

Abstract
The basal (constitutive) activity of G protein-coupled receptors allows for the measurement of inverse agonist activity. Some competitive antagonists turn into inverse agonists under conditions where receptors are constitutively active. In contrast, neutral antagonists have no inverse agonist activity, and they block both agonist and inverse agonist activity. The mu opioid receptor (MOR) demonstrates detectable constitutive activity only after a state of dependence is produced by chronic treatment with a MOR agonist. We therefore sought to identify novel MOR inverse agonists, and novel neutral MOR antagonists in both untreated and agonist-treated MOR cells. CHO cells expressing the cloned human mu receptor (hMOR-CHO cells) were incubated for 20 hr with medium (control) or 10 μM (2S,4aR,6aR,7R,9S,10aS,10bR)-9-(benzoyloxy)-2-(3-furanyl)dodecahydro-6a,10b-dimethyl-4,10-dioxo-2H-naphtho-[2,1-c]pyran-7-carboxylic acid methyl ester (herkinorin, HERK). HERK-treatment generates a high degree of basal signaling and enhances the ability to detect inverse agonists. [35S]-GTP-γ-S assays were conducted using established methods. We screened 21 MOR “antagonists” using membranes prepared from HERK-treated hMOR-CHO cells. All antagonists, including CTAP and 6β-naltrexol, were inverse agonists. However, LTC-2 7 4 ( (-)-3-cyclopropylmethyl-2,3,4,4aα,5,6,7,7aα-octahydro-1H-benzofuro[3,2-e]isoquinolin-9-ol)) showed the lowest efficacy as an inverse agonist, and, at concentrations less than 5 nM, had minimal effects on basal [35S]-GTP-γ-S binding. Other efforts in this study identified KC-2-009 ((+)-3-((1R,5S)-2-((Z)-3-Phenylallyl)-2-azabicyclo[3.3.1]nonan-5-yl)phenol hydrochloride) as an inverse agonist at untreated MOR cells. In HERK-treated cells, KC-2-009 had the highest efficacy as an inverse agonist. In summary, we identified a novel and selective MOR inverse agonist (KC-2-009), and a novel MOR antagonist (LTC-274) that shows the least inverse agonist activity among 21 MOR antagonists. LTC-274 is a promising lead compound for developing a true MOR neutral antagonist.
Introduction
G protein-coupled receptors (GPCRs) can demonstrate basal or spontaneous activity in the absence of an agonist. This basal or constitutive activity allows the measurement of inverse agonist activity. As reviewed by Kenakin (Kenakin, 2004) competitive antagonists will often act as inverse agonists under conditions where receptors are constitutively active. A neutral antagonist is a compound that can block both agonist and inverse agonist activity. The opioid receptors belong to the GPCR family, and consist of three genes coding for the μ (MOR), δ (DOR) and κ (KOR) opioid receptors (Kieffer and Evans, 2008). Among the opioid receptors, only the δ receptor (DOR) has readily detectable constitutive activity (Costa and Herz, 1989) under opioid naïve/control conditions. Agonist-treatment can generate a high degree of basal signaling and enhances the ability to detect inverse agonists and true neutral antagonists. Thus, until the discovery of compounds like (+)-3-((1R,5S)-2-((Z)-3-Phenylallyl)-2-azabicyclo[3.3.1]nonan-5-yl)phenol hydrochloride (KC-2-009), constitutively active μ opioid receptors (MOR) are typically observed only after a state of dependence is created by chronic treatment of cells or animals with a MOR agonist (Sadee et al., 2005; Wang et al., 2001; Wang et al., 2007; Xu et al., 2007; Xu et al., 2004). Among a number of classical μ opioid antagonists, only 6β-naltrexol and 6β-naltrexamide were reported to be neutral antagonists in membranes prepared from μ-agonist pretreated cells (Wang et al., 2007). Moreover, only a minimal degree of inverse agonism was observed in non-dependent HEK cells expressing the MOR (Wang et al., 2001). Since neutral antagonists are potential therapeutic agents (Sadee et al., 2005), and in light of the limited availability of compounds that demonstrate inverse agonist activity in control MOR cells, the major purpose of this study was to identify such compounds.
