

Abstract
Ovarian cancer is the most malignant gynecologic neoplasm. Although new chemotherapeutic agents have improved patients' 5-year survival rate, the overall mortality of ovarian cancer has remained largely unchanged in the past several decades. The main reason for the lack of success in effectively treating ovarian cancer is our limited understanding of its etiology and the very few molecular diagnostic markers and therapeutic targets known so far. Identification and characterization of ovarian cancer-associated genes are fundamental for unveiling the pathogenesis of its initiation and progression, especially the development of recurrent diseases. As there are a vast number of genes for which molecular genetic changes and aberrant gene expression have been reported in ovarian cancer, this review will only focus on summarizing those exemplified genes that have been demonstrated to have biological functions in promoting ovarian cancer development and potential clinical significance. The genes to be discussed include nuclear proteins (Notch3, HBXAP [Rsf-1], NAC1 and NFκB), cytoplasmic proteins (fatty acid synthase and apolipoprotein E) and cell surface/secretory proteins (mucin-4, mesothelin, claudin, HLA-G, kallikrein and folate receptor and osteopontin). Since the study of ovarian cancer-associated genes is complicated by several factors unique to ovarian cancer, we will also present our views on the limitations and challenges of current ovarian cancer research.
Keywords: ovarian cancer, TCGA, The Cancer Genome Atlas, tumor-associated gene
Ovarian cancer is a highly aggressive neoplastic disease in women. The high mortality rate is largely attributable to the fact that ovarian cancer develops without obvious symptoms, almost always resulting in advanced, widespread disease when patients are first diagnosed. Unfortunately, the molecular etiology of ovarian cancer remains elusive, and the attempts to develop effective early detection and treatment for ovarian cancer patients are empirical. Identification and characterization of important ovarian cancer-associated genes are fundamental steps toward elucidating its molecular etiology [1]. To this end, over the past several years, many research teams, including our own, have applied several genome-wide technologies, including high-throughput sequencing technology, digital karyotyping, SNP array, transcriptome profiling and proteomics, to study the molecular landscape of ovarian cancer [2,3]. As a result, a number of genes with molecular genetic changes and aberrant gene expression have been discovered in ovarian cancer, and several of those gene products may have clinical implications [4,5]. More recently, the US government-sponsored The Cancer Genome Atlas (TCGA) has provided a large amount of data on DNA and RNA copy number, mutation analysis and methylation profiles in a large series of ovarian carcinoma. These advances are expected to facilitate identification of candidate ovarian tumor-associated genes in the near future. However, the genes that participate in the development of ovarian cancer may represent only a small fraction of the ovarian cancer-associated genes, as many of them are the ‘by-stander’ molecules that are simply associated with ovarian cancer development but do not causally contribute to its initiation and progression. In this review, we will summarize the recent advances in identifying and characterizing ovarian cancer genes, with special emphasis on their roles in pathogenesis and their potential translational implications. As the ovarian cancer-associated genes are numerous, we select several upregulated genes as examples to demonstrate their roles in understanding the molecular etiology of ovarian cancer and their potential applications as diagnostic markers and therapeutic targets. Of note, there are a number of genes, such as p53, AKT2, cyclin E, ERRB2 (Her2) and CA125 (mucin-16), among many others, that have been extensively reviewed elsewhere [1,6,7], and they will not be discussed in this article. Table 1 lists the selected genes to be discussed and their molecular and clinical significance in ovarian cancer.
Table 1. Examples of ovarian cancer-associated genes.
