

Abstract
Racemic α-acylphosphinates and formylphosphinate hydrate were used directly as the substrates in a proline derivative-catalyzed cross aldol reaction with ketones. Because of the preexisting the phosphorus stereogenic center, a mixture of two diastereomers of the corresponding α-hydroxyphosphinates were obtained in this reaction. Good to high enantioselectivities (up to 99% ee) were obtained simultaneously for both of these two diastereomers in good yields. Good diastereoselectivities were also obtained when the reaction generates an additional carbon stereogenic center.
Introduction
α-Hydroxyphosphinic acid derivatives, such as α-hydroxyphosphinates, have been shown to be very important enzyme inhibitors.1 For example, α-hydroxyphosphinate-derived peptides have been used as the inhibitors of human immunodeficiency virus (HIV) protease2 and renin.3 Also these compounds have found use as GABA antagonists4 or herbicides.5,6 With the exception of phosphinic acids, the phosphoryl group in other phosphinic acid derivatives, such as phosphinates, generally are chiral because the pentavalent phosphorus atom has a tetrahedral structure. Recent study6 also revealed that phosphinate enantiomers due to such phosphorus chirality may have totally different biological activities: One enantiomer was found significantly more herbicidal than the other enantiomer or the racemic mixture.6 This finding evinces the importance of the phosphorus chirality in the biological activity of the phosphinate compounds. Thus, developing an asymmetric synthesis for α-hydroxyphoshinates that can fix both the stereochemistry of the α-hydroxy-substituted carbon and the phosphorus stereogenic centers during the synthesis is very important. Such a method is expected to have the potential of lowering production costs and use rates, reducing the side effects, and lessening the environmental burden during the manufacture and application of these materials.
Besides the enzymatic resolution of racemic α-hydroxyphosphinates7 and the synthesis starting with optically active α-hydroxyphosphonates8 or α-hydroxy-H-phosphinates,7 only a handful of chemical methods are available for the synthesis of optically active α-hydroxyphosphinates.8 Shibuya and coworkers have reported a synthesis via the diastereoselective reaction of chiral aldehydes and achiral phosphites (a phosphoaldol reaction).9a Alternatively, the phosphoaldol reaction may be carried out with achiral aldehydes and chiral phosphites.9b An enantioselective method based on the phosphoaldol reaction was as also reported;8c however, due to the intrinsic chirality of the phosphinate group, this method actually generates a mixture of diastereomers, which has not been analyzed for its ee values.9c While the above chemical methods produce useful degrees of stereocontrol at the hydroxy-substituted α-carbon center, none of them has addressed the stereochemistry of the phosphinate group. Simultaneously fixing the stereochemistry of both the α-carbon and phosphorus stereogenic centers during the synthesis of α-hydroxyphosphonates is still a great challenge for organic chemists.
Recently we reported the first organocatalytic cross aldol reaction10 of α-ketophosphonates and ketones for the synthesis of α-hydroxyphosphonates (Eq 1).11a,b In principle, this reaction may be used for the preparation of α-hydroxyphosphinates, since the structure of α-ketophosphinates and α-ketophosphonates are very similar. Nonetheless, unlike α-ketophosphonates, α-ketophosphinates are chiral due to the chirality of the phosphorus atom. To use a racemic starting material (A) in a catalytic enantioselective synthesis (Scheme 1) to get both diastereomeric products (B and C) in high ee values is highly challenging because 1) the preexisting stereogenic center in substrate A may interfere with the enantiofacial selectivity of the catalyst so that matched reactants generate diastereomers with higher enantiomeric excesses.12 2) the preexisting stereogenic center in substrate A may affect the reaction rates of the individual enantiomeric substrates and lead to a kinetic resolution.13 If the reaction rates k+ and k- is equal, a kinetic resolution is avoidable, and the reaction may be carried out with 100% conversion of the racemic substrate, which is the major advantage of this approach as compared with the normal kinetic resolution where the maximum conversion of the substrate is only 50%. Because it is difficult to meet both requirements at the same time, it is not surprising that only a few of such chemical reactions are known in the literature.14 Gotor and coworkers have classified this type of reactions into the stereodivergent parallel kinetic resolution.15 Nevertheless, it should be pointed out that this type of reactions does not always fulfill one of the guidelines laid down for parallel kinetic resolution by Eames:16 the reactions must afford distinct and easily separable products. While the diastereomers obtained in this type of reactions are distinct, they are not always easy to separate, as with our α-hydroxyphosphinate products (see below).