The numerous compounds submitted to our laboratory by our medicinal chemistry collaborators for evaluation at opioid receptors were used for this study. As described in previous papers (Kurimura et al., 2008), compounds are evaluated in opioid receptor binding assays and functional [35S]GTP-γ-S binding assays using CHO cells that express the cloned human μ opioid (MOR), δ opioid (DOR) or κ opioid (KOR) receptors. From this work we identified several compounds that were inverse MOR agonists using “standard” non-dependent hMOR-CHO cells (data not shown). To facilitate this work we developed a protocol that generates cells with a high degree of MOR constitutive activity, thereby allowing the robust measurement of MOR inverse agonist activity. hMOR-CHO cells were pretreated with the MOR agonist (2S,4aR,6aR,7R,9S,10aS,10bR)-9-(benzoyloxy)-2-(3-furanyl)dodeca-hydro-6a,10b-dimethyl-4,10-dioxo-2H-naphtho-[2,1-c]pyran-7-carboxylic acid methyl ester (herkinorin, HERK) for 20 hr and [35S]-GTP-γ—S binding performed. Previous studies from our laboratory (Xu et al., 2007) showed that treating hMOR-CHO cells with HERK produced more constitutively active MORs than chronic treatment with the MOR agonist [D-Ala2-MePhe4,Gly-ol5]enkephalin (DAMGO). Specifically, we showed that treating hMOR-CHO cells with HERK, but not DAMGO, increased basal “single-point” [35S]-GTP-γ-S binding, increased the BMAX of the agonist-responsive high affinity [35S]-GTP-γ-S binding site and suppressed forskolin-stimulated cAMP accumulation (see Fig. 3, Table 2 and Fig. 4 in (Xu et al., 2007)). These efforts identified KC-2-009 as an inverse agonist at both untreated and HERK-treated MOR cells, and a MOR antagonist (-)-3-cyclopropylmethyl-2,3,4,4aα,5,6,7,7aα-octahydro-1H-benzofuro[3,2-e]isoquinolin-9-ol) (LTC-274) that shows the least inverse agonist activity among 21 classical MOR antagonists.
Figure 3.

Ke value of LTC-274 for agonists and inverse agonists. The Ke values of LTC-274 for agonists and inverse agonists (Table 2) were pooled for statistical analysis. *p<0.01 when compared to agonists (unpaired Student’s t-test).
Table 2.
Profile of Inverse Agonists in HERK Treated Membranes
| Drug | EC50 (nM±SD) |
EMAX (Percent±SD) |
|---|---|---|
| LTC-274 | 1.6±0.4 | -37±2 |
| 6-β-Naltrexamide | 2.6±0.5 | -67±2 |
| BNTX | 3.1±0.43 | -82±2 |
| Diprenorphine | 0.26±0.06 | -84±3 |
| NorBNI | 32±6 | -86±3 |
| CTAP | 38±5 | -89±2 |
| Naltrexone | 0.96±0.13 | -90±2 |
| NTB | 6.6±1.3 | -90±3 |
| Naloxone | 1.8±0.1 | -91±1 |
| NTI | 17±4 | -94±4 |
| 6-β-naltrexol | 1.2±0.12 | -98±1 |
| KC-2-009 | 6.7±0.4 | -100±1 |
Inverse agonist dose-response curves (10 points per curve) were generated as described in Methods. The EMAX is normalized with 100% being the inhibition of [35S]-GTP-γ-S binding produced by 5 μM KC-2-009. The data of 3 experiments were meaned and the best-fit estimates of the EC50 and EMAX determined using nonlinear least squares curve fitting programs. Each value is ±SD.
Figure 4.

Effect of pretreatment drug on the efficacy of KC-2-009. Cells were treated for 20 hr in the absence and presence of 10 μM HERK, 10 μM DAMGO, 10 μM gedunin, and 1 μM fentanyl. KC-2-009 dose-response curves (10 data points each) were then generated as described in methods. The inset plots the EMAX values. *p<0.05 (ANOVA, Dunnett’s Multiple Comparison Test) when compared to control EMAX. Each value is the mean±SD (n=3).
Methods
Cell culture and membrane preparation
As described (Rothman et al., 2007), the recombinant hMOR-CHO cells were produced by stable transfection with human MOR cDNA, and provided by Dr. Larry Toll (SRI International, CA). The cells were grown on plastic flasks in DMEM/F-12 (50%/50%) medium containing 10% FetalClone II (HyClone) and Geneticin (G-418: 0.20-.25 mg/ml) (Invitrogen) under 95% air/5% CO2 at 37° C. Cell monolayers were harvested and cell pellets stored at -80° C.
For drug pretreatment cells were incubated in fresh medium with 10 μM HERK for 20 h. Cells were washed three times with phosphate-buffered saline (PBS, pH 7.4), harvested, and processed for use in various assays. This treatment produces a state of opioid tolerance (Xu et al., 2007).
[35S]GTP-γ-S binding assays
The [35S]-GTP-γ-S assays were conducted as described elsewhere (Rothman et al., 2007). In this description, buffer “A” is 50 mM Tris-HCl, pH 7.4, containing 100 mM NaCl, 10 mM MgCl2, 1 mM EDTA and buffer “B” is buffer A plus 1.67 mM DTT and 0.15% BSA. On the day of the assay, cells were thawed on ice for 15 min and homogenized using a polytron in 50 mM Tris-HCl, pH 7.4, containing 4 μg/mL leupeptin, 2 μg/mL chymostatin, 10 μg/mL bestatin and 100 μg/mL bacitracin. The homogenate was centrifuged at 30,000 × g for 10 min at 4 °C, and the supernatant discarded. The membrane pellets were resuspended in buffer B and used for [35S]GTP-γ-S binding assays. [35S]GTP-γ-S binding was determined as described previously. Briefly, test tubes received the following additions: 50 μL buffer A, 50 μL GDP in buffer A (final concentration = 40 μM), 50 μL drug in buffer A/0.1% BSA, 50 μL [35S]GTP-γ-S in buffer A (final concentration = 50 pM), and 300 μL of cell membranes (50 μg of protein) in buffer B. The final concentrations of reagents in the [35S]GTP-γ-S binding assays were: 50 mM Tris-HCl, pH 7.4, containing 100 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 1 mM DTT, 40 μM GDP and 0.1% BSA. Incubations proceeded for 3 hr at 25 °C. Nonspecific binding was determined using non-radioactive GTP-γ-S (40 μM). Bound and free [35S]-GTP-γ-S were separated by vacuum filtration through GF/B filters. The filters were punched into 24-well plates to which was added 0.6 mL LSC-cocktail (Cytoscint). Samples were counted, after an overnight extraction, in a Trilux liquid scintillation counter at 60% efficiency.
