

Abstract
Agrin is a multidomain heparan sulfate proteoglycan involved in postsynaptic differentiation at the neuromuscular junction. Binding of agrin to synaptic basal lamina is mediated by the N-terminal agrin (NtA) domain. The NtA domain of agrin is followed by a tandem of nine follistatin-like (FS) domains forming a rod-like spacer to the laminin G-like domains of the molecule. Here we report that the most C-terminal cysteine residue of NtA (Cys123) forms an interdomain disulfide bond with the FOLN subdomain of the FS module. Remarkably, this single cysteine is flanked by Leu117 and Val124, which are two essential β-branched amino acids forming the heterocomplex of NtA with the γ1 chain of laminin. Moreover, we show that this covalent linkage compensates for the seven amino acid residue splice insert at the very C-terminal helix H3 and causes a rigid interface between NtA and FS independent of the alternative mRNA splice event. These results suggest that the interdomain disulfide bond between the NtA and the first FS domain might be important for the proper folding of agrin.
Keywords: agrin, N-terminal agrin, follistatin, interdomain disulfide, FOLN subdomain, Kazal subdomain
Introduction
The extracellular matrix (ECM) is a network of extracellular macromolecules that provides a structural support between tissues in eukaryotic organisms. The ECM plays an important role in tissue development, cell differentiation, and signal transduction. It is made up of complex that has variety of polysaccharide and protein molecules, which are typically produced by the cells in the matrix.1 ECM proteins are mostly large, modular proteins, consisting of sets of frequently repeated domains. This allows for the acquisition of structural information from individual modules.2
The neuromuscular junction (NMJ) mediates the efficient communication between neurons and muscle fibers. It is composed of pre- and postsynaptic cells that are separated by a synaptic cleft containing the synaptic basal lamina. The ECM protein agrin is released by motor neurons and induces postsynaptic specializations at the NMJ, including clustering of acetylcholine receptors.3 Agrin is a multidomain heparan sulfate proteoglycan with an apparent molecular weight of 400–600 kDa on SDS-PAGE. The N-terminus of agrin is composed of the N-terminal agrin (NtA) domain, which is required for binding of laminin and localization of agrin at basal lamina. Remarkably, NtA is a target of alternative mRNA splicing that causes an insert of seven amino acid residues.4,5 The NtA domain is followed by nine repeats of follistatin-like (FS) domains.6,7 Mapping of the laminin-binding site of NtA revealed that two amino acid residues are critical for binding.8 Leu117 and Val124 form a heterotetrameric interface between the triple helical coiled coil of laminin and the C-terminal helix H3 of NtA. The C-terminus of agrin is composed of three laminin G-like domains, which function in the clustering of acetylcholine receptors.9
FS domains are found in proteins such as agrin, BM-40, follistatin, and ovomucoid-like chicken protein and are predominantly arranged as repetitive modules in tandem arrays forming rod-like structures.1,2 The structure of the FS domain of mouse BM-40 revealed an N-terminal β-hairpin with four cysteines linked 1-3 and 2-4 (FOLN subdomain), directly followed by a larger globular α/β-fold with six cysteines linked 5-9, 6-8, and 7-10 (Kazal subdomain).10 The separation of cysteines and their linkage pattern within the FOLN subdomain are reminiscent of epidermal growth factor.2 The Kazal domain is known as a member of the pancreatic secretory trypsin inhibitor family. A double-headed inhibitor of trypsin (on one site) and of chymotrypsin, subtilisin, and elastase (on the other site) is present in large amounts in submandibular glands and consists of two tandem Kazal type domains.11
In this work, we show in an integrated approach combining long-wavelength X-ray crystallography and mass spectrometry that there is an interdomain disulfide bridge between Cys123 of NtA and the following first FS domain in agrin. Intradomain disulfide bridges are commonly seen in ECM proteins and serve to stabilize the structural conformation of these molecules. Interdomain disulfide bridges are rare, and to date, only a few examples are known.12,13 We propose that this interdomain disulfide linkage in agrin plays an important role in stabilizing the overall structure as it is independent of an alternative mRNA splice event.