| Gene name | Subcellular location | Biological functions | Molecular change | Clinical significance | Potential targeted therapy | Ref. |
|---|---|---|---|---|---|---|
| Notch3 | Nucleus | Signaling receptor | Amplification; upregulation | Tumor recurrence | γ-secretase inhibitor | [8–10] |
| HBXAP (Rsf-1) | Nucleus | Chromatin remodeling | Amplification; upregulation | Poor OS | NA | [11,12] |
| NAC1 | Nucleus | Transcription regulator | Upregulation | Tumor recurrence | NA | [28–31] |
| NF-κB | Nucleus | Signal transduction | Upregulation | Poor PFS | PS1145 | [118–123] |
| Fatty acid synthase | Cytoplasm; secretory | Metabolism in fatty acid pathway | Upregulation | Poor OS | C93, enzyme inhibitor | [32–36,39] |
| Apo-E | Cytoplasm | Cell survival | Upregulation | Prolonged OS | NA | [70–76] |
| Mucin-4 (Muc4) | Surface | Surface marker with diverse functions | Upregulation | Not significant | Humanized Muc4 mAb | [50–55] |
| Mesothelin | Surface | Surface marker with diverse functions | Upregulation | Prolonged OS | Mesothelin vaccine Humanized mAB | [58–60] |
| Claudin | Surface | Tight junction proteins | Upregulation | Poor OS | CPE | [98–99] |
| Kallikrein | Surface; secretory | Protease | Upregulation | Generally with worse outcome serum marker | NA | [74–87] |
| Folate receptor-α | Surface | Receptor to bind to folic acid | Upregulation | Cell surface target | Humanized mAb BGC 945 | [112–116] |
| HLA-G | Surface; secretory | Immune evasion and suppression | Upregulation | Susceptible marker for chemotherapy | NA | [107–111] |
| Osteopontin | Surface; secretory | Cell survival, invasion | Upregulation | Serum marker | Humanized mAb | [126–131] |
CPE: Clostridium perfringens enterotoxin; mAB: Monoclonal antibody; NA: Not applicable; OS: Overall survival; PFS: Progression-free survival.
Molecular genetics in ovarian carcinomas
It has been well established that ovarian cancer is a heterogeneous disease that is comprised of different histological subtypes with distinct clinicopathological and molecular features. Ovarian tumors are traditionally classified by cell type into serous, mucinous, endometrioid and clear-cell tumors corresponding to different types of (Müllerian) epithelia in the organs of the female reproductive tract. The tumors in each of the categories are further subdivided into three groups based on their clinical behavior: benign (cystadenoma), malignant (carcinoma) and borderline (low-malignant-potential). Recent advances in molecular studies have established the molecular features unique to each specific histologic subtype of ovarian carcinoma (Figure 1) [1], and further underscore the importance of recognition of disease heterogeneity in ovarian cancer research. The main conclusion from these studies is that ovarian tumors have very different clinical, histopathological and molecular features, and their pathogenesis differs significantly. For example, high-grade serous carcinoma, the most common and malignant type of ovarian cancer, is characterized by very frequent TP53 mutations and CCNE1 (encoding cyclin E1) amplification, while low-grade serous carcinoma harbors either KRAS, BRAF or ERRB2 mutations in approximately two-thirds of cases. Clear-cell carcinoma is unique for its high percentage of PIK3CA activating mutations when purified tumor samples and cell lines are analyzed. Endometrioid carcinoma has unique aberration in a signaling pathway invovling somatic mutations of CTNNB1 (encoding β-catenin), PTEN and PIK3CA. Mucinous ovarian tumor has KRAS mutations in nearly half of the specimens. Since the molecular genetics of ovarian cancer has been recently summarized [1], in the following sections, we will mainly focus on reviewing upregulated genes in ovarian cancer in high-grade (conventional) serous carcinoma, the most common and lethal type of ovarian cancer. Their possible mechanisms in contributing to tumor progression in ovarian cancer are illustrated in Figure 2.
Figure 1. The main molecular genetic alterations in different types of ovarian epithelial carcinomas.
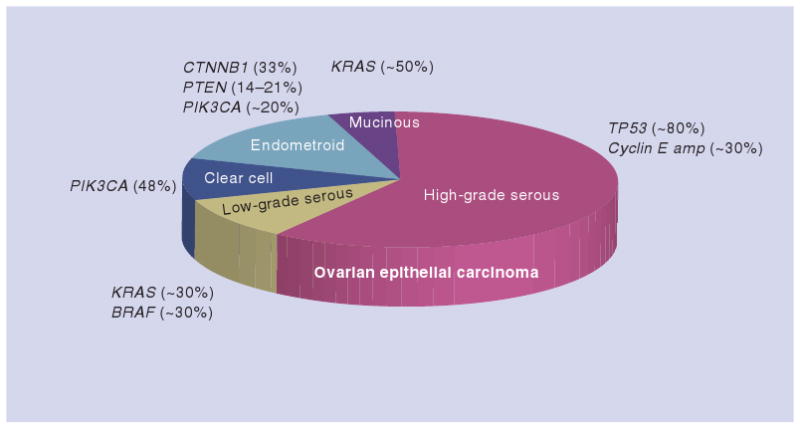
The percentage of each genetic change is estimated based on several published studies. The volume of different histologic types of ovarian cancer reflects their approximate prevalence in ovarian cancer patients.
