

Abstract
Quantitative prediction of protein–protein binding affinity is essential for understanding protein–protein interactions. In this article, an atomic level potential of mean force (PMF) considering volume correction is presented for the prediction of protein–protein binding affinity. The potential is obtained by statistically analyzing X-ray structures of protein–protein complexes in the Protein Data Bank. This approach circumvents the complicated steps of the volume correction process and is very easy to implement in practice. It can obtain more reasonable pair potential compared with traditional PMF and shows a classic picture of nonbonded atom pair interaction as Lennard-Jones potential. To evaluate the prediction ability for protein–protein binding affinity, six test sets are examined. Sets 1–5 were used as test set in five published studies, respectively, and set 6 was the union set of sets 1–5, with a total of 86 protein–protein complexes. The correlation coefficient (R) and standard deviation (SD) of fitting predicted affinity to experimental data were calculated to compare the performance of ours with that in literature. Our predictions on sets 1–5 were as good as the best prediction reported in the published studies, and for union set 6, R = 0.76, SD = 2.24 kcal/mol. Furthermore, we found that the volume correction can significantly improve the prediction ability. This approach can also promote the research on docking and protein structure prediction.
Keywords: structure-derived statistical potential, potential of mean force, knowledge-based potential, protein–protein interactions, prediction of binding affinity
Introduction
Protein–protein interactions participate in an extremely wide range of life processes, including cellular metabolism of matter and energy, signal transduction, and so on. Thus, understanding protein–protein interactions is a very important issue in biology. However, satisfactory solutions to many problems in this field have not been obtained yet, including predictions of protein–protein affinity and protein–protein structure. All of them require a precise energy function. Many efforts have been made to develop such functions but the achieved accuracy still need to be improved in practice.1–3 In this article, we focus on structure-derived statistical potentials to predict protein–protein affinity.
Structure-derived statistical potentials have been widely applied not only in protein structure prediction and design but also in protein complexes studies, such as protein–ligand affinity prediction (the ligand can be protein, peptide, DNA, RNA, or other molecules), mutation-induced changes in protein stability, and rational drug design.4–13 In those approaches, the potential is extracted by statistically analyzing known three-dimensional structure data of biomolecules. Therefore, they were also termed knowledge-based potentials. One kind of them, potential of mean force (PMF), is derived from the statistical mechanics of simple liquids,14–16 which converts particle pair distribution of distance into distance-dependent potential function. PMF has been frequently used in affinity prediction and structure scoring, because its physical meaning and function curve are similar to those of the “true” energy potential, which in principle can be derived from fundamental analysis of the forces between particles,10,17 such as quantum chemical calculations. Therefore, PMF was also called as energy-like potential or quantity.
Volume correction must be considered when PMF is applied in protein systems. It is one of the key factors that can improve the precision of prediction and the reasonableness of potential function. Since PMF was introduced into the studies of protein systems, the understanding and the application of volume correction (or frequency correction) have undergone a series of development.
Sippl18 observed the frequency of the alpha-C of a residue pairs and normalized it with the average frequency over all residue pairs. Then, the normalized frequency was transformed into potential directly without considerations of the frequency correction. This traditional PMF approach was the mainstream method in early researches.19,20
Subsequently, some approaches to calculate PMF are based on the radial distribution function (RDF) in the statistical mechanics of simple liquids.14–16 In those approaches, the frequency was normalized in the manner of dividing occurrence numbers in a sphere volume without any correction. However, the occupied volume in a more complex system, such as in a protein system, is not a whole sphere. Therefore, when normalizing the occurrence frequency of atom pairs, the whole sphere volume is not a good indicator of the actual occupied volume. For example, Bahar and Jernigan21 considered the theoretical basis of PMF as the RDF. They normalized the occurrence numbers with the numbers in a whole sphere volume (4πr2dr). They further analyzed in detail the distribution tendency of the occurrence numbers of residue pairs in protein systems with increasing distance and compared it with the occurrence numbers in a whole sphere (Fig. 2 in Ref. 21, the tangent in this figure corresponds to the distribution of numbers in a whole sphere). Form this figure, we can get the hint of correcting the distribution of the occurrence numbers in a whole sphere with a function to obtain the better approximation to the distribution in protein systems. Mitchell et al.22 found that the factor of a whole sphere (4πr2dr) gives an average potential that is weakly repulsive over the entire distance range with no attractive region at typical interaction distances. They thought that this abnormality is due to the occupied volume of atoms in protein complexes deviating significantly from r2 proportionality.
Figure 2.
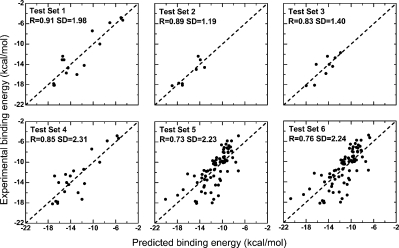
The predicted binding affinity by improved approach fitting with experimental data for six test sets. The linear correlation coefficient (R) and standard deviation (SD) were calculated. The results of the statistical analysis are given in Table I.
