and therapy. Diabetes. 1996;45(6):705–710. doi: 10.2337/diab.45.6.705. [DOI] [PubMed] [Google Scholar]
- 20.Wright DE, Bowman EP, Wagers AJ, Butcher EC, Weissman IL. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J Exp Med. 2002;195(9):1145–1154. doi: 10.1084/jem.20011284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhattacharya D, Rossi DJ, Bryder D, Weissman IL. Purified hematopoietic stem cell engraftment of rare niches corrects severe lymphoid deficiencies without host conditioning. J Exp Med. 2006;203(1):73–85. doi: 10.1084/jem.20051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL. The aging of hematopoietic stem cells. Nat Med. 1996;2(9):1011–1016. doi: 10.1038/nm0996-1011. [DOI] [PubMed] [Google Scholar]
- 23.Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, Weissman IL. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. 2005;102(26):9194–9199. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rossi DJ, Seita J, Czechowicz A, Bhattacharya D, Bryder D, Weissman IL. Hematopoietic stem cell quiescence attenuates DNA damage response and permits DNA damage accumulation during aging. Cell Cycle. 2007;6(19):2371–2376. doi: 10.4161/cc.6.19.4759. [DOI] [PubMed] [Google Scholar]
- 25.Uchida N, Weissman IL. Searching for hematopoietic stem cells: evidence that Thy-1.1lo Lin- Sca-1+ cells are the only stem cells in C57BL/Ka-Thy-1.1 bone marrow. J Exp Med. 1992;175(1):175–184. doi: 10.1084/jem.175.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ikuta K, Kina T, MacNeil I, Uchida N, Peault B, Chien YH, Weissman IL. A developmental switch in thymic lymphocyte maturation potential occurs at the level of hematopoietic stem cells. Cell. 1990;62(5):863–874. doi: 10.1016/0092-8674(90)90262-d. [DOI] [PubMed] [Google Scholar]
- 27.Spilianakis CG, Lalioti MD, Town T, Lee GR, Flavell RA. Interchromosomal associations between alternatively expressed loci. Nature. 2005;435(7042):637–645. doi: 10.1038/nature03574. [DOI] [PubMed] [Google Scholar]
- 28.Miyamoto T, Weissman IL, Akashi K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc Natl Acad Sci U S A. 2000;97(13):7521–7526. doi: 10.1073/pnas.97.13.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kamel-Reid S, Letarte M, Sirard C, Doedens M, Grunberger T, Fulop G, Freedman MH, Phillips RA, Dick JE. A model of human acute lymphoblastic leukemia in immune-deficient SCID mice. Science. 1989;246(4937):1597–1600. doi: 10.1126/science.2595371. [DOI] [PubMed] [Google Scholar]
- 30.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. Nature. 1994;367(6464):645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 31.McCune JM, Namikawa R, Kaneshima H, Shultz LD, Lieberman M, Weissman IL. The SCID-hu mouse: murine model for the analysis of human hematolymphoid differentiation and function. Science. 1988;241(4873):1632–1639. doi: 10.1126/science.241.4873.1632. [DOI] [PubMed] [Google Scholar]
- 32.Jamieson CH, Gotlib J, Durocher JA, Chao MP, Mariappan MR, Lay M, Jones C, Zehnder JL, Lilleberg SL, Weissman IL. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc Natl Acad Sci U S A. 2006 doi: 10.1073/pnas.0601462103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Passegue E, Jochum W, Schorpp-Kistner M, Mohle-Steinlein U, Wagner EF. Chronic myeloid leukemia with increased granulocyte progenitors in mice lacking junB expression in the myeloid lineage. Cell. 2001;104(1):21–32. doi: 10.1016/s0092-8674(01)00188-x. [DOI] [PubMed] [Google Scholar]
- 34.Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR, 3rd, Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423(6938):448–52. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- 35.Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423(6938):409–414. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- 36.Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, Sawyers CL, Weissman IL. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351(7):657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 37.Fialkow PJ, Jacobson RJ, Papayannopoulou T. Chronic myelocytic leukemia: clonal origin in a stem cell common to the granulocyte, erythrocyte, platelet and monocyte/macrophage. Am J Med. 1977;63(1):125–130. doi: 10.1016/0002-9343(77)90124-3. [DOI] [PubMed] [Google Scholar]
- 38.Weissman I. Stem cell research: paths to cancer therapies and regenerative medicine. Jama. 2005;294(11):1359–1366. doi: 10.1001/jama.294.11.1359. [DOI] [PubMed] [Google Scholar]
- 39.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]