Results
Long-wavelength structure of NtA
The crystal structure of chicken NtA was determined to 2.3-Å resolution by molecular replacement using the previously solved chicken NtA core domain as search template (PDB code: 1PXU).14 The structure is composed of five β-strands forming a canonical shaped β-barrel domain flanked by α-helices at both termini [H1 and H3; Fig. 1(A)]. The most extraordinary feature of the crystal structure is the high flexibility of Cys123 located in the C-terminal α-helix H3, implying a potential disulfide linkage [Fig. 1(B)]. The use of soft X-rays with a wavelength of λ =1.9995 Å (6.213 kEv) and the possibility to collect highly redundant diffraction data were crucial to study the conformational flexibility of Cys123 of the C-terminal Helix H3 in NtA. In contrast to data collection protocols at λ = 1.54 Å, we could show for the first time an inherent structural flexibility of this free accessible thiol group. This finding is particularly striking based on the nonreducing milieu within the ECM and the close spatial neighborhood of the proceeding first FS domain.
Figure 1.
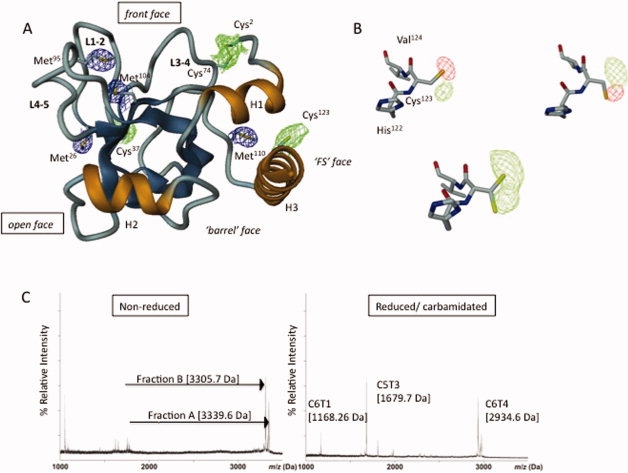
Long-wavelength X-ray structure of NtA and MS mapping of the interdomain disulfide linkage. (A) Ribbon diagram of the NtA structure. The β-strands are in blue and α-Helices (H1-H3) are in gold; loops L1-2, L3-4, and L4-5 connecting individual β-strands strands are oriented to the same surface of the protein. The 4σ Fo-Fc electron density maps for individual sulfur atoms in methionine thioether groups (in blue) and cysteine thiol moieties (in blue) are highlighted. (B) The alternate conformations adopted by Cys123. Upper row: calculated Fo-Fc electron density map (4σ contour level) around Cys123 is indicated each by means of the positive (green) and negative (red) electron density peak. The diffraction data were measured at λ = 1.54 Å. Lower row: a 4σ contour level Fo-Fc difference map (diffraction data collected at 1.6995 Å) indicating the free degree of rotation at κ angle at the thiolgroup of Cys123. (C) Left panel: MALDI-TOF spectrum of nonreduced fractions A (3339.6 Da) and B (3305.7 Da) obtained by sequential cleavage of NtA-FS by CNBr and trypsin. Right panel: the corresponding MALDI-TOF spectra of reduced and carbamidated trypsin digests of fraction A (C5T3 and C6T1) and B (C6T4). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
NtA is disulfide bridged with the FOLN subdomain of FS via C-terminal Cys123
To elucidate a possible disulfide linkage status of Cys123, proteolytic fragments of NtA-FS were generated by treatment with cyanogen bromide (CNBr), followed by reverse-phased high performance liquid chromatography (RP-HPLC) separation. The fragments obtained after separation on RP-HPLC were subjected to MALDI-TOF mass spectroscopy (MS; for nomenclature and molecular masses of the fragments see Materials and Methods and Tables II and III). Six fragments (C1–C6) are predicted from CNBr digestion of NtA-FS, from which four contain cysteine residues (Supporting Information-Table II). The disulfide bridge between Cys2 and Cys74 in the crystal structure [Fig. 1(A)] suggests that CNBr fragments C1 and C2 are disulfide linked. Although C1 contains a single cysteine residue (Cys2), fragment C2 contains two cysteine residues (Cys37 and Cys74). The crystal structure of recombinant NtA expressed in eukaryotic cells revealed that Cys37 is free, oriented inside the β-barrel core domain.4 MS confirmed the presence of corresponding fragments, and the observed data agree with the predicted masses (data not shown).
Table II.