Imperfections in the aforementioned studies show that in systems as complicated as proteins, the occupied volume is not proportional to a whole sphere. In contrast to in simple liquids system, the normalized frequency of atom pairs (or residue pairs) can work well15 using f(r) = N(r)/volume(r), here occupied volume is a whole sphere: volume(r) = 4πr2dr.
Since then to obtain the real occupied volume in protein systems, the volume correction has been developed along two ways, one of which is based on correction functions and the other on structural statistics. The first way corrects occupied volume with a certain function to get the better approximation than a whole sphere volume 4πr2dr. Zhou and Zhou23 established DFIRE approach, which corrected volume with rα. The exponent α is a constant, whose empirical value was first found equal to 1.5723 and refined to 1.6124 subsequently. DFIRE was applied in the affinity prediction of protein complexes later.25 In this article, we tested our approach on the test set from DFIRE. Shen and Sali26 went a step further. They analytically derived a statistical potential termed DOPE for decoy discrimination of single protein structure. The DOPE corrects volume with a correction factor of rα(r). The effective exponent α(r) is a function of interparticle distance r, which results in a more flexible application.
It should be noted that these approaches above corrected volume with a uniform factor to all atom types. In other words, they used the same correction factor for distinct atoms. But in fact, each of the atom types is different on occupied volume. Therefore, a distinct volume correction should be used for each of the atom types.
This problem is naturally solved by the second type approach of volume correction, which acquires the volume correction factors directly from statistics to structures. This type of approach, unlike the first one, is independent of a certain function form to correct occupied volume. Moreover, in contrast to the first way, it is able to distinguish different atom types surrounded by distinct environments, by generating a unique volume correction for each atom type. Therefore, a more accurate correction can be acquired. Muegge and Martin27 corrected the volume based on structural statistics. In their approach, each atom type is treated with a different volume correction. Their approach performed well in the prediction of protein–ligand binding affinity. However, the implementation of their approach is very complicated in practice, which obstructed its popularity.
The approach presented in this article belongs to the second type of approach, but we circumvented the complicated step of volume correction process. The volume correction was achieved using a novel and very simple frequency correction. More reasonable potentials were obtained, and the prediction to protein–protein binding affinity on six test sets from five literatures also showed good performance of our approach.
Results and Discussions
Details of the pair potentials
Three pair potentials chosen as representative examples are shown in Figure 1. These potentials are backbone–backbone (B-B) potential 1-1, backbone-side chain (B-S) potential 7-4, and side chain–side chain (S-S) potential 8-6. The potentials calculated by Eq. (2), that is, our approach, are represented by the solid curve. For comparison, the potentials from Eq. (1), that is, the traditional approach, are represented by the dashed curve. The numbers 1-1, 7-4, and 8-6 are labels of atom pairs, whose atom types are defined in Table III.
Figure 1.
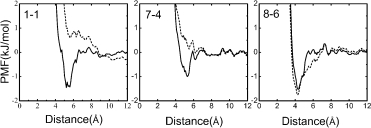
Examples of potential. 1-1 is backbone-backbone (B-B) potential, 7-4 is backbone-side chain (B-S) potential, and 8-6 is side chain-side chain (S-S) potential. The numbers in 1-1, 7-4, and 8-6 represent the atom types defined in Table II. The solid curves represent the potentials from improved approach, and the dashed curves represent the potentials from traditional approach.
Table III.