Scheme 1.

Catalytic Enantioselective Reaction with a Racemic Substrate
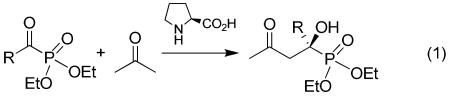
It is our contention that the enantiofacial selectivity of the aldol reaction of α-ketophosphonates is mainly determined by the size difference between the phosphonate and the alkyl group of the α-ketophosphonate,11b while the actual stereochemistry of the phosphorus atom should have minimum influence on the enantiofacial selectivity (Scheme 2). Furthermore, the phosphonate group is pointing away from the reaction center in the favored transition state11 and, therefore, its stereochemistry should not have a major influence on the reaction rate. Thus, we hypothesized that such a cross aldol reaction may be used for the high enantioselective synthesis of both diastereomers of the α-hydroxyphosphinates from racemic α-ketophosphinates. Herein, we wish to report a catalytic and highly enantioselective synthesis of both diastereomers of α-hydroxyphosphinates with simultaneous fixing of both the hydroxy-substituted α-carbon and the phosphorus stereogenic centers in the products through a proline derivative-catalyzed cross aldol reaction of racemic acylphosphinates and ketones.
Scheme 2.

Proposed Transition States for the Cross Aldol Reaction of α-Ketophosphonates
Results and Discussion
Ethyl benzoylphenylphosphinate (5a) and acetone (6a) were used as the model compounds to study the reaction conditions. We screened several readily available proline-derivatives as the catalyst (Figure 1). The results are summarized in Table 1. The cross aldol reaction went smoothly at room temperature with all these catalysts and excellent yields of the aldol product were obtained. Although l-prolinamide (1), (S)-N-tosylprolinamide (2), and l-proline tetrazole (3) are very reactive catalysts, only moderate enantioselectivities were obtained for the two diastereomeric products 7a and 8a (entries 1-3). Nevertheless, the results of l-proline (4) were promising: Using a 20 mol % loading of 4 and acetone as the solvent at room temperature, 7a and 8a were obtained in a total yield of 89% in a ratio of 53:47. The ee values of these two diastereomeric products were determined to be 95% and 74%, respectively (entry 4). Other solvents, such as THF, CH2Cl2, DMF, DMSO, and an ionic liquid, proved to be inferior to acetone (entries 5-9). To our pleasure, by lowering the reaction temperature to -30 °C, the enantioselectivities of these two products were further improved to 99% and 91% ee, respectively, although the reaction was slower at this temperature and the total yield was lower (entry 10). It should be pointed out that, under all these conditions, diastereomeric products 7a and 8a were obtained in a ratio of roughly 50:50 with different degree of conversions of the substrate (from around 60% to over 90%), which hints that the reaction rates of both enantiomeric substrates of 5a are almost equal.
Figure 1.

Catalysts Screened for the Cross Aldol Reaction
Table 1.
Cross aldol reaction of ethyl (benzoylphenyl)phosphinate (5a) and acetone (6a) catalyzed by L-proline derivativesa
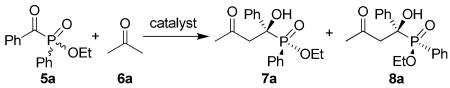 | |||||||
|---|---|---|---|---|---|---|---|
| entry | catalyst | loadingb | solvent | yield (%)c | ee (%)d | 7a/8ae | |
| 7a | 8a | ||||||
| 1f | 1 | 10 | acetone | 93 | 72 | 47 | 55:45 |
| 2f | 2 | 10 | acetone | 63 | 45 | 28 | 58:42 |
| 3f | 3 | 10 | acetone | 83 | 82 | 62 | 54:46 |
| 4f | 4 | 20 | acetone | 89 | 95 | 74 | 53:47 |
| 5 | 4 | 20 | THF | 90 | 95 | 67 | 52:48 |
| 6 | 4 | 20 | CH2Cl2 | 79 | 94 | 65 | 50:50 |
| 7 | 4 | 20 | DMSO | 81 | 52 | 32 | 48:52 |
| 8 | 4 | 20 | DMF | 81 | 69 | 39 | 58:42 |
| 9g | 4 | 20 | acetone | 87 | 91 | 59 | 59:41 |
| 10f,h | 4 | 20 | acetone | 58 | 99 | 91 | 52:48 |
Unless otherwise indicated, all reactions were carried out with the ketophosphinate 5a (0.50 mmol), acetone (0.5 mL), and the catalyst in the specified solvent (1.0 mL) at rt for 24 h.
mol %.