Calculated Masses of the Predicted CNBr Fragments of NtA-FS
| Fragment | Sequence | Mass (Da) |
|---|---|---|
| C1 | NC2PERELQRREEEANVVLTGTVEEIM | 3044.48 |
| C2 | NVDPVHHTYSC37KVRVWRYLKGKDIVTHEILLDGGNKVVIGGFGDPLIC74 DNQVSTGDTRIFFVNPAPQYM | 7741.93 |
| C3 | WPAHRNELM | 1153.56 |
| C4 | LNSSLM | 664.33 |
| C5 | R*ITLR*N LEEVEHC123VEEHR*K*LLADKPNSYFTQTPPTPR*DAC150RGM | 5050.52 |
| C6 | LC155GFGAVC161ER*SPTDPSQASC173VC175K*K*TAC180PVVVAPVC188GSDYSTYSNEC199 ELEK*AQC206 NQQR*R*IK*VISK*GPC220GSK*DPC226AEVTR*SHHHHHH | 9194.25 |
The peptide nomenclature is described in Experimental Procedures. The molecular weights of the predicted CNBr fragments are given as the monoisotopic masses of the singly protonated peptides. Cysteines are highlighted as superscripts, while Ala132 and Cys150, which mark the last and the first residue of the NtA and the FS domain, respectively, are underlined. Stars indicate putative trypsin cleavage sites in C5 and C6.
Table III.
Trypsin Cleavage Products of the CNBr Fragments C5 and C6
| Peptide fraction | Peptide mass (nonreduced)a | Peptide mass (reduced)a | Δ Mass | Trypsin cleavage fragment | Sequence | Peptide mass (reduced)a | Peptide mass (reduced/ alkylated)a | Δ Mass |
|---|---|---|---|---|---|---|---|---|
| A | 3339.6 | 3343.5 | 4 | C5T3 | NLEEVEHC123VEHR | 1622.72 | 1679.72 | 57.0 |
| C5T6 | DAC150RGM | 667.2b | ND | |||||
| C6T1 | LC155GFGAVC161ER | 1053.5 | 1168.26 | 115.1 | ||||
| B | 3305.7 | 3312.5 | 7 | C6T4 | TAC180PVVVAPVC188 GSDYTYSNEC199ELK | 2763.2 | 2934.55 | 171.34 |
| C6T9 | GPC220GSK | 547.2 | ND | |||||
| C6T2c | SPTDPSQASC173V C175K | 1321.6 | ||||||
| C6T5c | AQC206NQQR | 846.4 |
All Peptide masses are given in Da. Individual fragments are labeled sequentially. Fraction A consists of C5T3, C5T6, and C6T1, whereas fraction B comprises C6T4 and C6T9, respectively. The peptides C6T2 and C6T5 containing cysteine residues Cys173, Cys175, and Cys206 could not be isolated. The calculated masses are given as monoisotopic values of the singly protonated peptides.
Molecular mass was calculated based on combined theoretical mass of individual trypsin fragments.
The oxidation and subsequent hydroxymethylation of the C-terminal Met adds +16Da to the peptide mass of 651.25 Da.
CNBr-trypsin fragments that could not be isolated.
Further analysis of the remaining fragments (C3–C6) revealed a rather broad peak in the mass spectrum. Reduction with dithiothreitol (DTT) showed that the observed mass corresponds to the calculated mass of C6, however, no precise measurement for fragment C5 could be obtained, which is attributable to incomplete cleavage by CNBr that resulted in a fragment of C3-C4-C5. Heterogeneous N-glycosylation on C3 might explain the lack of a well-defined mass spectrum of the C3-C4-C5 fragment.6,7 However, cleavage of NtA-FS by CNBr followed by reduction demonstrates that C5 is linked via disulfide bridges to C6. The connection can only be mediated by cysteines present on C5 (Cys123 and/or Cys150) because fragments C3 and C4 do not possess any cysteine residues. Therefore, a disulfide linkage between NtA (Cys123) and the first cysteine residue of FS (Cys150) can be excluded.
The next strategy to resolve disulfide bridging was to digest the CNBr-fragments C5-C6 with trypsin, followed by MALDI-TOF analysis before and after reduction with DTT and alkylation with iodacetoamide. In the nonreduced state, two peaks were identified as peptides A and B with observed molecular masses of 3339.6 Da (fraction A) and 3305.7 Da (fraction B), respectively [Fig. 1(C), left panel].
The observed mass of 3339.6 Da (fraction A) corresponds to the combined theoretical mass of C5T3 (1622.72 Da) and C5T6 (667.27 Da) disulfide bonded to C6T1 (1053.48 Da; SI-Table 3). A difference of 4 Da in molecular mass between the nonreduced (3339.6 Da) and sum of reduced (3343.47 Da) fragments is indicative for the formation of two disulfide bridges. The peptide was resolved by reduction with DTT and carbamidation with iodoacetamide into two peptides with masses of 1679.7 Da and 1168.3 Da, respectively [Fig. 1(C), right panel]. The mass of 1679.4 Da corresponds to C5T3 with one carbamidated cysteine (Cys123), whereas 1168.3 Da corresponds to C6T1 with two carbamidated cysteine residues (Cys155 and Cys161). However, the peptide fragment corresponding to C5T6 (Cys150) could not be identified on reduction and alkylation because of its small size.