Atom Type Definition for Heavy Atoms of the Standard Amino Acids
| Atom type | Type definition |
|---|---|
| 1 | Cα (all amino acids, except Gly) |
| 2 | Gly-Cα |
| 3 | N (all amino acids, except Pro) |
| 4 | C (all amino acids) |
| 5 | O (all amino acids) |
| 6 | Val-Cγ1, Val-Cγ2, Leu-Cδ1, Leu-Cδ2, Ile-Cγ2, Ile-Cδ, Thr-Cγ |
| 7 | Leu-Cγ, Ile-Cγ1, Gln-Cγ, Lys-Cγ, Lys-Cδ, Glu-Cγ, Arg-Cγ |
| 8 | Cβ (all amino acids, except Pro, Ser, Thr, Cys) |
| 9 | Met-Sδ |
| 10 | Pro-N |
| 11 | Phe-Cγ, Tyr-Cγ |
| 12 | Phe-Cδ1, Phe-Cδ2, Phe-Cɛ1, Phe-Cɛ2, Phe-Cζ, Tyr-Cδ1, Tyr-Cδ2, Tyr-Cɛ1, Tyr-Cɛ2 |
| 13 | Trp-Cγ |
| 14 | Trp-Cɛ2 |
| 15 | Ser-Cβ |
| 16 | Ser-Oγ, Thr-Oγ |
| 17 | Thr-Cβ |
| 18 | Asn-Nδ2, Gln-Nɛ2 |
| 19 | Cys-Sγ |
| 20 | Lys-Nζ |
| 21 | Arg-Cζ |
| 22 | Arg-Nη1, Arg-Nη2 |
| 23 | His-Cγ |
| 24 | His-Cδ2 |
| 25 | His-Nɛ2 |
| 26 | His-Cɛ1 |
| 27 | Asp-Cγ, Glu-Cδ |
| 28 | Asp-Oδ1, Asp-Oδ2, Glu-Oɛ1, Glu-Oɛ2 |
| 29 | Cys-Cβ |
| 30 | Met-Cɛ |
| 31 | Tyr-Cζ |
| 32 | Pro-Cδ |
| 33 | Asn-Cγ, Gln-Cδ |
| 34 | Asn-Oδ1, Gln-Oɛ1 |
| 35 | Lys-Cɛ |
| 36 | Arg-Nɛ |
| 37 | Arg-Cδ |
| 38 | His-Nδ1 |
| 39 | Trp-Nɛ1 |
| 40 | Tyr-Oη |
| 41 | OXT (the extra oxygen at the carboxyl terminal) |
| 42 | Pro-Cβ |
| 43 | Pro-Cγ |
| 44 | Met-Cγ |
| 45 | Trp-Cɛ3, Trp-Cζ2, Trp-Cζ3, Trp-Cη2 |
| 46 | Trp-Cδ1 |
| 47 | Trp-Cδ2 |
For pair potentials 1-1 and 7-4 from traditional approach (dashed curves in Fig. 1), repulsion at all distances can be observed. Potential 8-6 from traditional approach is very similar to that from improved approach; the two curves share a normal shape without strong repulsion at all distances.
The solid curves in Figure 1 represent the potentials from our improved approach as previously mentioned, have classic picture of nonbonded atom pair interaction as Lennard-Jones potential. They exhibit strong repulsion at short distances, followed by one or several valleys with local minimums, representing the interaction preference at certain distances. When the distance between atom pairs is increased, the values of potentials trend zero, which means the atom pairs have very little interaction at such a long distance.
In short, strong repulsive interactions can be observed at all distances in B-B and B-S potentials from traditional approach (dashed curves in Fig. 1). Corresponding to our results, the potentials calculated from other traditional approaches in literatures also exhibit similar curves.21,22 However, in potentials from improved approach, this strong repulsion is weakened (solid curves in Fig. 1). These potentials from improved approach are more reasonable and show a classical picture of nonbonded atom pair interaction as Lennard-Jones potential. This indicates better accordance of our approach with acknowledged theories.
In traditional approach, the abnormal repulsions at all distances of B-B and B-S potentials can be attributed to the shortage of observed frequency of atom pairs. The main reason for the shortage is the less exposure of the backbone atoms than the side chain atoms in protein–protein interface. As the space around backbone atom cannot be filled with atoms of the other chain, observed frequency of B-B and B-S atom pairs remains low.
Binding affinity prediction of protein–protein complexes for six test sets
To evaluate the prediction ability of our approach to the affinity prediction of protein–protein complexes, we collected as much test data as possible from the literature of binding affinity prediction and discarded none of them. Because there have not been an authoritative benchmark of test sets for binding affinity prediction of protein–protein complexes, we collected test data from published studies, which predicted protein–protein binding affinity using various approaches not just PMF. Then, we compared prediction ability of their methods with ours according to linear correlation between predicted affinity and experimental data. It should be noted that we discard none of the test data in the literature, because the correlation coefficient (R) could be significantly increased artificially by an additional restriction of included test data.
Finally, affinity predictions on six test sets (Table I) were done. The potentials for predictions were trained from 127 PDB entries (Table II). A total of 47 atom types for all the heavy atoms of the 20 amino acids were defined (Table III). Finally, 86 protein-protein complexes (Table IV) were predicted. Test sets 1–5 come from five published articles. Test set 6 is the union set of sets 1–5, which means it contains all nonrepeated data of the first five sets. For all the six test sets, we evaluated prediction ability using linear correlation coefficient (R) and standard deviation (SD) of fitting predicted affinity to experimental data, and then, we compared R and SD of our prediction with the linear correlation results reported in the literatures. The criterion of the better prediction should be larger R and smaller SD in absolute value contemporaneously. All of the literatures reported R, whereas only two of them reported SD yet. The results are presented in Table I and Figure 2.
Table I.