Total yield of the inseparable diastereomers (7a and 8a) isolated after column chromatography.
Determined by HPLC analyses.
The ratio of 7a/8a; determined by 1H NMR analyses.
2.0 mL of acetone was used.
Ionic liquid 1-butyl-3-methylimidazolium terafluoroborate (300 mg) was added.
Carried out at -30 °C for 96 h.
The scope of this reaction was then evaluated, and the results are collected in Table 2. It was found that, besides ethyl benzoylphenylphosphinate (entry 1), substituted benzoylphosphinates were also good substrates for this reaction. For example, 98% and 91% ee were obtained for the two diastereomeric products of 4-fluorobenzoylphosphinate (5b) in 67% total yield under the optimized reaction conditions (entry 2). Also good ee values (98% and 80% ee, respectively) were obtained for the product of 3-fluorobenzoylphosphinate (5c, entry 3). Similarly, 4-bromo- (5d), 4-chloro- (5e) and 3-chloro- (5f) substituted derivatives also produce excellent results (entries 4-6). Substrates substituted with electron-donating groups (i.e., Me, OMe, 5g and h) react more slowly, but the enantioselectivities obtained for the products are very good (entries 7-8). A heteroarene-substituted (thiophen-2-yl) α-ketophosphinate (5i) also leads to good enantioselectivity for one of the diastereomer (84% ee), whereas the enantioselectivity for the other diastereomer is slightly lower (62% ee, entry 9). Alkyl-substituted α-ketophosphinates react in a similar manner. For example, ethyl acetylphenylphosphinate (5j) gives 95% and 87% ee of the two diastereomeric products, respectively (entry 10). Similar results were also obtained for ethyl phenylpropionylphosphinate (5k, entry 11). From the results in Table 2, it is evident that the stereochemistry of the phosphinate group does have some influence on the enantioselectivity of this reaction: The ee value of one of the two diastereomers is always slightly higher than the other. Nevertheless, the reactivity of the two enantiomers of the racemic starting acylphosphinates are almost the same since the ratio of the two diastereomeric products 7 and 8 maintained around 50:50 in all cases.
Table 2.
L-Proline-Catalyzed Aldol Reaction of Racemic α-Ketophosphinates and Acetonea
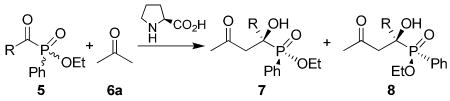 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R | T (°C) | time (h) | yield (%)b | ee (%)c | drd | |
| 7 | 8 | ||||||
| 1 | Ph(a) | -30 | 96 | 58 | 99 | 91 | 52:48 |
| 2 | 4-F-C6H4(b) | -30 | 120 | 67 | 98 | 91 | 50:50 |
| 3 | 3-FC6H4(c) | 0 | 36 | 89 | 96 | 80 | 52:48 |
| 4 | 4-BrC6H4(d) | -30 | 120 | 70 | 97 | 87 | 54:46 |
| 5 | 4-ClC6H4(e) | -30 | 120 | 65 | 94 | 73 | 52:48 |
| 6 | 3-ClC6H4(f) | 0 | 36 | 92 | 99 | 89 | 53:47 |
| 7 | 4-MeC6H4(g) | -30 | 120 | 45 | 90 | 80 | 54:46 |
| 8 | 4-MeOC6H4(h) | rt | 40 | 87 | 93 | 71 | 56:44 |
| 9 | rt | 48 | 85 | 84 | 62 | 46:54 | |
| 10 | Me(j) | rt | 24 | 88 | 95 | 87 | 44:56 |
| 11 | Et(k) | rt | 40 | 90 | 87 | 61 | 45:55 |
Unless otherwise indicated, all reactions were carried out with the racemic ketophosphinate (0.50 mmol) in dry acetone (2.0 mL), with L-proline (0.10 mmol, 20 mol %) as the catalyst for the specified reaction time and temperature.
Total yield of the inseparable diastereomers (7 and 8) isolated after column chromatography.
Enantioselectivity was determined by HPLC analyses, due to the overlap some of the peaks, the error limits maybe is considerably larger than normal in those cases.