In summary, the data presented show that intrafragment disulfide linkages within C5 (between Cys123 and Cys150) and within C6T1 (between Cys155 and Cys161) are not formed. Furthermore, they show that C5T3 and C5T6 are associated to C6T1 via two disulfide bonds and provide evidence for disulfide bridges between Cys123 and Cys150 with Cys155 or Cys161. Therefore, it can be concluded that NtA is disulfide linked via Cys123, located at the C-terminal α-helix H3, with the FOLN subdomain of FS. However, the order of linkage between the fragments remains elusive (see Discussion).
The observed mass of 3305.7 Da (fraction B) corresponds to the theoretical mass of C6T4 (2764.22 Da) disulfide linked to C6T9 (548.25) with a mass difference of 7 Da between nonreduced and reduced fragments [Fig. 1(C), left panel and Table II]. The peptide was resolved on DTT reduction and alkylation with iodoacetamide into a peptide (C6T4) with a mass of 2934.6 Da that corresponds to three carbamidated cysteine residues [Cys180, Cys188, and Cys199; Fig. 1(C), right]. The smaller peptide corresponding to C6T9 (Cys220) could not be identified following reduction and alkylation. These data imply disulfide bridge formation between Cys180, Cys188, or Cys199 with Cys220 including an intrafragment disulfide bond within C6T4. All attempts to isolate fragments containing Cys173, Cys175 (C6T2), and Cys206 (C6T5) failed, most probably because they do not bind to the reverse-phase support during HPLC purification.
Discussion
Many ECM modules contain disulfide bridges, most probably to increase stability of relatively small domains and to protect against proteolysis. Cysteines are the most important identifiers of a particular module by sequence analysis.1 Almost all conserved cysteine residues in various ECM proteins are involved in formation of disulfide bridges, mostly intramodular ones. Exceptions of this rule are rare and have so far only been observed for Ig-like and FN-III domains. The 3D structures of several cell surface proteins containing Ig-like domains clearly demonstrate the switch of disulfide bridges between strands.15 Domains of the FN-III type usually do not contain disulfide bridges; however, the structure of neuroglian reveals a disulfide linkage.16 Cole et al.12 demonstrated an intermodular disulfide linkage in the soluble Interleukin-6R domain, revealing a bridge between Cys6 of the N-terminal Ig-domain to Cys174 of the following first FN-III domain.
Crystal structures of FS domains within BM-40 and follistatin reveal a nonglobular structure that is stabilized by a small hydrophobic core in its larger, C-terminal portion (Kazal subdomain) and by a total of five disulfide bonds.10,17 Remarkably, in both X-ray structures, the N-terminal FOLN subdomain shows the largest degree of mobility. The subdivision of FS is reflected in a disulfide linkage pattern of two nonoverlapping sets (1-3 and 2-4) and (5-9, 6-8, and 7-10; Fig. 2).
Figure 2.
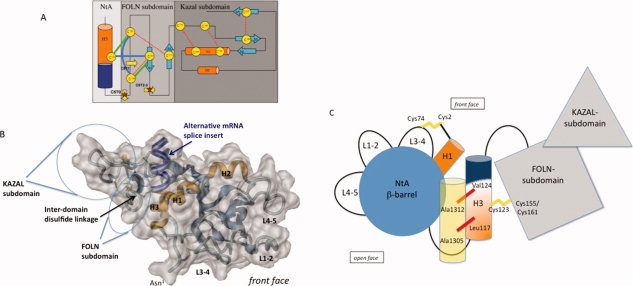
The proposed NtA-FS tandem. (A) Topology model of interdomain disulfide bond formation between NtA and FS domain. Secondary structural elements (α-helices in orange and β-strands in blue) and conserved disulfide bond pattern within the FS domain (thin red dotted lines) are highlighted. Cysteine residues are shown in circles labeled sequentially. The FS domain is assembled from two rather weakly interacting subunits, a highly twisted N-terminal β-hairpin (FOLN subdomain) and a pair of antiparallel α-helices connected to a small three-stranded antiparallel β-sheet (Kazal subdomain). Arrows mark trypsin cleavage sites, and stars represent isolated, reduced, and alkylated fragments (see also Table III). Possible new interdomain disulfide bonds between Cys123 and Cys155 or Cys161, as well as the new intradomain disulfide bond between Cys150 and Cys155, are highlighted as thick green (model 1) and blue (model 2) lines. In any case, the disulfide bridge between Cys155 and Cys173 has to be reshuffled. A theoretical third model, comprising a free Cys150 within C5 has to be excluded, based on the fact that a separate CNBr-trypsin fraction before reduction via DTT containing Cys150 only, as well as a consequent disulfide bridge between 2 and 4 could not be detected. (B) Surface representation of the modeled NtA-FS tandem based on a FTDOCK approach. Color scheme and labels as in Fig.1. Individual cysteine residues are shown in render mode. The alternative mRNA splice insert in H3 is highlighted in darkblue. (C) Topology diagram of the NtA-FS tandem. Individual domains of the tandem are highlighted in different color schemes according to previous diagrams. Loop regions extending the β-barrel (L1-2, L3-4, and L4-5) and the 9-residue linker between the barrel and α-helix H3, as well as the 15-residue long connection between the C-terminus of NtA and the N-terminus of FS, are shown as black lines. Disulfide bridges are highlighted in yellow. The alternative mRNA splice insert at H3 is shown in blue. For simplicity reasons, only the γ1-chain of laminin is shown in yellow transparent with key interface residues marked with red bars. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