Linear Correlation Between Experimental Binding Affinity and Predicted Affinity for Six Test Sets
|
R |
SD |
|||||
|---|---|---|---|---|---|---|
| Test set | Ref. a | No. of complexes | Oursb | Literaturec | Oursb | Literaturec |
| 1 | a | 15 | 0.91 | 0.96d | 1.98 | NAe |
| 2 | b | 8 | 0.89 | 0.74 | 1.19 | 1.5 |
| 3 | c | 9 | 0.83 | 0.70 | 1.40 | 2.0 |
| 4 | d | 21 | 0.85 | 0.75 | 2.31 | NAe |
| 5 | e | 82 | 0.73 | 0.73 | 2.23 | NAe |
| 6 | f | 86 | 0.76 | – | 2.24 | – |
The results from our approach.
The results from literature.
In the literature, for the polar and apolar components, the correlation coefficient is 0.63 and 0.77, respectively. When the two terms are added together and weighted by two free parameters α and β, the correlation extends to 0.96.
NA, no available standard deviation (SD) was reported in the literature.
Table II.
The 127 PDB Entries for Training Potentials
| 12gs | 1a09 | 1a14 | 1a1n | 1a2c | 1a2k | 1a2x | 1a2y | 1a3b | 1a3r |
| 1a46 | 1a4w | 1a5g | 1a5s | 1a61 | 1a9e | 1ab9 | 1abo | 1abr | 1abw |
| 1agd | 1ak4 | 1an1 | 1aqc | 1aqd | 1aqv | 1avw | 1axd | 1axi | 1bd2 |
| 1bhf | 1bj1 | 1bnd | 1brb | 1brc | 1bt6 | 1dkz | 1dzb | 1e4x | 1e96 |
| 1eay | 1eer | 1efn | 1efu | 1exf | 1fdl | 1flt | 1gbb | 1gc1 | 1gg2 |
| 1gl1 | 1got | 1gua | 1gux | 1gzs | 1h0d | 1h2s | 1he1 | 1hwg | 1ikf |
| 1ir3 | 1itb | 1jhg | 1jhl | 1kip | 1lck | 1ld9 | 1lfd | 1mct | 1mel |
| 1mlc | 1nmc | 1oak | 1oby | 1obz | 1oey | 1oga | 1ogt | 1ohz | 1okk |
| 1okv | 1ol5 | 1osp | 1osz | 1qew | 1qja | 1qls | 1qo3 | 1rst | 1rsu |
| 1scn | 1sfi | 1shd | 1sib | 1slg | 1slu | 1sm3 | 1smf | 1spp | 1taf |
| 1tbg | 1tec | 1tx4 | 1tze | 1upt | 1uzx | 1vad | 1wej | 1www | 1x11 |
| 1ycs | 1zfp | 2cbl | 2fib | 2h1p | 2hrp | 2jel | 2prg | 2seb | 2tgp |
| 2trc | 2vaa | 3cyh | 3nse | 3sgb | 3sic | 5csm |
Table IV.
The PDB Interfaces and Experimental Affinities in Six Test Sets
| PDB ID | Interface | Affinity (kcal/mol) | PDB ID | Interface | Affinity (kcal/mol) |
|---|---|---|---|---|---|
| 2ptc | E/I | −18.1 | 1tpa | E/I | −17.8 |
| 2kai | AB/I | −12.4 | 4cpa | blank /I | −10.0 |
| 3cpa | blank/S | −5.3 | 3sgb | E/I | −12.7 |
| 2sec | E/I | −13.1 | 1cse | E/I | −13.1 |
| 1cho | E/I | −14.6 | 2tpi | Z/I | −18.1 |
| 2tpi | Z/S | −5.8 | 2tgp | Z/I | −18.2 |
| 2sni | E/I | −15.8 | 4tpi | Z/I | −17.7 |
| 1tec | E/I | −14.0 | 4sgb | E/I | −11.7 |
| 2sic | E/I | −12.7 | 2er6 | E/I | −9.8 |
| 1acb | E/I | −16.1 | 1tbq | JK/S | −17.3 |
| 1atn | A/D | −11.8 | 3tpi | Z/S | −7.8 |
| 4htc | HL/I | −15.4 | 1bth | HL/P | −16.5 |
| 1dfj | E/I | −18.0 | 1avw | A/B | −12.3 |
| 1stf | E/I | −13.5 | 3hfl | LH/Y | −14.5 |
| 1vfb | AB/C | −11.4 | 1nsn | HL/S | −11.8 |
| 1igc | HL/A | −12.7 | 1ahw | AB/C | −11.5 |
| 1wej | HL/F | −9.5 | 1mel | M/B | −10.5 |
| 1nmb | HL/N | −10.0 | 1fdl | HL/Y | −11.4 |
| 2jel | HL/P | −11.5 | 1jhl | HL/A | −11.8 |
| 3hfm | HL/Y | −13.3 | 1mlc | AB/E | −9.7 |
| 1bql | HL/Y | −14.5 | 4ins | AB/CD | −7.4 |
| 1hbs | ABCD/EFGH | −4.8 | 1brs | B/E | −17.3 |
| 1tce | A/B | −5.8 | 1lck | A/B | −7.0 |
| 1lcj | A/B | −7.8 | 2pld | A/B | −9.0 |
| 1sps | A/D | −9.1 | 1b46 | A/B | −7.2 |
| 1b3l | A/B | −8.0 | 1b9j | A/B | −8.1 |
| 1b58 | A/B | −9.0 | 1jeu | A/B | −9.3 |
| 1jev | A/B | −9.4 | 1b5i | A/B | −9.6 |
| 1b32 | A/B | −9.7 | 1b40 | A/B | −9.9 |
| 1qkb | A/B | −10.0 | 1b4z | A/B | −7.1 |
| 2olb | A/B | −7.6 | 1qka | A/B | −8.1 |
| 1b3g | A/B | −9.2 | 1b3f | A/B | −9.4 |
| 1b05 | A/B | −9.7 | 1b52 | A/B | −9.7 |
| 1jet | A/B | −9.8 | 1b51 | A/B | −10.0 |
| 1b5j | A/B | −10.1 | 1ola | A/B | −9.5 |
| 1dkz | A/B | −9.1 | 1dkg | AB/D | −10.3 |
| 2pcc | A/B | −10.0 | 1gua | A/B | −10.1 |
| 1ycs | A/B | −10.3 | 1efn | A/B | −16.6 |
| 1fss | A/B | −14.9 | 1mda | HL/A | −7.3 |