The ratio of 7:8 or vice versa; determined by 1H NMR analyses.
Methyl α-ketophosphinates also participate in this reaction. However, determining the enantiomeric excess values of the products is highly challenging, as the enantiomers cannot be separated by the columns we have (data not shown).
To determine the absolute configuration of the major enantiomers formed in this reaction, the two diastereomeric products 7 and 8 have to be separated. While compounds 7 and 8 obtained in this reaction cannot be separated by column chromatography, we did achieve separation through recrystallization in a few cases, and suitable crystals for the X-ray crystallographic analysis were obtained for the enantiomer of products 7d (ent-7d) with D-proline as the catalyst. As shown in Scheme 3, ethyl (4-bromobenzoyl)phenylphosphinate (5d) was allowed to react with acetone using D-proline as the catalyst under the optimized conditions identified for L-proline. The diastereomeric mixture of ent-7d and ent-8d obtained this way was separated by careful recrystallization from a mixture solvent of EtOAc and hexane (The diastereomeric products were dissolved in a minimum amount of EtOAc and then diluted with hexane. The solution was kept at an open system in such a way that the solvents evaporated very slowly. After 24 h at rt, about 70% of compound ent-7d crystallized out.). The crystalline compound obtained was identified to be ent-7d, whereas the mother liquor contains mainly the remaining ent-8d. The absolute configuration of ent-7d was determined to be S for both the α-carbon and the phosphorus stereogenic centers (SC,SP) by X-ray crystallography.17 To determine the absolute configuration of the other diastereomers, separate HPLC analyses of the racemic product, the product obtained from L-proline (Table 2, entry 1) and D-proline (Scheme 3), and pure ent-7d were undertaken.18 From the results of the racemic sample, it is clear that the stereoisomers with retention times of 48.1 min and 113.8 min are a pair of enantiomers, and those with retention times 27.2 min and 117.1 min of are the other pair of enantiomers (please see page S-61 of the Supporting Information18). Pure ent-7d, which has been determined to be SCSP by X-ray crystallography, shows a retention time of 48.1 min (page S-62). Thus, the stereoisomer with the retention time of 113.8 min should have the stereochemistry of RCRP since it is the enantiomer of ent-7d (that is, it is 7d). With these results at hand, it is relatively easy to determine the stereochemistry of the other two enantiomers by using the HPLC analysis of the compounds 7d/8d obtained in Table 2, entry 1. In this HPLC spectrum (page S-64), one major peak is at 111.2 min, which was assigned to RCRP as described above. The other major peak is at 115.1 min, which must be RCSP, because most of the R-configured phosphinate group has been already fixed in compound 7d (retention time 111.2 min). Thus, the peak at 27.2 min must be SCRP. On the basis of the above analysis, compound 8d should have a stereochemistry of RC,SP (retention time 117.1 min in S-60), which is also consistent with the reaction mechanism.
Scheme 3.

Determination of the Absolute Configuration of the Reaction Products
On the basis of the aforementioned results, a mechanism is proposed to account for the enantioselectivity outcome (Scheme 4). Just as we expected, it does not matter whether the stereochemistry of the phosphorus atom in acylphosphinate 5a is R (TS-I and II) or S (TS-III and IV), the attack of the enamine onto the si-face of this substrate is always favored (TS-I and TS-III in Scheme 4), because the unfavorable steric interactions between the large axial phosphinate group and the axial enamine methyl group (see TS-II and TS-IV) are avoided in these two transition states. These favored transition states lead to the formation of RCRP-7a and RCSP-8a diastereomers as the major products, as observed in the experiment. These results are also in accord with those of the aldol reaction of acylphosphonate derivatives catalyzed by similar catalysts (Scheme 2).11
Scheme 4.