In Vivo Antagonism Experiments
Male ICR mice (Harlan) weighing between 25-35 grams were used for all in vivo studies. Mice were weighed and baselined in the 55°C tail-flick assay (Wells et al., 2001). Subcutaneous (sc) injections of vehicle (saline) or LTC-274 (0.01-1.0 mg/kg) were administered 10 minutes prior to an intracerebroventricular (icv) injection of an approximate A90 dose of morphine (10 nmol). Mice were retested for tail-flick latencies at 10 and 20 minutes post morphine injection, a time corresponding to peak morphine effect. Mean percent antinociception was calculated for each group using the following formula: % Antinociception = (test latency — baseline latency)/(10-baseline latency) × 100. The antagonist dose-response curves for LTC-274 were calculated using linear regression software (FlashCalc, Dr. Michael Ossipov, University of Arizona).
Data Analysis and Statistics
These methods are described elsewhere (Rothman et al., 2007; Xu et al., 2001; Xu et al., 2007). For [35S]GTP-γ-S binding experiments, the percent stimulation of [35S]GTP-γ-S binding was calculated according to the following formula: (S — B)/B × 100, where B is the basal level of [35S]GTP-γ-S binding and S is the stimulated level of [35S]GTP-γ-S binding. Agonist, or inverse agonist, dose-response curves (ten points/curve) are generated, and the data of several experiments, typically 3, are pooled. The EC50 and Emax values are determined using either the program MLAB-PC (Civilized Software, Bethesda, MD), KaleidaGraph (Version 3.6.4, Synergy Software, Reading, PA) or Prism 4.0 (GraphPad Software, Inc, San Diego, CA). In most cases, the stimulation produced by an agonist is expressed as a percent of the stimulation produced by 10 μM DAMGO. Similarly, the inhibition produced by an inverse agonist is reported as a percent of the inhibition produced by 5 μM KC-2-009 ((+)-3-((1R,5S)-2-((Z)-3-Phenylallyl)-2-azabicyclo[3.3.1]nonan-5-yl)phenol hydrochloride). For determination of antagonist Ke values we used the “shift” experimental design. Agonist or inverse agonist dose-response curves (ten points/curve) were generated, in the absence and presence of an antagonist. The data of several experiments, typically 3, were pooled, and the Ke values were calculated according to the equation: [antagonist]/(EC50-2/EC50-1 — 1), where EC50-2 is the EC50 value in the presence of the antagonist and EC50-1 is the EC50 value in the absence of the antagonist.
Sources
[35S]GTP-γ-S (SA = 1,250 Ci/mmol) was obtained from DuPont NEN (Boston, MA). Various opioid peptides were provided by Multiple Peptide System via the Research Technology Branch, NIDA. HERK was synthesized as described previously (Harding et al., 2005). Gedunin ((1S,3aS,4aR,4bS,5R,6aR,10aR,10bR,12aS)-5-(acetyloxy)-1-(3-furanyl)-1,5,6,6a,7,10a,10b,11,12,12a-decahydro-4b,7,7,10a,12a-pentamethyl-oxireno[c]phenanthro[1,2-d]pyran-3,8(3aH,4bH)-dione) was isolated as described previously (Brandt et al., 2008). The synthesis of LTC-274, the (-)-isomer of a racemate reported in 1981 (Ciganek, 1981) by Dr. Hsin’s group and KC-2-009 by Dr. Rice’s group, will be described in other papers. GTP-γ-S and GDP were obtained from Sigma Aldrich Chemical Co. (St. Louis, MO). Various opioid compounds were obtained from the NIDA Drug Supply program or commercial sources. The sources of other agents are published (Xu et al., 2004).