By sequential cleavage of NtA-FS using CNBr and trypsin coupled to RP-HPLC purification, we have isolated fragments C5T3, C5T6, C6T1, C6T4, and C6T9. MALDI-TOF analysis of the reduced and carbamidated peptide fragments revealed that Cys123 from NtA is linked via a disulfide bridge with the FOLN subdomain of the following FS. This connection can be obtained either via its linkage to Cys155 or to Cys161, which leaves two possibilities for the formation of the disulfide bridge between NtA and FS [Fig. 2(A)]. The first possibility requires only one new disulfide bond between Cys123 and Cys155, while the conserved 1-3 bridge (Cys150-Cys161) remains intact. This model is supported by a sequence alignment of all nine FS domains of chicken agrin, showing that five disulfide bonds are highly conserved, with the exception of the third FS domain, where two disulfide bonds, 2-4 and 5-9, are replaced (Fig. 3). The first disulfide bridge (1-3) seems to be strictly required, however, a second connection within FOLN (2-4) and link to the Kazal subdomain (5-9) are not. In contrast, model 2 would require the formation of two new disulfide bridges between Cys123-Cys161 and Cys150-Cys155, respectively. Therefore, this possibility is less likely because (i) it would require reshuffling of two disulfide bridges within the FS domain (1-3 and 2-4) and (ii) the short distance of only 4 amino acids and the stereochemical constraints at the N-terminal tip of the β-hairpin make a disulfide bridge between Cys150 and Cys155 very unlikely.
Figure 3.

Sequence alignment of all nine FS domains in chicken agrin. The first FS domain starts with residue number 148. The upper scheme aligns the first four cysteine residues of the FOLN subdomain (disulfide linkage pattern between cysteine residues 1-3 and 2-4), whereas the Kazal subdomain with six cysteines is shown below (disulfide linkage pattern between cysteine residues 5-9, 6-8, and 7-10). Conserved residues within the Kazal subdomain are highlighted. Remarkably, the disulfide linkage between 2nd and 3rd as well as 5th and 9th cysteine residue is interrupted in the FS-3 domain.
Taken together, only models having a free cysteine at position four (Cys173) are possible, and a disulfide reshuffling within the FS domain is still required. Based on the limited set of data concerning disulfide bridging within the Kazal subdomain, a possible role of a free Cys173 remains elusive. However, superposition of both FS domains from BM-40 and follistatin revealed a hingelike motion between FOLN and Kazal subdomains.17 If both Kazal-like moieties are superimposed, the respective FOLN subdomains are bent and axially rotated by 45° relative to one another. Together with the observed variability in the positioning of cysteines 2, 4, 5, and 9, the interface between both subdomains is likely to be flexible. This flexibility might be important for the different spatial orientations in the long-range tandem arrays of nine FS domains in agrin [(Fig. 2(C)].
A particularly striking feature is the fact that the NtA domain has significant structural similarity with TIMP-1, an endogenous inhibitor of matrix metalloproteinases (MMPs). These enzymes are secreted as inactive zymogens and have to be activated by selective promatrixin proteinases.18 A possible interference of the Kazal inhibitor domain with this activation step might explain the stabilization effect of agrin within NMJ and the diminished MMP-activities if agrin is released inside the synaptic cleft.19 Agrin remains associated for weeks with the synaptic basal lamina in vivo, and mice that overexpress an NtA-containing agrin construct at the NMJ show increased stability of postsynaptic structures. These authors19 showed that regulated inhibition of MMP3-activity results in changes in structure and function of the NMJ. Consistent with its modular organization, different regions of agrin have been associated with distinct functional properties.20 An alternate function for agrin's FS domains is suggested by their homology with Kazal-type proteinase inhibitors. Studies in vitro have shown that agrin inhibits several proteinases, including a membrane-bound plasmin-like activity.21 In addition to their structural role, the FS domains may also act as protease inhibitors. Interestingly, a proposed intermodular disulfide linkage between NtA and the FOLN-subdomain of the following FS module underlines this hypothesis.