Proposed Mechanism for the Cross Aldol Reaction of Acylphosphinates
Next the aldol reaction of the hydrate of ethyl formylphosphinate (9) was studied. This reaction is expected to generate secondary α-hydroxyphosphinates, which are closer analogues of proteinogenic α-amino acids. The results of this reaction are summarized in Table 3. When acetone was used as the substrate at room temperature, L-proline (4) failed to generate any desired product (entry 1), probably because compound 9 is not compatible with the acidity of 4. These results are not surprising, since previously we obtained similar results with formylphosphonate hydrate.11b When less acidic L-prolinamide (1) was used as the catalyst under the above conditions (entry 2), the desired product was obtained in 89% yield with ee values of 90% and 79% for SCRP (10a) and SCSP (11a) products, respectively, in a ratio of 50:50. (The absolute configuration of products was assigned on the basis of the proposed mechanism in Scheme 4). When the reaction was carried out at 0 °C, the ee values of these two products were further improved to 93% and 87%, respectively (entry 3). Cyclopentanone may also be used in this reaction (entry 4). Although this reaction potentially can generate 8 diastereomers, it was found that only the syn diastereomers (referring to the newly formed carbon stereogenic centers)19 was formed during the reaction (>99:1 dr), and the ee value of the two diastereomers syn-10b (configuration: SC,SC,RP) and syn-11b (configuration: SC,SC,SP) obtained at room temperature are 97% and 62%, respectively. When the reaction was carried out at 0 °C, the ee value of syn-11b was further improved to 87% (entry 5). Cyclohexanone and 4-oxacyclohexanone are also suitable substrates for this reaction; nevertheless, the reactions are less diastereoselective (entries 6 and 7). For cyclohexanone an anti/syn ratio of 65:35 were obtained, and the four diastereomers 10c and 11c were obtained in 98%, 95%, 89%, and 93% ee, respectively (entry 6). Similarly, 4-oxacyclohexanone yielded an anti/syn ratio of 60:40, and the ee values for the four diastereomers 10d and 11d are 99%, 94%, 26%, and 99% ee, respectively (entry 7). The low diastereoselectivities observed for six-membered cyclic ketones versus cyclopentanone are in-line with our previous report on cross-aldol reaction of the α-formylphosphonate hydrate derivative11b and are probably due to steric reasons, although the exact reason is not clear at this moment. Since these products are inseparable liquid compounds by column chromatography, it is impossible to assign the ee values to the corresponding structures without ambiguity. Nevertheless, it is clear from Table 3 that the ratio of 50:50 were achieved in each entry for compound containing RP and SP stereogenic centers, which indicates that both enantiomers of the racemic substrate 9 react efficiently under the reaction conditions.
Table 3.
Aldol Reaction of Racemic Formylphosphinate Hydrate and Ketonesa
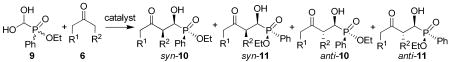 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| entry | 6 | catalyst | 10 and 11 | time (h) | yield (%)b | 10/11c | syn/antid | ee (%)e | ||||
| R1 | R2 | syn-10 | syn-11 | anti-10 | anti-11 | |||||||
| 1f | H | H | 4 | a | 24 | 0 | --- | --- | --- | --- | --- | --- |
| 2f | H | H | 1 | a | 1.5 | 89 | 50:50 | --- | 90g | 79g | --- | --- |
| 3 | H | H | 1 | a | 7 | 91 | 50:50 | --- | 93g | 87g | --- | --- |
| 4f | -(CH2)2- | 1 | b | 24 | 79 | 50:50 | >99:1 | 97 | 62 | --- | --- | |
| 5 | -(CH2)2- | 1 | b | 30 | 83 | 50:50 | 99:1 | 96 | 87 | --- | --- | |
| 6 | -(CH2)3- | 1 | c | 32 | 88 | 50:50 | 35:65 | 98h | 95h | 89h | 93h | |
| 7 | -(CH2OCH2)- | 1 | d | 30 | 91 | 50:50 | 40:60 | 99h | 94h | 26h | 99h | |
Unless otherwise indicated, all reactions were carried out with the racemic formylphosphinate hydrate (0.50 mmol) in dry ketone (0.5 mL) with the catalyst (0.10 mmol, 5 mol %) for the specified reaction time at 0 °C.
Total yield of the inseparable diastereomers (10 and 11) isolated after column chromatography.
Determined by 1H NMR analyses; the value refers to the ratio of the products containing RP and SP stereochemistry.
Determined by 1H NMR analyses, syn and anti refer to the stereochemistry of the two newly generated carbon stereogenic centers.
Enantioselectivity was determined by HPLC analyses.
The reaction was carried out at room temperature.
This compound does not has syn or anti stereochemistry.
The assignments of the ee values to the structures are arbitrary in these cases. Due to partial overlaps of some of the peaks, the error limit of the ee values is considerably larger than normal.