Results
In preliminary [35S]GTP-γ-S binding experiments, we screened 21 compounds with known MOR antagonist activity in control cell membranes and further identified their inverse agonist activity using HERK-treated membranes. These single point assays (10 μM) identified LTC-274 as having the least apparent inverse agonist efficacy. Other initial experiments identified KC-2-009 as a potent and efficacious inverse MOR agonist in control cells. These two compounds are part of a structure-activity study involving a large number of compounds (manuscripts in preparation). Some results for LTC-274 and KC-2-009 are reported here. Table 1 reports the structure of LTC-274 and KC-2-009 and their KI values for the MOR, DOR and KOR. KC-2-009 binds with modest affinity and selectively to the MOR (KI = 26 nM). LTC-274 displayed high affinity for the MOR (KI = 0.08 nM), but also displayed sub-nM affinity for the KOR. Other experiments determined that the Ke value of LTC-274 against the MOR agonist DAMGO was 0.22 nM and that LTC-274 is a partial KOR agonist (EC50 = 2.3±0.7 nM, EMAX = 23±1%) (data not shown). Fig. 1 reports the concentration-response curves with these compounds. As described in Fig. 1A, DAMGO displayed full MOR agonist effects in control cell membranes (EC50 = 28±6 nM, EMAX = 283±10 % stimulation). LTC-274 showed minimal agonist activity (EC50 = 1.5±0.9 nM, EMAX = 34±2 % stimulation). When normalized to the stimulation produced by 10 μM DAMGO in the same curve, the EMAX value of LTC-274 was 9.4±0.01 % control, indicating insignificant partial agonist activity. In contrast, KC-2-009 was a potent inverse agonist (EC50 = 12±4 nM, EMAX = -24±1.3 %). In HERK-treated membranes (Fig. 1B), DAMGO remained a full agonist, but, as expected, the EC50 value (94±17 nM) was shifted to the right, and the absolute EMAX value of DAMGO (152±6%) was reduced, as compared to the results obtained in control membranes. HERK-treatment substantially increased the inverse agonist efficacy of KC-2-009 but decreased the potency (EC50 = 67±5 nM, EMAX = -63±1.0%). HERK-treatment revealed inverse agonist activity for LTC-274, but the efficacy (EMAX = -18±1.0 %) was much reduced as compared with KC-2-009. In light of the substantial efficacy of KC-2-009 as an inverse MOR agonist in HERK-treated membranes, we defined the inhibition of [35S]-GTP-γ-S binding produced by 5 μM KC-2-009 as 100% when generating dose-response curves with other inverse agonists. Similarly, we used 10 μM DAMGO as the standard when generating agonist dose-response curves in the control and HERK-treated membranes
Table 1.
KI Values of Selected Compounds at the Cloned Human Opioid Receptors
| Drug | MOR (nM±SD) |
DOR (nM±SD) |
KOR (nM±SD) |
|---|---|---|---|
KC-2-009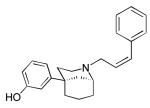
|
26 ± 2 | 454 ± 25 | 123 ± 4 |
LTC-274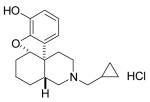
|
0.08± 0.05 | 2.0±0.1 | 0.10±0.40 |
Radioligand binding assays using [125I]IOXY were conducted using published methods (Hiebel et al., 2007). Each value is the mean±SD (n=3).
Figure 1.

Effect of DAMGO, HERK and LTC-274 in control (Panel A) and HERK-treated cells (Panel B). Each value is the mean±SD (n=3). The EC50 and EMAX values are reported in the Results section.
These and other experiments (see Table 2) demonstrate that HERK-treated membranes can be used to detect and characterize inverse MOR agonists with sensitivity and reliability. Although the initial screening experiments, and the experiments described in Fig. 1, did not identify a MOR antagonist totally lacking inverse agonist activity in HERK-treated membranes, LTC-274, when used at a concentrations of 1 nM and 5 nM, had minimal effect on [35S]-GTP-γ-S binding, rendering it suitable for determining antagonist Ke values for both agonists and inverse agonists, as described below.
The next series of experiments determined the Ke value of LTC-274 using a variety of agonists and inverse agonists using membranes prepared from HERK-treated cells. Fig. 2 shows representative “shift” experiments with an agonist (Panel A, dynorphin A(1-13)) and with an inverse agonist (Panel B, CTAP). Although dynorphin A(1-13) is usually considered a KOR agonist, it is a weaker but efficacious MOR agonist as well, rendering it suitable for studying the MOR in a cell line that does not express the KOR. The results showed that LTC-274 caused a dose-dependent rightward shift for both dynorphin A(1-13) and CTAP. As reported in Table 3, for the agonist drugs (dynorphin A(1-13), DAMGO, morphine), the calculated Ke value was ∼ 0.2 nM using both concentrations of LTC-274. For the inverse agonist drugs (CTAP, diprenorphine, KC-2-009), the calculated Ke value ∼0.3 nM was similar with both concentrations of LTC-274. To determine if LTC-274 has a different Ke value for agonists and inverse agonists, we pooled the six Ke values from each class of ligand (Table 3). The results (Fig. 3) showed that the Ke value for agonists (0.17±0.06 nM) was significantly lower (p<0.01) than for inverse agonists (0.29±0.06 nM) (unpaired Student’s t-test).
Figure 2.

Representative “shift experiments” in membranes prepared from HERK-treated cells with an agonist (dynorphin A(1-13), Panel A) and an inverse agonist (CTAP, Panel B). Each value is the mean±SD (n=4 for Panel A, n=3 for Panel B). The Ke values are reported in Table 3.
Table 3.