The interdomain disulfide bridging between Cys123 and the FOLN subdomain of FS confirms the previously proposed model of NtA-laminin interaction, which clearly indicates a predominant role of residues at the “barrel face” of helix H3 (interaction interface) and a “FS face” at the opposite side. Cys123 on the FS face marks the linker for the interaction between NtA and the following first FS domain (see above). Disulfide bridges are well known to stabilize noncovalent conformations and are thus important and, usually, conserved structural features. The transition from an intradomain to an interdomain cysteine bridge may offer the advantage of a defined spatial orientation of the NtA and the first FS domain in agrin.
The C-terminal α-helix (H3) of chicken NtA contains a seven-residue splice insert comprising residues Glu126–Ala132. Motor neurons in developing spinal cord contain agrin transcripts that include the splice insert. However, the majority of agrin mRNA in nonneuronal tissues is characterized by the absence of the seven-residue insert.6,22 We suggest that the additional disulfide bridge between NtA and FS performs a further stabilization effect, which is required to accommodate the loss of seven amino acid residues in the C-terminal α-helix H3 [Fig. 2(B)]. These findings are supported by the fact that alternative mRNA splice events are species specific. Only chicken NtA shows the insert of seven amino acid residues, whereas mouse, human, and rat NtA domains do not. However, all species reveal the same pattern of synapse formation and stabilization.
Despite the fact that the linker-region between NtA and FS (residues Asp133–Ala149) remains the same, a loss of the seven-residue insert within α-helix H3 would cause a remarkable effect on the spatial orientation of both domains. The intermodular disulfide bond would be able to stabilize a required relative orientation to each other and preserve functional activity in the protein.
To solve this question, the 3D structure of the NtA-FS tandem would be needed. Work in this direction is in progress.
Materials and Methods
Protein purification and CNBr cleavage
The NtA-FS was dialyzed overnight against 1% acetic acid, followed by speed vac drying. The resulting pellet was resuspended in 70% formic acid and digested overnight with CNBr. The digest was again speed vac dried, the pellet was taken up in 0.1% trifluoroacetic acid (TFA), and the resulting peptide fragments were purified by RP-HPLC on a Vydac TP52 C18 (2.1 × 250 mm) column.
Trypsin cleavage
Fractions obtained after RP-HPLC were subjected to speed vac drying to evaporate acetonitrile. The pH was adjusted to 8.0, and digestion with trypsin (Promega) was performed at an enzyme/substrate ratio of 1:25 at 37°C for 2 h. The reaction was terminated by the addition of TFA to a final concentration of 0.1%. The samples were desalted by absorption on C18 Zip Tips (P10 size, Millipore) washed with 0.1% TFA and desorbed with 2 mL of 50% acetonitrile/0.1% TFA. The samples were analyzed by MALDI-TOF as described below.
Reduction and alkylation
Reduction was carried out by 10 mM DTT in 100 mM Tris-HCl pH 8.0 for 1 h. Following reduction, cysteines were alkylated using 50 mM iodoacetamide and incubated for a further 15 min at room temperature. The reaction was stopped by the addition of 1/10 volume of 10% TFA. The samples were desalted as described above and analyzed on MALDI-TOF.
MALDI-TOF mass spectrometry
Three hundred nanoliters of peptide solution were deposited onto anchor spots of a 36-sample support target (Bruker Daltonik), and the droplet was allowed to dry at room temperature. Mass spectra were recorded on a Bruker Reflex III instrument (Bruker Daltonik). Peptides were analyzed in linear mode. Assignment of the CNBr or tryptic peptides of NtA-FS was done by comparing the measured masses with those predicted by the protein sequence of NtA-FS.
Peptide nomenclature
The first letter and number of the fragment name indicates the cyanogen cleavage products (C1 to C6). Peptides of CNBr fragments C5 (C5T3 and C5T6) and C6 (C6T4 and C6T9) generated by trypsin cleavage are labeled with an additional T. The peptides are numbered sequentially according to their position based on the amino acid sequence (Tables I and II).
Table I.