Conclusions
In summary, a novel method for the highly enantioselective synthesis of the biologically significant α-hydroxyphosphinates was developed on the basis of a proline derivative-catalyzed asymmetric aldol reaction of ketones and α-ketophosphinates or α-formyphosphinate hydrate. Both the newly formed carbon stereogenic centers and the phosphorus stereogenic center were fixed during the aldol reaction.
Experimental Section
General experimental procedure for the synthesis of tertiary α-hydroxyphosphinate via aldol reaction of racemic α-ketophosphinate
To a stirred solution of the α-ketophosphinate (0.5 mmol) and dry acetone (2.0 mL) was added L-proline (0. 10 mmol, 20 mol %) at -30 °C. The reaction mixture was stirred at this temperature for the time as specified in Table 1 (monitored by TLC) and then quenched by few drops of water. The mixture was extracted with ethyl acetate (3 × 10 mL), and the combined extracts were washed with brine solution (2 mL), dried over MgSO4, and then evaporated to give the crude product, which was purified by column chromatography over silica gel (4:1 ethyl acetate/hexane) to furnish the desired α-hydroxyphosphinate as a pure compound (a mixture of two inseparable diastereomers).
Ethyl (1-hydroxy-3-oxo-1-phenylbutyl)(phenyl)phosphinate (7a/8a)
white solid, mp 91-92 °C; 1H NMR (500 MHz, CDCl3) δ (mixture of two diastereomers) 1.13 and 1.36 (t, J = 7.0 Hz, 3 H), 2.05 and 2.17 (s, 3 H), 3.05 and 3.08 (d, J = 8.0 Hz, 0.5 H), 3.75-4.15 (m, 2 H), 4.88 and 5.06 (br s, 1 H), 7.15-7.66 (m, 10 H); 13C NMR (125 MHz, CDCl3) δ (mixture of two diastereomers) 16.6 (q), 32.5 (q), 46.7 (q), 62.2 (q), 76.2 and 76.4 (d), 126.2 and 126.6 (d), 126.8 (q), 127.6 and 127.8 (d), 127.9 and 128.0 (d), 128.1 (q), 132.6 and 132.7 (d), 133.4 and 133.7 (d), 139.2 (q), 210.1 (q); 31P NMR (CDCl3) δ (mixture of two diastereomers) 36.3, 37.4; νmax (neat, cm-1): 3212, 1710, 1598, 1510, 1475, 1439, 1411; Anal. Calcd. for C18H21O4P: C, 65.05; H, 6.37; Found: C, 64.75; H, 6.45.
General procedure for the synthesis of secondary α-hydroxyphosphinate via aldol reaction of racemic α-formylphosphinate hydrate
To a stirred solution of the formylphosphinate hydrate (108 mg, 0.5 mmol) in the ketone (0.5 mL) in dry CH2Cl2 at 0 °C was added L-prolinamide (2.9 mg, 0.025 mmol, 5 mol %). The reaction mixture was stirred at this temperature for 24-32 h (depending on the substrate). The solvent was evaporated under vacuum and the residue was purified by flash chromatography (EtOAc) over silica gel to furnish the desired secondary α-hydroxyphosphinate as a pure compound (a mixture of inseparable diastereomers).
Ethyl (1-hydroxy-3-oxobutyl)(phenyl)phosphinate (10a/11a)
Colorless oil; 1H NMR (500 MHz, CDCl3) δ (mixture of two diastereomers) 1.28 (2 t, J = 7.0 Hz, 3 H), 2.14 (s, 3H), 2.68-2.84 (m, 2 H), 3.91-3.99 (m, 1 H), 4.07-4.14 (m, 1 H), 4.47-4.58 (m, 1 H); 13C NMR (125 MHz, CDCl3) δ (mixture of two diastereomers) 16.7(q), 30.9 (d), 44.1 (q), 61.9 (q), 65.8 and 66.5 (d, JCP = 121.6 and 120.1 Hz), 127.4 and 128.4 (d, JCP = 25.8 and 25.3 Hz), 128.8 (q), 132.8 (q), 133.0 (d), 207.2 (q); 31P NMR (CDCl3) δ (mixture of two diastereomers) 39.6, 40.1; νmax (neat, cm-1): 3251, 1715, 1592, 1479, 1439, 1394 1362; Anal. Calcd. for C12H17O4P: C, 56.25; H, 6.69; Found: C, 56.11; H, 6.88.