| 1ak4 | A/D | −6.5 | 1ebp | A/C | −11.7 |
| 1hwg | C/A | −13.0 | 3hhr | B/A | −13.6 |
| 3ssi | Symmetry | −16.0 | 1avz | AB/C | −6.4 |
| 1a0o | A/B | −8.1 | 1gla | G/F | −6.7 |
Test set 1 (Table I) contains 15 protein–protein complexes from Ref. 28. In this literature, the affinity is described as the sum of solvation terms based on atomic solvation parameter (ASP) and an energy term to account for the loss of translational and rotational entropy. For the polar and apolar solvation components, the correlation coefficients (R) are 0.63 and 0.77, respectively. When they revised their function by adding the two terms together weighted by two newly introduced free parameters, α and β, the correlation extends to 0.96. And, no SD values were reported in the literature. In our prediction, R = 0.91 and SD = 1.98 kcal/mol (Fig. 2).
Test set 2 (Table I) contains eight protein–protein complexes from Ref. 29. In this literature, they used a method constructed from molecular surface preferences. For set 2, R = 0.74 and SD = 1.5 kcal/mol were reported. In our prediction, R = 0.89 and SD = 1.19 kcal/mol.
Test set 3 (Table I) contains nine protein–protein complexes from Ref. 30. In this literature, they used a method based on MJ potential. For set 3, R = 0.70 and SD = 2.0 kcal/mol were reported. In our prediction, R = 0.83 and SD = 1.40 kcal/mol (Fig. 2).
Test set 4 (Table I) contains 21 protein–protein complexes from Ref. 31, in which the method is based on ASP. R = 0.75 with no SD value was reported in the literature. In our prediction, R = 0.85 and SD = 2.31 kcal/mol (Fig. 2).
Test set 5 (Table I) contains 82 complexes from Ref. 25 predicted by DFIRE, which is the only test set predicted by PMF method. Our prediction performed equally well as the literature in term of correlation coefficients (R = 0.73).25 The SD of our prediction is 2.23 kcal/mol. The literature did not report the SD.
Test set 6 contains all data in sets 1–5, adding up to 86 complexes. The R and the SD of our prediction are 0.76 and 2.24 kcal/mol, respectively. It is particularly worth noting that although set 6 contains more test data (86) than set 5 (82), but in our predictions R = 0.76 of set 6 is better than R = 0.73 of set 5. The reason is that these test sets were extracted from published articles directly and have not been refined. For example, test set 5 contains 20 OppA-peptide complexes, whose peptides have highly similar sequences, resulting in a biased prediction toward this type of complexes. Test set 6 contains more data, which partly balance out this effect.
A good test set served as a benchmark should be nonredundant or at least with restricted numbers of similar complexes. However, until now, there is no authoritative test set served as benchmark for binding affinity prediction of protein–protein complexes. We plan to build such a benchmark in our future studies.
Overall, test sets 1, 2, 3, and 4 contain less test data, so predictions can easily obtain good linear correlation (Table I) as in both our predictions and literature. Moreover, for all test sets except set 1, correlations of our prediction are better than report in the literature. Nevertheless, the meaning of the correlation for small test set should not be overestimated, as it is unstable. If one or a few test data in these small sets are changed, the correlation (R and SD) might be significantly changed. Therefore, performance on the four sets may not say much about prediction ability. For test set 5, a larger set, our prediction obtained as good correlation as literature (0.73). For set 6, which contains all data in sets 1–5, our prediction obtained even better R (0.76) than for set 5 (0.73).