Figure 2. Possible functions of ovarian cancer-associated genes as discussed in this article.

The overexpressed genes in ovarian cancer contribute to tumor progression by participating in several cellular processes including proliferation (cell-cycle progression), prevention of apoptosis, tumor invasion and metastasis, escape from anoikis (anchorage dependent growth), immune evasion, DNA damage repair and lipid synthesis. Several gene products, including HLA-G, mucin-4, FASN, folate receptor-α and osteopontin are secretory, and they may serve as serum biomarkers for cancer detection. Moreover, some upregulated proteins (Table 1) are potential therapeutic targets, as the reagents that inhibit their gene functions are available for current and future clinical applications. The arrow indicates a positive regulation and the block a negative regulation. The dashed lines denote nuclear translocation of the cytoplasmic or membrane proteins.
FASN: Fatty acid synthase; HLA-G: Human leukocyte antigen-G; NICD3: Intracellular domain of Notch3.
Notch3
Notch protein is an evolutionarily conserved membrane receptor involved in several fundamental biological processes including cell fate regulation, cell proliferation and cell death during development [8]. The basic molecular constituents in the Notch pathway are DSL ligands, Notch receptors and nuclear effectors (reviewed in [9]). In mammals, there are four members of receptor, Notch1–4, and five ligands including Jagged1–2, and δ-like 1, 3 and 4. Interaction between a ligand and the N-terminal EGF-repeat region of the Notch extracellular domain (ECD) initiates a conformational change in the receptor, triggering two sequential proteolytic cleavages by ADAM-family metalloproteases and γ-secretase at the juxta-membrane and intra-membrane sites of the receptor. This results in protease cleavages and the release of the Notch intracellular domain (NICD), which is then translocated into the nucleus where it cooperates with the DNA-binding protein CSL to activate transcription of Notch downstream effectors (Figure 3).
Figure 3. Correlation of 11q13.5 amplification and overall survival in ovarian serous carcinoma patients.
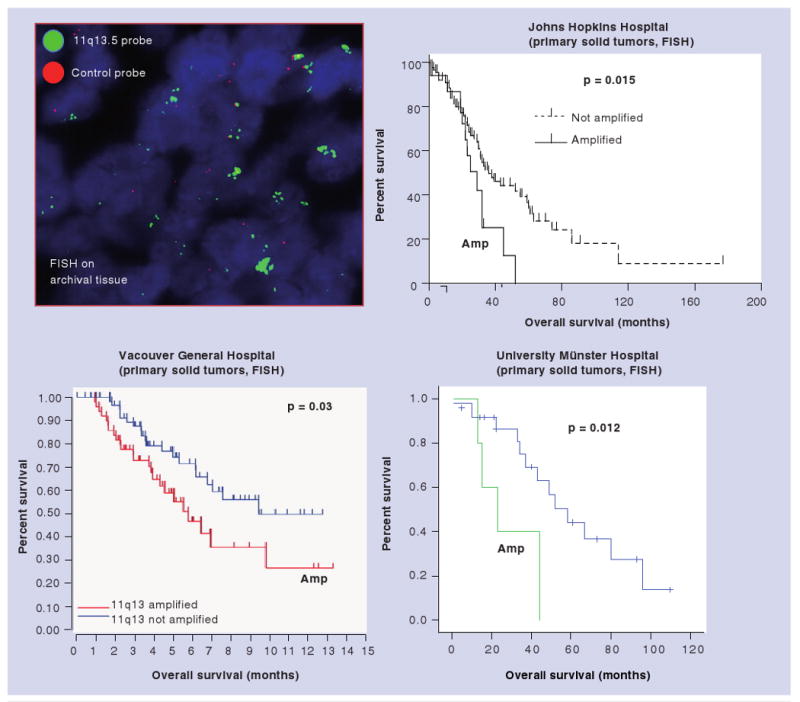
The 11q13.5 amplicon contains several genes including HBXAP (Rsf-1). Three independent studies apply the fluorescent in situ hybridization probes to detect the copy number of the 11q13.5 locus and demonstrate a significantly shorter overall survival in patients whose tumors harbor 11q13.5 amplification.