Ke Values of LTC-274 Determined with Agonists and Inverse Agonists in HERK-treated Membranes
| Drug | EC50 (nM) EMAX (%) |
Ke (nM±SD) (1 nM LTC-274) |
Ke (nM±SD) (5 nM LTC-274) |
|---|---|---|---|
| Agonists | |||
| Dynorphin A(1-13) | 179±14 90±2% |
0.18±0.02 | 0.33±0.04 |
| DAMGO | 53±3 98±1% |
0.20±0.02 | 0.25±0.02 |
| Morphine | 32±11 74±5% |
0.10±0.02 | 0.10±0.01 |
| Inverse Agonists | |||
| CTAP | 16±1 -89±2 |
0.19±0.02 | 0.31±0.05 |
| Diprenorphine | 0.26±0.06 -84±0.03 |
0.28±0.06 | 0.32±0.05 |
| KC-2-009 | 4.1±0.5 -100±1 |
0.37±0.05 | 0.20±0.24 |
Ke values of LTC-274 determined with agonists and inverse agonists in HERK-treated membranes. As described in Methods, agonist or inverse agonist dose-response curves were generated in the absence and presence of 1 and 5 nM LTC-274. The EMAX values are normalized to the effect of DAMGO or KC-2-009. Each value is the mean±SD (n=3).
In light of reports that CTAP acts as a neutral MOR antagonist (Bilsky et al., 1996), we were surprised to observe that CTAP acts as an inverse agonist under our assay conditions. We therefore determined the inverse agonist activity of wide range of compounds that are MOR antagonists. Thus, we also included drugs that act preferentially at other opioid receptors, but which nevertheless are MOR antagonists. The results (Table 2) indicated that all compounds demonstrate inverse agonism at the MOR receptor using HERK-treated membranes. Among these compounds, LTC-274 had the lowest EMAX.
As noted in the Introduction, previous studies (Xu et al., 2007) showed that treating hMOR-CHO cells with HERK produced more constitutively active MORs than chronic treatment with the MOR agonist, [D-Ala2-MePhe4,Gly-ol5]enkephalin (DAMGO). This observation could in principle be due residual HERK dissolved in the membranes despite the washing procedures used, a possibility supported by the fact that HERK is a highly lipophilic compound. To test this hypothesis, we compared the effect of treating cells with HERK, DAMGO, fentanyl and gedunin on the inverse agonist efficacy of KC-2-009. Fentanyl (logP = 3.8) was chosen because it is more lipophilic than HERK (logP = 3.2). Gedunin is structurally related to HERK with a similar lipophilicity (logP = 3.1) but has no affinity for opioid receptors (unpublished data). This compound was included to control for nonspecific effects of HERK. DAMGO was included as a general control MOR agonist. Note that the logP values were calculated. The results (Fig. 4) clearly showed that treating hMOR-CHO cells with HERK resulted in the greatest inverse agonist efficacy (EMAX = 71.3±1.5% as compared with 54.4±1.6% for the control condition). Pretreatment with DAMGO (EMAX = 49.3±1.4%) and gedunin (EMAX = 58.5±1.6%) had small but statistically significant effects on the EMAX of KC-2-009. Fentanyl did not significantly change the efficacy of KC-2-009 (EMAX= 53.0±1.2%).
Pretreatment with increasing s.c. doses of LTC-274 produced dose-related antagonism of the CNS antinociceptive effects of i.c.v. morphine (Fig. 5). The estimated potencies of LTC-274 as an antagonist were 0.19 (0.12-0.29) mg/kg and 0.19 (0.13-0.30) mg/kg, for the 10 and 20 min post-morphine time points, respectively.
Figure 5.

Opioid antagonist activity of LTC-274 in the 55°C tail flick assay. Mice were pretreated at t = -10 min with vehicle or doses of s.c. LTC-274. At t = 0 min, all mice received an i.c.v. injection of morphine (10 nmol). LTC-274 pretreatment produced dose-related antagonism of the morphine antinociceptive effects at t = 10 and 20 min (times corresponding to agonist peak effect).