Supplementary Material
Acknowledgments
The authors thank the NIH-NIGMS (Grant No. 1SC1GM082718-01A1) for the generous financial support of this research and the Welch Foundation (Grant No. AX-1593) for the financial support in getting the preliminary data of this research. The authors also thank Dr. Derong Ding for help with the synthesis of some of the α-ketophosphonate starting materials and Dr. Edward R. T. Tiekink for help with the X-ray analysis of compound ent-7d.
Footnotes
Supporting Information Available. Full citation of references 4a and 4b, HPLC conditions, characterization data and NMR spectra for all new compounds, HPLC analysis spectra, and CIF data for compound ent-7d. This material is available free of charge via Internet at http://pubs.acs.org.
References
- 1.For reviews, see: Kuhkar VP, Hudson HR. Aminophosphonic and Aminophosphinic Acids. John Wiley; Chichester: 2000. Kafarski P, Lejczak B. Curr Med Chem-Anti-Cancer Agents. 2001;1:301–312. doi: 10.2174/1568011013354543.Berlicki L, Kafarski P. Curr Org Chem. 2005;9:1829–1850.Redmore D. In: Topics in Phosphorus Chemistry. Griffith EJ, Grayson M, editors. Vol. 8. John Wiley; New York: 1976. pp. 515–585.
- 2.Stowasser B, Budt KH, Li JQ, Peyman A, Ruppert D. Tetrahedron Lett. 1992;33:6625–6628. [Google Scholar]
- 3.Patel DV, Rielly-Gauvin K, Ryono DE, Free CA, Rogers WL, Smith SA, DeForrest JM, Oehl RS, Petrillo EW., Jr J Med Chem. 1995;38:4557–4569. doi: 10.1021/jm00022a022. [DOI] [PubMed] [Google Scholar]
- 4.(a) Froestl W, Mickel SJ, Hall RG, et al. J Med Chem. 1995;38:3297–3312. doi: 10.1021/jm00017a015. [DOI] [PubMed] [Google Scholar]; (b) Froestl W, Mickel SJ, von Sprecher G, et al. J Med Chem. 1995;38:3313–3331. doi: 10.1021/jm00017a016. [DOI] [PubMed] [Google Scholar]
- 5.Rosen RE, Weaver DG, Cornille JW, Spangler LA. US Patent 5,500,405. 1996
- 6.Spangler LA, Mikołajczyk M, Burdge EL, Kiełbasiński P, Smith HC, Łyzwa P, Fisher JD, Omelańczuk J. J Agric Food Chem. 1999;47:318–321. doi: 10.1021/jf980527j. [DOI] [PubMed] [Google Scholar]
- 7.Yamagishi T, Miyamae T, Yokomatsu T, Shibuya S. Tetrahedron Lett. 2004;45:6713–6716. [Google Scholar]
- 8.Patel DV, Rielly-Gauvin K, Ryono DE. Tetrahedron Lett. 1990;31:5591. [Google Scholar]
- 9.(a) Yamagishi T, Suemune K, Yokomatsu T, Shibuya S. Tetrahedron Lett. 2001;42:5033–5036. [Google Scholar]; (b) Cai J, Zhou Z, Zhao G, Tang C. Heteroatom Chem. 2003;14:312–315. [Google Scholar]; (c) Yamagishi T, Yokomatsu T, Suemune K, Shibuya S. Tetrahedron. 1999;55:12125–12136. [Google Scholar]; For a review, see: Shibuya S. Yakugaku Zasshi. 2004;124:725–749. doi: 10.1248/yakushi.124.725.
- 10.For reviews on organocatalyzed cross aldol reactions, see: List B. Synlett. 2001:1675–1686.List B. Tetrahedron. 2002;58:5573–5590.List B. Acc Chem Res. 2004;37:548–557. doi: 10.1021/ar0300571.Notz W, Tanaka F, Barbas CF., III Acc Chem Res. 2004;37:580–597. doi: 10.1021/ar0300468.. For examples on the cross aldol of activated ketone substrates, see: Enders D, Grondal C. Angew Chem, Int Ed. 2005;44:1210–1212. doi: 10.1002/anie.200462428.Luppi G, Cozzi PG, Monari M, Kaptein B, Broxterman QB, Tomasini C. J Org Chem. 2005;70:7418–7421. doi: 10.1021/jo050257l.Shen Z, Li B, Wang L, Zhang Y. Tetrahedron Lett. 2005;46:8785–8788.Tokuda O, Kano T, Gao WG, Ikemoto T, Maruoka K. Org Lett. 2005;7:5103–5105. doi: 10.1021/ol052164w.Tang Z, Cun LF, Cui X, Mi AQ, Jiang YZ, Gong LZ. Org Lett. 2006;8:1263–1266. doi: 10.1021/ol0529391.Samanta S, Zhao CG. Tetrahedron Lett. 2006;47:3383–3386.Bøgevig A, Kumaragurubaran N, Jørgensen KA. Chem Commun. 2002:620–621. doi: 10.1039/b200681b.. (h) References 11a-c.