The figures from the Vancouver General Hospital (BC, Canada) and the University of Münster (Münster, Germany) are courtesy of data from Dr Huntsman and Dr Staebler, respectively.
FISH: Fluorescent in situ hybridization.
Among all Notch receptors, Notch3 has been found to participate in the development of ovarian serous carcinoma. Based on both digital karyotyping [2] and SNP array analyses [3] that assess genome-wide DNA copy number changes, Park et al. have identified Notch3 gene (19p13.12) amplification in 19.5% of ovarian high-grade serous carcinomas [10]. Notch3 DNA copy number was positively correlated with Notch3 protein expression based on immunohistochemistry and fluorescent in situ hybridization (FISH) studies [10]. Functional inactivation of Notch3 by either γ-secretase inhibitor or Notch3-specific siRNA resulted in suppression of cell proliferation and induction of apoptosis in the cell lines that overexpressed Notch3, but had no effect in cell lines that expressed minimal amounts of Notch3 [10]. These studies suggest that constitutive Notch3 expression due to gene amplification or epigenetic activation participates in the development of ovarian serous carcinomas. The Notch3 signaling pathway is amenable to targeted therapy. This can occur in multiple steps, including disruption of Notch3 and ligand binding using decoy proteins and inhibition of γ-secretase by small compound inhibitors. Future clinical trials have been planned to address if γ-secretase inhibitor has any clinical benefit in ovarian cancer patients.
HBXAP (Rsf-1)
Amplification of the chromosome 11q13.5 locus is frequently detected in several types of human cancer, including ovarian serous carcinoma, of which 11q13.5 amplification was associated with significantly shorter overall survival in ovarian cancer patients [11]. In order to explore the molecular mechanisms by which amplification of this locus contributes to disease aggressiveness, investigators have screened the candidate genes within this amplicon for their contribution to drug resistance [12]. Hepatitis B virus x-associated protein (HBXAP [Rsf-1]) was found to be the only gene in which knockdown sensitized tumor cells to paclitaxel. They also found that HBXAP (Rsf-1) was upregulated in paclitaxel-resistant ovarian cancer cell lines and HBXAP (Rsf-1) immunoreactivity in primary ovarian carcinoma tissues correlated with in vitro paclitaxel resistance [12]. Overexpression of HBXAP (Rsf-1) significantly enhanced paclitaxel resistance in ovarian cancer cells. Downregulation of hSNF2H or disruption of hSNF2H and HBXAP (Rsf-1) interaction enhanced paclitaxel sensitivity in tumor cells with HBXAP (Rsf-1) upregulation. HBXAP (Rsf-1) expression regulated expression in several genes and activated certain signaling pathways that may contribute to drug resistance.
HBXAP encodes a cellular nuclear protein that contains several distinct structure motifs including the PHD domain [13]. HBXAP was first identified as a novel cellular protein that bound to the pX nuclear protein of hepatitis B virus [14]. Almost at the same time as when HBXAP was identified, an independent research group also identified the full-length HBXAP protein as a subunit (Rsf-1) in a chromatin assembly factor, called ISWI-containing factor remodeling and spacing factor (RSF) [15]. The human RSF complex is composed of two subunits: the ATPase hSNF2H and p325 (Rsf-1) [15,16]. Recent studies have indicated that HBXAP (Rsf-1) plays a role in chromatin remodeling [15] and transcriptional regulation [13,14]. HBXAP has been shown to function as a histone chaperone in the nuclei, while its binding partner, hSNF2H, possesses nucleosome-dependent ATPase activity [17]. The HBXAP/hSNF2H complex (RSF complex) mediates ATP-dependent chromatin remodeling, which alters the chromatin structure or positioning of nucleosomes. At the cellular level, RSF participates in chromatin remodeling by mobilizing nucleosomes in response to a variety of growth-modifying signals and environmental cues. Such nucleosome remodeling is essential for transcriptional activation or repression [18], DNA replication [19] and cell-cycle progression [20].