Discussion
According to the extended ternary complex model described by Samama (Samama et al., 1994) and expounded upon by Kenakin (Kenakin, 2004), inverse agonists preferentially inhibit constitutively receptors, whereas agonists preferentially activate “inactive” receptors. As reviewed by Kenakin, inverse agonists inhibit the activity of constitutively active GPCRs. In the absence of constitutive activity, inverse agonist ligands act as pure competitive “neutral” antagonists, i.e. ligands with zero efficacy (Kenakin, 2004). As the level of constitutive activity increases, neutral antagonists may therefore demonstrate inverse agonist activity. The data in Table 2 nicely illustrate this point, as all of the MOR antagonists were potent and efficacious inverse agonists. In selecting compounds for Table 2, we included both classical MOR antagonists (6-β-Naltrexamide, 6-β-naltrexol, CTAP, naltrexone, naloxone) as well as other compounds that act preferentially at other opioid receptors, but which nevertheless are MOR antagonists (NTB, NorBNI), so as to provide structural diversity. The results show that the task of developing a neutral antagonist would appear to be a difficult one. As noted above, every classic MOR antagonist showed substantial inverse agonist efficacy, including compounds previously described as being neutral antagonists (6-β-naltrexol) (Raehal et al., 2005). LTC-274 stood out as a compound with the lowest efficacy as an inverse agonist. Indeed, at doses ≤ 5 nM, LTC-274 had minimal effect on [35S]-GTP-γ-S binding. Thus, we believe that LTC-274 is an “almost neutral” antagonist and is a promising lead compound for developing a true neutral MOR antagonist. Such compounds may have several promising therapeutic applications (Sadee et al., 2005) in the long-term treatment of opioid addiction and narcotic overdose. As noted in the Results, LTC-274 is a low efficacy partial agonist at the KOR. It’s potency at KOR (EC50 = 2.3 nM) is ∼ 10-fold greater than its Ke as a MOR antagonist. Given the adverse effects that result from KOR agonism (Roth et al., 2002), we believe that analogs of LTC-274 should be sought that are neutral MOR antagonists and which have much greater selectivity for MOR as compared to KOR, or which are KOR antagonists. Since the MOR is develops a high degree of constitutive activity in vivo, neutral antagonists produce a less severe withdrawal syndrome than antagonists such as naloxone (Raehal et al., 2005). Thus, neutral antagonists may be helpful in treating narcotic overdose and addiction.
As predicted from the in vitro results, LTC-274 was a potent antagonist of a CNS-mediated effect of morphine (e.g., antinociception) (Fig. 5). The calculated potencies of LTC-274 were comparable to those of the prototypic opioid antagonist naltrexone examined under roughly similar conditions (Raehal et al., 2005). We plan additional studies to further characterize the antagonist effects of LTC-274 in vivo, with a focus on examining the propensity of this compound to precipitate opioid withdrawal in acute and chronic dependence models.
Previous studies indicated that constitutively active MORs are most readily observed using μ-agonist pretreated cells (Sadee et al., 2005; Wang et al., 2001; Wang et al., 2007; Xu et al., 2007; Xu et al., 2004). Thus, in the present study we treated hMOR-CHO cells with HERK, a salvinorin A analog with MOR agonist activity (Harding et al., 2005), since we showed that treating hMOR-CHO cells with HERK produced more constitutively active MORs than chronic treatment with DAMGO (Xu et al., 2007). The ability of HERK pretreatment to induce substantial constitutive activity could in principle result from residual HERK dissolved in the membranes despite the washing procedures used. This idea is supported by the fact that HERK is a highly lipophilic compound. To test this hypothesis, we compared the effect of treating cells with HERK and various MOR agonists, chosen for their particular properties, on the inverse agonist potency and efficacy of KC-2-009. Fentanyl was chosen because it is more lipophilic than HERK. Gedunin, an analog of HERK with a similar lipophilicty that is inactive at opioid receptors (unpublished data), was included to control for nonspecific effects of HERK. DAMGO was included as a general control MOR agonist. The results (Fig. 4) clearly showed that treating hMOR-CHO cells with HERK resulted in the greatest inverse agonist efficacy (EMAX = 71.3±1.5% as compared with 54.4±1.6% for the control condition). The other drug treatments produced little change in the efficacy, supporting the hypothesis that the ability of HERK pretreatment to induce substantial constitutive activity is not a non-specific effect and is not related to the lipophilicity of this compound. It seems likely that the ability of HERK pretreatment to induce substantial constitutive activity is related to its inability to induce MOR internalization (Groer et al., 2007).
The data reported in Table 2 also indicate that HERK-induced MOR constitutive activity is not due to residual drug. If HERK-induced increases in basal [35S]-GTP-γ-S binding were due to residual drug, then the inverse agonist activity observed with the various antagonists reported in Table 2 would simply reflect competitive inhibition of HERK-stimulated [35S]-GTP-γ-S binding. In this case, all antagonists should be able to inhibit HERK-stimulated [35S]-GTP-γ-S binding to the same degree. In other words, the EMAX values for all antagonists should be the same. However, this is not the case, since the EMAX values range from -37% for LTC-274 to -100% for KC-2-009.
The development of an assay system that provides a robust signal for measurement of inverse agonist activity allowed us to test, in a preliminary way, the hypothesis that the Ke value of a MOR receptor antagonist is the same whether tested with an agonist or inverse agonist. As far as we can determine, this hypothesis has not been systematically studied. In an early report of Costa and Herz (Costa and Herz, 1989), the pA2 value (Ke) of MR2266, which acted a neutral δ antagonist, was similar when tested with a delta agonist (DADLE) or an inverse agonist (ICI174,864) in the GTPase assay. However, the agonist and inverse agonist GTPase assays were done under markedly different assay conditions: high Na+/low K+ and low Na+/high K+, respectively, making a direct comparison of the Ke values problematic.