The volume correction is very important for the prediction of binding affinity
We found that the introduction of volume correction makes the pair potentials more reasonable and results in great improvement on prediction ability for the binding affinity of protein–protein complexes.
Above in Figure 1, we have shown that B-B potential 1-1, B-S potential 7-4 from traditional approach without volume correction have strong repulsive interactions at all distances (dashed curves in Fig. 1). But, in potentials from our approach considering volume correction, this strong repulsion is weakened and the attractive valley appears (solid curves in Fig. 1). A classic picture of nonbonded atom pair interaction as Lennard-Jones potential was shown. It represents that volume correction can obtain more reasonable potential. In comparison, S-S potential 8-6 already has reasonable shape and has not large change after volume correction.
Correct understanding to the interactions of B-B and B-S atom pairs is very important for the binding affinity prediction, because B-B and B-S atom pair interactions make a large percentage contribution in protein–protein interactions. We analyzed the percentage contribution of B-B, B-S, and S-S pair based on 127 protein–protein complexes in the training set. The components of B-B, B-S, and S-S make up 23.5, 50.1, and 26.4% of the total interaction pairs, respectively. Therefore, if inaccurate estimates of B-B and B-S pair potentials are used to predict binding affinity, the results will be affected significantly. Figure 3 shows the prediction for 86 protein–protein complexes in test set 6. The traditional approach without volume correction obtained linear R = 0.07 and SD = 3.45 kcal/mol. Meanwhile, our approach considering volume correction obtained R = 0.76, SD = 2.24 kcal/mol. Above showed the introduction of volume correction is very important for the improvement of prediction ability.
Figure 3.
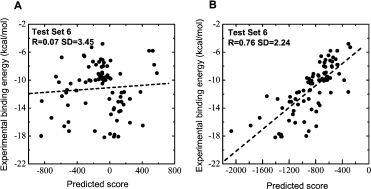
The binding affinity prediction to test set 6. The linear correlation coefficient (R) and standard deviation (SD) were calculated. (A) Prediction from traditional approach. (B) Prediction from improved approach considering volume correction.
A web server for the binding affinity prediction of protein–protein complexes
We developed a web server PPEPred (http://www.bioinfo.tsinghua.edu.cn/~suyu/ppepred/) for the binding affinity prediction of protein–protein (protein–peptide) complexes from three-dimensional structure data, based on the approach in this article. The parameters a and b for the prediction in Eq. (6) are 0.007850 and −4.491 kcal/mol from the linear fitting to test set 6. The inputs of PPEPred server are the structure name, two chains name, and user needs to upload the structure data of Protein Data Bank (PDB) file or user file in PDB format. The output is the affinity of this complex. The web server is free and open to everyone.
Materials and Methods
Potential of mean force
Traditional approaches of PMF were widely applied in the studies of protein structure and protein–protein interaction. Here, the traditional potential between two atoms of type i and type j with distance r can be described by the function:
| (1) |
where kB is the Boltzmann constant and T is the absolute temperature. qij(r) is the normalized frequency between atom pairs of type i and type j, and qxx(r) represents average normalized frequency covered all atom pairs, which are defined in Eq. (4). Other traditional approaches consider qij(r) and qxx(r) as density in a whole sphere volume (4πr2dr). Both of them can obtain similar results in calculation.
However, there are some problems with traditional approaches mentioned earlier. qij(r) and qxx(r) have distinct distribution, corresponding the average density in calculating the RDF in the theory of simple liquids. But, qij(r)/qxx(r) ignored the distinct distribution between them. To get more reasonable potential, this deviation ought to be corrected in an improved approach.
Up till now, all the improved approaches corrected the deviation based on the average density on volume, named as volume correction. But, in comparison with the simple liquid systems, proteins are extremely complicated soft matter. In protein systems, these approaches were deduced in an extremely complicated manner, and the steps of implement also contain many details in practice, such as the approximation in acquiring the distributions of atom volume and adopting diverse width of bins at different distances in statistics.25–27
On the contrary, in our approach, we directly correct the volume effect based on frequency, which can achieve the same goal of correction and largely simplify the process of implement in practice. The improved approach of PMF in our work is given by:
| (2) |
where kB is the Boltzmann constant and T is the absolute temperature. qij(r) and qxx(r) are the normalized frequency of atom pairs, which are defined in Eq. (4). fcor is the correction factor, derived from smoothing qxx(r)/qij(r) ranging from 0 to 12 Å, by a moving window of 3.5 Å width with bin of width 0.1 Å.
To obtain the stable potentials in statistics, we considered the potentials only when the total occurrence number of atom pairs was larger than 1000. If the total occurrence number of atom pairs of type i and type j was smaller than 1000, we set Aij(r) = 0. That is, we ignored the contribution of a particular pair type if it had not sufficient data in statistics.