In ovarian cancer, 11q13.5 amplification in ovarian serous carcinomas contributes to shorter overall survival as compared with those without 11q13.5 amplification (Figure 2). It is likely that tumors with increased DNA copy number of Rsf-1 overexpress Rsf-1 protein that renders the de novo paclitaxel resistance. The HBXAP (Rsf-1) expressing residual tumors after tumor cytoreduction surgery may survive better during chemotherapy and tend to recur earlier, and thus they are likely related to poor clinical outcome in ovarian cancer patients. This finding that paclitaxel resistance in HBXAP (Rsf-1) upregulated cells is mediated by the chromatin remodeling activity from the HBXAP (Rsf-1)/hSNF2H complex may have translational implications for new cancer therapy, as enhancing the sensitivity of chemotherapy can be achieved by inactivating the HBXAP (Rsf-1)/hSNF2H or by interrupting the complex formation in cancer cells [12]. Third, hSNF2H has been shown to be required for cell proliferation and survival in HBXAP (Rsf-1) expressing OVCAR3 cells, but not in SKOV3 cells with or without Rsf-1 induction. This finding is probably because OVCAR3 cells with HBXAP (Rsf-1) gene amplification and constitutive upregulation have become molecularly ‘addicted’ to HBXAP (Rsf-1)/hSNF2H expression, while SKOV3 cells that have a very low level of endogenous HBXAP (Rsf-1) expression are less sensitive to hSNF2H knockdown [11].
NAC1
NAC1 encoded by NACC1 belongs to the BTB/POZ domain gene family and contains the BEN domain that potentially mediates protein–DNA and protein–protein interactions during chromatin organization and transcription [21]. NAC1 was first identified and cloned as a novel transcript in which the gene is induced in the nucleus accumbens, a unique forebrain structure involved in reward motivation and many addictive behaviors [22–26]. In human cancer, the role of NAC1 has been recently revealed [27–31]. NAC1 is significantly overexpressed in several types of carcinomas, including ovarian serous carcinomas. NAC1 proteins are localized in discrete nuclear bodies. Furthermore, Nakayama et al. demonstrate that intense NAC1 immunoreactivity in primary ovarian tumors predicts early recurrence. NAC1 immunointensity was significantly higher in post-chemotherapy recurrent ovarian carcinomas than primary pre-chemotherapy specimens [28,31]. Furthermore, NAC1 expression in 62 post-chemotherapy effusions predicts poor progression-free survival [31]. More recently, we have identified an increase in the DNA copy number at the NACC1 locus (chromosome 19p13) that encodes NAC1 in approximately a third of ovarian serous carcinomas using SNP arrays and FISH analysis [3], suggesting that increased gene copy number is one of the mechanisms for NAC1 overexpression in ovarian carcinomas.
Like several BTB/POZ family members, NAC1 proteins homodimerize through the BTB/POZ domain. Induced expression of the NAC1 deletion mutant (N130), exclusively containing the BTB/POZ domain, disrupts NAC1 nuclear bodies, prevents tumor formation and promotes tumor cell apoptosis in a mouse tumor xenograft model. Overexpression of full-length NAC1 is sufficient to enhance tumorigenecity of ovarian surface epithelial cells and NIH3T3 cells in athymic nu/nu mice [28]. Nucleotide sequencing in purified ovarian cancer tissues fails to reveal somatic mutations in the NAC1 open reading frame [Shih I-M, Johns Hopkins University School of Medicine. Unpublished data]. The above studies on ovarian cancer suggest that NAC1 is an ovarian tumor recurrence-associated gene with oncogenic potential, and the interaction between BTB/POZ domains of NAC1 proteins is essential for tumor cell proliferation and survival in those tumors with NAC1 overexpression. The molecular mechanism by which NAC1 contributes to cellular survival and growth awaits further determination. Preliminary studies have demonstrated that NAC1 negatively regulated the components of the Gadd45 tumor suppressor pathway, including Gadd45α and its binding protein, Gadd45gip1 [29,30].