Data Collection and Refinement Statistics
| Data collection at 1.9995 Å | |
| Resolution (Å) (last shell) | 28–2.3 (2.44–2.3) |
| Observed reflections | 1.130368 |
| Unique reflections | 8736 |
| Completeness | 97.3 (97.3) |
| Redundancy | 31.1 |
| Rsyma | 8.6 (33.9) |
| Unit cell dimensions (Å, °) | |
| a/b and c | 80.23/50.71 and 54.19 |
| β | 117.094 |
| Refinement statistics | |
| R-factorb (%) | 22.1 (42.7) |
| Rfree (%) | 27.6 (45.8) |
| Bond lengthc (Å) | 0.006 |
| Bond anglesc (°) | 1.3 |
| Ramachandran plotd | 87.5/12.5/0/0 |
Rsym = 100* Σ|I − 〈I〉|/ΣI. Values for the final resolution shells are in parenthesis.
Rfactor = 100* Σ||Fobs| − |Fcalc||/Σ|Fobs|.
Root mean square error.
Percentage of residues in most favored/additionally allowed/generously allowed, and disallowed regions.
Structure determination of NtA
The NtA domain was produced in HEK 293 cells and purified from conditioned media as described previously.8,14 To obtain crystals in a quality to perform long-wavelength X-ray diffraction experiments, cocoamidoprylbetaine was added to the original crystallization condition.5,23
The long-wavelength data set were collected at a wavelength λ = 1.9995 Å at the Canadian light source. Diffraction images were processed using program suite MOSFLM, and the structure factors were scaled and reduced using SCALA from the CCP4 package.24 The statistics of the merged data and the refined structure are given in Table I. Molecular replacement was performed using the AMORE program of the CCP4 package. The core of the five stranded β-barrel OB-fold of NtA was used as search model, comprising amino acid residues 18–110. Loop regions connecting β-strands S1-S2 (L1-2), S3-S4 (L3-4), S4-5 (L4-5) and all three helical segments (H1-H3) were truncated. Refinement with Canadian light source was alternated with manual electron density refitting of side chains and terminal regions.25 The target parameters of Engh and Huber, overall anisotropic B-factor scaling, and bulk solvent corrections were used. Finally, water molecules were added chosen by distance criteria and hydrogen bonding geometry and were tested for position in spherical density, reasonable temperature factors, real space R-values, and improvement of the R-factors. The coordinates have been deposited at Brookhaven database with the PDB-code 3I7O.
Molecular modeling
Docking of NtA (PDB code:1PXU) with the FS domain (PDB code:1BMO) was performed using FTDOCK.26 The homology model of the FS domain was generated using SWISS-Model. The lowest energy docked structures were subjected to 100 cycles of unrestrained Powell minimization using CNS.25 Harmonic restraints were imposed on the protein atoms (3 kcal/mol Å2) with increased weight (20 kcal/mol Å2).
References
- 1.Hohenester E, Engel J. Domain structure and organisation in extracellular matrix proteins. Matrix Biol. 2002;21:115–128. doi: 10.1016/s0945-053x(01)00191-3. [DOI] [PubMed] [Google Scholar]
- 2.Bork P, Downing AK, Kieffer B, Campbell ID. Structure and distribution of modules in extracellular proteins. Q Rev Biophys. 1996;29:119–167. doi: 10.1017/s0033583500005783. [DOI] [PubMed] [Google Scholar]
- 3.Ruegg MA, Bixby JL. Agrin orchestrates synaptic differentiation at the vertebrate neuromuscular junction. Trends Neurosci. 1998;21:22–27. doi: 10.1016/s0166-2236(97)01154-5. [DOI] [PubMed] [Google Scholar]
- 4.Stetefeld J. The laminin-binding domain of agrin is structurally related to N-TIMP-1. Nat Struct Biol. 2001;8:705–709. doi: 10.1038/90422. [DOI] [PubMed] [Google Scholar]
- 5.Stetefeld J, Ruegg MA. Structural and functional diversity generated by alternative mRNA splicing. Trends Biochem Sci. 2005;30:515–521. doi: 10.1016/j.tibs.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Denzer AJ, Gesemann M, Schumacher B, Ruegg MA. An amino-terminal extension is required for the secretion of chick agrin and its binding to extracellular matrix. J Cell Biol. 1995;131:1547–1560. doi: 10.1083/jcb.131.6.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denzer AJ, Hauser DM, Gesemann M, Ruegg MA. Synaptic differentiation: The role of agrin in the formation and maintenance of the neuromuscular junction. Cell Tissue Res. 1997;290:357–365. doi: 10.1007/s004410050941. [DOI] [PubMed] [Google Scholar]