The Ke experiments (Table 3), which used a structurally diverse set of compounds, demonstrated that the Ke value of LTC-274 was similar for both concentrations tested, indicating that the assay system followed classical pharmacological principles. However, the pooled Ke value for inverse agonists (0.29 nM) was ∼70% higher than the pooled Ke value for agonists (0.19 nM) (Fig. 3). This preliminary finding confirms the hypothesis that the Ke of a MOR antagonist can be different for agonists as compared to inverse agonists. However, it should be noted that since MOR receptors normally display minimal constitutive activity, it was necessary to generate constitutively active MOR by treating cells with HERK, a MOR agonist. It is possible that different results would have been obtained using a cell line that naturally expresses constitutively active MOR, or that has been pretreated with a different selective MOR agonist. Moreover, it is possible that the different Ke values derive from the fact that LTC-274 is not a pure “neutral” antagonist under these assay conditions. Resolving this issue will obviously require the development of a neutral antagonist. Finally, it should be noted that the pooled Ke values were not strikingly different, and that future studies will need to examine a greater number of compounds to verify these results.
In summary, we report here two novel compounds. To our knowledge, few drugs that act as inverse agonists at non-dependent MOR have been reported. Thus, the identification of KC-2-009 as a moderate potency and selective MOR inverse agonist should provide a tool to determine the effect of inverse MOR agonists in vivo. We also identified a MOR antagonist (LTC-274) that shows the least inverse agonist activity among 21 MOR antagonists. We believe that LTC-274, as an “almost neutral” antagonist, is a promising lead compound for developing a true neutral MOR antagonist. Such compounds could have several promising therapeutic applications, including the treatment of narcotic overdose and assisting in the maintenance of abstinence (Sadee et al., 2005).
Acknowledgements
The authors thank Matthew Schmidt and Gary Brandt for technical assistance.
This work was supported in part by the Intramural Research Program, National Institute on Drug Abuse, NIH, DHHS and NIH grant DA018151 (TEP).
List of non-standard abbreviations
- Gedunin
1S,3aS,4aR,4bS,5R,6aR,10aR,10bR,12aS)-5-(acetyloxy)-1-(3-furanyl)-1,5,6,6a,7,10a,10b,11,12,12a-decahydro-4b,7,7,10a,12a-pentamethyloxireno[c]phenanthro[1,2-d]pyran-3,8(3aH,4bH)-dione
- Herkinorin, HERK
(2S,4aR,6aR,7R,9S,10aS,10bR)-9-(benzoyloxy)-2-(3-furanyl)dodecahydro-6a,10b-dimethyl-4,10-dioxo-2H-naphtho-[2,1-c]pyran-7-carboxylic acid methyl ester
- CHO cells expressing the cloned human mu receptor
hMOR-CHO cells
- LTC-274
(-)-3-Cyclopropylmethyl-2,3,4,4aα,5,6,7,7aα-octahydro-1H-benzofuro[3,2-e]isoquinolin-9-ol)
- CTAP
D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2
- DADLE
[D-Ala2,D-Leu5]enkephalin
- DAMGO
[D-Ala2-MePhe4,Gly-ol5]enkephalin
- KC-2-009
(+)-3-((1R,5S)-2-((Z)-3-Phenylallyl)-2-azabicyclo[3.3.1]nonan-5-yl)phenol hydrochloride
- MOR
μ opioid receptor
- DOR
δ opioid receptor
- KOR
κ opioid receptor
References
- Bilsky EJ, Bernstein RN, Wang Z, Sadee W, Porreca F. Effects of naloxone and D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2 and the protein kinase inhibitors H7 and H8 on acute morphine dependence and antinociceptive tolerance in mice. J Pharmacol Exp Ther. 1996;277:484–490. [PubMed] [Google Scholar]
- Brandt GE, Schmidt MD, Prisinzano TE, Blagg BS. Gedunin, a novel hsp90 inhibitor: semisynthesis of derivatives and preliminary structure-activity relationships. J Med Chem. 2008;51(20):6495–6502. doi: 10.1021/jm8007486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciganek E. 2,3,4,4a,5,6,7,7a-Octahydro-1H-benzofuro[3,2-e]isoquinoline: a new morphine fragment. J Am Chem Soc. 1981;103(20):6261–6262. [Google Scholar]
- Costa T, Herz A. Antagonists with negative intrinsic activity at delta opioid receptors coupled to GTP-binding proteins. Proc Natl Acad Sci USA. 1989;86:7321–7325. doi: 10.1073/pnas.86.19.7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, Bohn LM. An opioid agonist that does not induce mu-opioid receptor beta-arrestin interactions or receptor internalization. Mol Pharmacol. 2007;71(2):549–557. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding WW, Tidgewell K, Byrd N, Cobb H, Dersch CM, Butelman ER, Rothman RB, Prisinzano TE. Neoclerodane diterpenes as a novel scaffold for mu opioid receptor ligands. J Med Chem. 2005;48(15):4765–4771. doi: 10.1021/jm048963m. [DOI] [PubMed] [Google Scholar]