- 11.For some recent examples of asymmetric synthesis of α-hydroxyphosphonates, see: Samanta S, Zhao CG. J Am Chem Soc. 2006;128:7442–7443. doi: 10.1021/ja062091r.Dodda R, Zhao CG. Org Lett. 2006;8:4911–4914. doi: 10.1021/ol062005s.Liu J, Yang Z, Wang Z, Wang F, Chen X, Liu X, Feng X, Su Z, Hu C. J Am Chem Soc. 2008;130:5654–5655. doi: 10.1021/ja800839w.Pawar VD, Bettigeri S, Weng SS, Kao JQ, Chen CT. J Am Chem Soc. 2006;128:6308–6309. doi: 10.1021/ja060639o.Gondi VB, Hagihara K, Rawal VH. Angew Chem Int Ed. 2009;48:776–779. doi: 10.1002/anie.200804244.Abell JP, Yamamoto H. J Am Chem Soc. 2008;130:10521–10523. doi: 10.1021/ja803859p.Pogatchnik DM, Wiemer DF. Tetrahedron Lett. 1997;38:3495–3498.Cermak DM, Du Y, Wiemer DF. J Org Chem. 1999;64:388–393.Skropeta D, Schmidt RR. Tetrahedron: Asymmetry. 2003;14:265–273.Wroblewski AE, Balcerzak KB. Tetrahedron: Asymmetry. 2001;12:427–431.Yokomatsu T, Yamagishi T, Shibuya S. Tetrahedron: Asymmetry. 1993;4:1401–1404.Rowe BJ, Spilling CD. Tetrahedron: Asymmetry. 2001;12:1701–1708.Arai T, Bougauchi M, Sasai H, Shibasaki M. J Org Chem. 1996;61:2926–2927. doi: 10.1021/jo960180o.Sasai H, Bougauchi M, Arai T, Shibasaki M. Tetrahedron Lett. 1997;38:2717–2720.Saito B, Katsuki T. Angew Chem, Int Ed. 2005;44:4600–4602. doi: 10.1002/anie.200501008.Groaning MD, Rowe BJ, Spilling CD. Tetrahedron Lett. 1998;39:5485–5488.
- 12.Adam W, Humpf HU, Roschmann KJ, Saha-Möller CR. J Org Chem. 2001;66:5796–5800. doi: 10.1021/jo010350j. [DOI] [PubMed] [Google Scholar]
- 13.For reviews, see: Hoveyda AH, Didiuk Mary T. Curr Org Chem. 1998;2:489–526.Robinson Diane EJE, Bull Steven D. Tetrahedron: Asymm. 2003;14:1407–1446.
- 14.(a) Reynolds NT, Rovis T. Tetrahedron. 2005;61:6368–6378. [Google Scholar]; (b) Bocknack BM, Wang LC, Krische M. J Proc Nat Acad Sci USA. 2004;101:5421–5424. doi: 10.1073/pnas.0307120101. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a review on enzymatic reactions, see reference 15
- 15.For review, see: Dehli JR, Gotor V. Chem Soc Rev. 2002;31:365–370. doi: 10.1039/b205280f.
- 16.Eames J. Angew Chem Int Ed. 2000;39:885–888. doi: 10.1002/(sici)1521-3773(20000303)39:5<885::aid-anie885>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 17.For the ORTEP drawing of compound ent-7d, please see the Supporting Information.
- 18.For details, please see the Supporting Information.
- 19.The syn/anti stereochemistry was determined by the NOE experiments on the products syn-10b/syn-11b and anti-10c/anti-11c (for details, please see Pages S-59 and S-60 of the Supporting Information). The results are also in-line with those of the corresponding α-hydroxyphosphonates (please see reference 11b).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.