Later, we show how to obtain the normalized frequency qij(r) and qxx(r) statistically.
First, according to statistics, we obtain Nij(r), the occurrence numbers of atom pairs ij at a certain distance r, in a training database of protein–protein complexes, ranging from 0 to 12 Å at 0.1 Å intervals (but the occurrence numbers in which atom pairs distance is below 2.5 Å were set zero as unrealistically short for heavy atom pairs):
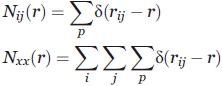 |
(3) |
where δ(x) is δ function, which is equal to 1 if its argument is zero, and zero otherwise. The subscript p designates that the summation cover all protein–protein complexes in the training database. The subscript i and j designate that the summation cover all atom pairs.
Then, we normalize the occurrence numbers to get the relative frequency:
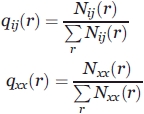 |
(4) |
where the summation is on atom pairs ranging from 0 to 12 Å.
Scoring and fitting experimental binding affinity
The scoring function to a protein–protein complex is defined as the summation over all atom pair interactions of the protein–protein complex:
| (5) |
where rcutoff is the cutoff distance between atoms i and j. Here, 12 Å is used.
To relate the score above to an absolute binding affinity, we fit it to binding affinity in a linear manner:
| (6) |
The training set
The Brookhaven Protein Data Bank32 was used to get the training data set in deriving the potential. We included only X-ray structures of protein–protein and protein–peptide complexes with resolutions better than 2.5 Å. Based on these criteria, 438 entries were yielded. To eliminate the structure similarity, we further filtered these entries based on molecular information in PDB entry and the cited literature in REMARK, with the aid of the molecule graph software (RasMol). For the same structure, we only reserved the entry of the best resolution. Finally, the training set contained 178 interfaces (in Supporting Information) from 127 PDB entries (Table II).
Atom type definition for heavy atoms of the standard amino acids
We defined 47 atom types for all the heavy atoms of the 20 amino acids (Table III). The definition of atom type is based on its physicochemical property, connectivity, and environment, derived from 40 atom types in Ref. 33. To obtain more details of interactions, it would be better that we define as many atom types as possible. On the other hand, to obtain statistically sufficient data, we could not define too many atom types. Therefore, the number of atom types was a compromise between the two considerations.
Conclusions
We present a novel PMF considering volume correction. In the prediction of protein–protein binding affinity, six test sets were tested and good performance was shown. This approach circumvents the complicated step of volume correction process and is extremely easy to implement in practice.
In this article, our approach is used to predict protein–protein binding affinity. But in respect of methodology, the statistics and calculation of this approach do not specialize in protein–protein complexes. Therefore, it can be applied to other fields, in which traditional approaches of PMF have been widely applied, such as protein–ligand docking and protein threading in structure prediction. It is expected to have a good performance.
References
- 1.Ajay, Murcko MA. Computational methods to predict binding free energy in ligand-receptor complexes. J Med Chem. 1995;38:4953–4967. doi: 10.1021/jm00026a001. [DOI] [PubMed] [Google Scholar]
- 2.Gohlke H, Klebe G. Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew Chem Int Ed. 2002;41:2644–2676. doi: 10.1002/1521-3773(20020802)41:15<2644::AID-ANIE2644>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 3.Gilson MK, Zhou HX. Calculation of protein-ligand binding affinities. Annu Rev Biophys Biomol Struct. 2007;36:21–42. doi: 10.1146/annurev.biophys.36.040306.132550. [DOI] [PubMed] [Google Scholar]
- 4.Sippl MJ. Boltzmanns principle, knowledge-based mean fields and protein folding. An approach to the computational determination of protein structures. J Comput-Aided Mol Des. 1993;7:473–501. doi: 10.1007/BF02337562. [DOI] [PubMed] [Google Scholar]
- 5.Sippl MJ. Knowledge-based potentials for proteins. Curr Opin Struct Biol. 1995;5:229–235. doi: 10.1016/0959-440x(95)80081-6. [DOI] [PubMed] [Google Scholar]
- 6.Jernigan RL, Bahar I. Structure-derived potentials and protein simulations. Curr Opin Struct Biol. 1996;6:195–209. doi: 10.1016/s0959-440x(96)80075-3. [DOI] [PubMed] [Google Scholar]
- 7.Jones DT, Thornton JM. Potential energy functions for threading. Curr Opin Struct Biol. 1996;6:210–216. doi: 10.1016/s0959-440x(96)80076-5. [DOI] [PubMed] [Google Scholar]