- Hiebel AC, Lee YS, Bilsky E, Giuvelis D, Deschamps JR, Parrish DA, Aceto MD, May EL, Harris LS, Coop A, Dersch CM, Partilla JS, Rothman RB, Cheng K, Jacobson AE, Rice KC. Probes for narcotic receptor mediated phenomena. 34. Synthesis and structure-activity relationships of a potent mu-agonist delta-antagonist and an exceedingly potent antinociceptive in the enantiomeric C9-substituted 5-(3-hydroxyphenyl)-N-phenylethylmorphan series. J Med Chem. 2007;50(16):3765–3776. doi: 10.1021/jm061325e. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Efficacy as a vector: the relative prevalence and paucity of inverse agonism. Mol Pharmacol. 2004;65(1):2–11. doi: 10.1124/mol.65.1.2. [DOI] [PubMed] [Google Scholar]
- Kieffer BL, Evans CJ. Opioid receptors: From binding sites to visible molecules in vivo. Neuropharmacology. 2009;56(Supl 1):205–212. doi: 10.1016/j.neuropharm.2008.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimura M, Liu H, Sulima A, Hashimoto A, Przybyl AK, Ohshima E, Kodato S, Deschamps JR, Dersch CM, Rothman RB, Lee YS, Jacobson AE, Rice KC. Probes for narcotic receptor mediated phenomena. 37. Synthesis and opioid binding affinity of the final pair of oxide-bridged phenylmorphans, the ortho- and para-b-isomers and their N-phenethyl analogues, and the synthesis of the N-phenethyl analogues of the ortho- and para-d-isomers. J Med Chem. 2008;51(24):7866–7881. doi: 10.1021/jm800913d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Lowery JJ, Bhamidipati CM, Paolino RM, Blair JR, Wang D, Sadee W, Bilsky EJ. In vivo characterization of 6beta-naltrexol, an opioid ligand with less inverse agonist activity compared with naltrexone and naloxone in opioid-dependent mice. J Pharmacol Exp Ther. 2005;313(3):1150–1162. doi: 10.1124/jpet.104.082966. [DOI] [PubMed] [Google Scholar]
- Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc Natl Acad Sci U S A. 2002;99(18):11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, Murphy DL, Xu H, Godin JA, Dersch CM, Partilla JS, Tidgewell K, Schmidt M, Prisinzano TE. Salvinorin A: allosteric interactions at the mu-opioid receptor. J Pharmacol Exp Ther. 2007;320(2):801–810. doi: 10.1124/jpet.106.113167. [DOI] [PubMed] [Google Scholar]
- Sadee W, Wang D, Bilsky EJ. Basal opioid receptor activity, neutral antagonists, and therapeutic opportunities. Life Sci. 2005;76(13):1427–1437. doi: 10.1016/j.lfs.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Samama P, Pei G, Costa T, Cotecchia S, Lefkowitz RJ. Negative antagonists promote an inactive conformation of the beta 2-adrenergic receptor. Mol Pharmacol. 1994;45(3):390–394. [PubMed] [Google Scholar]
- Wang D, Raehal KM, Bilsky EJ, Sadee W. Inverse agonists and neutral antagonists at mu opioid receptor (MOR): possible role of basal receptor signaling in narcotic dependence. J Neurochem. 2001;77(6):1590–1600. doi: 10.1046/j.1471-4159.2001.00362.x. [DOI] [PubMed] [Google Scholar]
- Wang D, Sun X, Sadee W. Different effects of opioid antagonists on mu-, delta-, and kappa-opioid receptors with and without agonist pretreatment. J Pharmacol Exp Ther. 2007;321(2):544–552. doi: 10.1124/jpet.106.118810. [DOI] [PubMed] [Google Scholar]
- Wells JL, Bartlett JL, Ananthan S, Bilsky EJ. In vivo pharmacological characterization of SoRI 9409, a nonpeptidic opioid mu agonist/delta antagonist that produces limited antinociceptive tolerance and attenuates morphine physical dependence. J Pharmacol Exp Ther. 2001;297:597–605. [PubMed] [Google Scholar]
- Xu H, Lu YF, Thomas JB, Carroll FI, Rice KC, Rothman RB. Opioid peptide receptor studies. 15. Relative efficacy of 4-[(N-allyl-3-methyl-4-piperidinyl)phenylamino]-N,N-diethylbenzamide and related compounds at the cloned human delta-opioid receptor. Synapse. 2001;40(4):269–274. doi: 10.1002/syn.1049. [DOI] [PubMed] [Google Scholar]
- Xu H, Partilla JS, Wang X, Rutherford JM, Tidgewell K, Prisinzano TE, Bohn LM, Rothman RB. A comparison of noninternalizing (herkinorin) and internalizing (DAMGO) mu-opioid agonists on cellular markers related to opioid tolerance and dependence. Synapse. 2007;61(3):166–175. doi: 10.1002/syn.20356. [DOI] [PubMed] [Google Scholar]
- Xu H, Wang X, Wang J, Rothman RB. Opioid peptide receptor studies. 17. Attenuation of chronic morphine effects after antisense oligodeoxynucleotide knock-down of RGS9 protein in cells expressing the cloned mu opioid receptor. Synapse. 2004;52(3):209–217. doi: 10.1002/syn.20019. [DOI] [PubMed] [Google Scholar]