- 8.Mascarenhas J. Mapping of the laminin-binding site of the N-terminal agrin domain (NtA) Embo J. 2003;22:529–536. doi: 10.1093/emboj/cdg041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gesemann M. Alternative splicing of agrin alters its binding to heparin, dystroglycan, and the putative agrin receptor. Neuron. 1996;16:755–767. doi: 10.1016/s0896-6273(00)80096-3. [DOI] [PubMed] [Google Scholar]
- 10.Hohenester E, Maurer P, Timpl R. Crystal structure of a pair of follistatin-like and EF-hand calcium-binding domains in BM-40. Embo J. 1997;16:3778–3786. doi: 10.1093/emboj/16.13.3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van de Locht A. Two heads are better than one: Crystal structure of the insect derived double domain Kazal inhibitor rhodniin in complex with thrombin. Embo J. 1995;14:5149–5157. doi: 10.1002/j.1460-2075.1995.tb00199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cole AR. Disulfide bond structure and N-glycosylation sites of the extracellular domain of the human interleukin-6 receptor. J Biol Chem. 1999;274:7207–7215. doi: 10.1074/jbc.274.11.7207. [DOI] [PubMed] [Google Scholar]
- 13.Muralidhara BK, Hirose M. Structural and functional consequences of removal of the interdomain disulfide bridge from the isolated C-lobe of ovotransferrin. Protein Sci. 2000;9:1567–1575. doi: 10.1110/ps.9.8.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mascarenhas JB. Structure and laminin-binding specificity of the NtA domain expressed in eukaryotic cells. Matrix Biol. 2005;23:507–513. doi: 10.1016/j.matbio.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 15.Bork P, Holm L, Sander C. The immunoglobulin fold. Structural classification, sequence patterns and common core. J Mol Biol. 1994;242:309–320. doi: 10.1006/jmbi.1994.1582. [DOI] [PubMed] [Google Scholar]
- 16.Huber AH, Wang YM, Bieber AJ, Bjorkman PJ. Crystal structure of tandem type III fibronectin domains from Drosophila neuroglian at 2.0 Å. Neuron. 1994;12:717–731. doi: 10.1016/0896-6273(94)90326-3. [DOI] [PubMed] [Google Scholar]
- 17.Innis CA, Hyvonen M. Crystal structures of the heparan sulphate-binding domain of follistatInsights into ligand binding. J Biol Chem. 2003;278:39969–39977. doi: 10.1074/jbc.M211284200. [DOI] [PubMed] [Google Scholar]
- 18.Nagase H, Woessner JF. Matrix metalloproteinases. J Biol Chem. 1999;274:21491–21494. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- 19.Werle MJ, VanSaun M. Activity dependent removal of agrin from synaptic basal lamina by matrix metalloproteinase 3. J Neurocytol. 2003;32:905–913. doi: 10.1023/B:NEUR.0000020631.69804.f5. [DOI] [PubMed] [Google Scholar]
- 20.Smith MA, Hilgenberg LG. Agrin in the CNS: A protein in search of a function? Neuroreport. 2002;13:1485–1495. doi: 10.1097/00001756-200208270-00001. [DOI] [PubMed] [Google Scholar]
- 21.Biroc SL, Payan DG, Fisher JM. Isoforms of agrin are widely expressed in the developing rat and may function as protease inhibitors. Brain Res Dev Brain Res. 1993;75:119–129. doi: 10.1016/0165-3806(93)90071-h. [DOI] [PubMed] [Google Scholar]
- 22.Tsen G, Halfter W, Kroger S, Cole GJ. Agrin is a heparan sulfate proteoglycan. J Biol Chem. 1995;270:3392–3399. doi: 10.1074/jbc.270.7.3392. [DOI] [PubMed] [Google Scholar]
- 23.Ozbek S, Muller J, Figgemeier E, Stetefeld J. Favourable mediation of crystal contacts by cocoamidopropylbetaine (CAPB) Acta Crystallogr D Biol Crystallogr. 2005;61:477–480. doi: 10.1107/S0907444905001204. [DOI] [PubMed] [Google Scholar]
- 24.CCP4, Collaborative Computing Project No. 4. The CCP4 suite: Programs for protein crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 25.Brunger AT. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 26.Gabb HA, Jackson RM, Sternberg M. Modelling protein docking using shape complementarity, electrostatics and biochemical information. J Mol Biol. 1997;272:106–120. doi: 10.1006/jmbi.1997.1203. [DOI] [PubMed] [Google Scholar]