- 8.Vajda S, Sippl MJ, Novotny J. Empirical potentials and functions for protein folding and binding. Curr Opin Struct Biol. 1997;7:222–228. doi: 10.1016/s0959-440x(97)80029-2. [DOI] [PubMed] [Google Scholar]
- 9.Moult J. Comparison of database potentials and molecular mechanics force fields. Curr Opin Struct Biol. 1997;7:194–199. doi: 10.1016/s0959-440x(97)80025-5. [DOI] [PubMed] [Google Scholar]
- 10.Lazaridis T, Karplus M. Effective energy functions for protein structure prediction. Curr Opin Struct Biol. 2000;10:139–145. doi: 10.1016/s0959-440x(00)00063-4. [DOI] [PubMed] [Google Scholar]
- 11.Gohlke H, Klebe G. Statistical potentials and scoring functions applied to protein-ligand binding. Curr Opin Struct Biol. 2001;11:231–235. doi: 10.1016/s0959-440x(00)00195-0. [DOI] [PubMed] [Google Scholar]
- 12.Buchete N, Straub JE, Thirumalai D. Development of novel statistical potentials for protein fold recognition. Curr Opin Struct Biol. 2004;14:225–232. doi: 10.1016/j.sbi.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 13.Skolnick J. In quest of an empirical potential for protein structure prediction. Curr Opin Struct Biol. 2006;16:166–171. doi: 10.1016/j.sbi.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 14.McQuarrie DA. Statistical mechanics. New York: Harper and Row; 1976. [Google Scholar]
- 15.Chandler D. Equilibrium theory of polyatomic fluids. New York: North-Holland; 1982. pp. 275–340. [Google Scholar]
- 16.Ben-Naim A. Statistical thermodynamics for chemists and biochemists. New York: Plenum Press; 1992. [Google Scholar]
- 17.Mitchell JBO, Laskowski RA, Alex A, Forster MJ, Thornton JM. Bleep: potential of mean force describing protein-ligand interactions. II. Calculation of binding energies and comparison with experimental data. J Comput Chem. 1999;20:1177–1185. [Google Scholar]
- 18.Sippl MJ. Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J Mol Biol. 1990;213:859–883. doi: 10.1016/s0022-2836(05)80269-4. [DOI] [PubMed] [Google Scholar]
- 19.Jiang L, Gao Y, Mao F, Liu Z, Lai L. Potential of mean force for protein-protein interaction studies. Proteins. 2002;46:190–196. doi: 10.1002/prot.10031. [DOI] [PubMed] [Google Scholar]
- 20.Hoppe C, Schomburg D. Prediction of protein thermostability with a direction- and distance-dependent knowledge-based potential. Protein Sci. 2005;14:2682–2692. doi: 10.1110/ps.04940705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bahar I, Jernigan RL. Inter-residue potentials in globular proteins and the dominance of highly specific hydrophilic interactions at close separation. J Mol Biol. 1997;266:195–214. doi: 10.1006/jmbi.1996.0758. [DOI] [PubMed] [Google Scholar]
- 22.Mitchell JBO, Laskowski RA, Alex A, Thornton JM. Bleep: potential of mean force describing protein-ligand interactions. I. Generating potential. J Comput Chem. 1999;20:1165–1176. [Google Scholar]
- 23.Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002;11:2714–2726. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2003;12:2121. doi: 10.1110/ps.0217002. Erratum. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang C, Liu S, Zhu Q, Zhou Y. A knowledge-based energy function for protein-ligand, protein-protein, and protein-DNA complexes. J Med Chem. 2005;48:2325–2335. doi: 10.1021/jm049314d. [DOI] [PubMed] [Google Scholar]
- 26.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muegge I, Martin YC. A general and fast scoring function for protein-ligand interactions: a simplified potential approach. J Med Chem. 1999;42:791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 28.Horton N, Lewis M. Calculation of the free energy of association for protein complexes. Protein Sci. 1992;1:169–181. doi: 10.1002/pro.5560010117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wallqvist A, Jernigan RL, Covell DG. A preference-based free energy parameterization of enzyme-inhibitor binding: applications to hiv-1 protease inhibitor design. Protein Sci. 1995;4:1881–1903. doi: 10.1002/pro.5560040923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang C, Vasmatzis G, Cornette JL, DeLisi C. Determination of atomic desolvation energies from the structures of crystallized proteins. J Mol Biol. 1997;267:707–726. doi: 10.1006/jmbi.1996.0859. [DOI] [PubMed] [Google Scholar]
- 31.Zhou H, Zhou Y. Stability scale and atomic solvation parameters extracted from 1023 mutation experiments. Proteins. 2002;49:483–492. doi: 10.1002/prot.10241. [DOI] [PubMed] [Google Scholar]
- 32.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melo F, Feytmans E. Novel knowledge-based mean force potential at atomic level. J Mol Biol. 1997;267:207–222. doi: 10.1006/jmbi.1996.0868. [DOI] [PubMed] [Google Scholar]