Fatty acid synthase
Fatty acid synthase (FASN) encodes an approximately 260-kD cytoplasmic enzyme that is responsible for all biochemical processes involving the de novo fatty acid synthesis by catalyzing the NADPH-dependent condensation of malonyl-CoA and acetyl-CoA to palmitate [32]. Only a minimal amount of FASN is expressed in normal adult tissues due to the presence of abundant dietary lipids. In contrast, elevated FASN levels are detected in a variety of neoplastic diseases including breast, colon, ovary and prostate cancer [33–39], as tumor cells become less sensitive to regulatory nutritional signals and prefer the de novo lipogenesis pathway. Furthermore, FASN expression occurs early in cancer development, and its level is highly correlated with the clinical aggressiveness of the tumors [33,34,40–43].
Several reports have provided cogent evidence that FASN is upregulated in ovarian serous carcinomas [32,34,44]. The most remarkable finding of FASN expression in ovarian cancer is its association with recurrent disease and overall survival [34] [Ueda S, Shih IM: Proteomic analysis of NAC1 regulated proteins in ovarian cancer. Manuscript submitted]. FASN protein levels increase in recurrent serous carcinomas as compared with their matched primary tumor specimens [Ueda S, Shih IM. Manuscript submitted]. Multivariate analysis showed that a higher FASN staining score in serous carcinomas was associated with a worse overall survival time. By analyzing the proteomes of SKOV3 cells with and without expression of a dominant-negative NAC1 (N130) that inhibits NAC1 homodimerization and thus inactivates NAC1-mediated functions, Ueda et al. recently found that FASN is one of the downstream target genes regulated by NAC1 [Ueda S. Unpublished data]. Similar to the protein level, the FASN transcript level in SKOV3 cells was significantly reduced by N130 induction or by NAC1 knockdown. Immunohistochemistry showed that NAC1 and FASN immunointensity in ovarian serous carcinoma tissues had a highly significant correlation. Moreover, C93, a new FASN inhibitor, induced massive apoptosis in carboplatin-/paclitaxel-resistant ovarian cancer cells. The recent studies provide new evidence that NAC1 and its dimerization are essential for FASN expression in ovarian serous carcinomas, since the FASN level significantly correlates with tumor recurrence and disease aggressiveness in these cells. The dependence of drug-resistant tumor cells on FASN suggests a potential application of FASN-based therapeutics for recurrent ovarian cancer patients. The mechanism by which FASN participates in tumor progression remains unclear. It is likely that promotion of de novo synthesis of long-chain fatty acid is important for those tumor cells overexpressing FASN [45]. Besides, a recent study demonstrated that the antitumor effects of FASN inhibitors in ovarian cancer cells may also be in part mediated by modulating other signaling pathways, including AMP-activated kinase, AKT and ErbB2 [46–49], suggesting an alternative role of the metabolism-independent pathway involved in FASN-mediated tumor development (Figure 2).
Mucin-4
Mucin-4 (Muc4) has become an emerging cancer marker for diagnosis and a novel target for therapy. Muc4 is a heavily glycosylated protein composed of noncovalently linked heterodimeric Muc4α and Muc4β subunits that are derived from a single gene product after proteolysis during biosynthesis. In adult normal tissues, Muc4 expression is detected on the apical surface of many epithelial tissues, such as the female reproductive tract, airway, breast and colon. However, Muc4 overexpression has been found in a variety of carcinomas, including breast, ovarian, lung, salivary gland, pancreatic and bile duct carcinomas. An association between Muc4 expression and poor survival has been reported in certain types of carcinomas [50]. Ecotpic expression of Muc4 induces oncogenic transformation of NIH3T3 cells in vitro [51]. Recent studies have demonstrated that Muc4 is involved in tyrosine kinase receptor-related signal transduction [52], and Muc4 expression can inhibit apoptosis induced by multiple insults including chemotherapeutic agents and the loss of cellular adhesion via ErbB2-dependent and ErbB2-independent mechanisms [53]. In ovarian cancer, Muc4 was detected in almost all ovarian carcinoma samples including cancer effusions (Figure 3), while normal ovarian surface epithelium and reactive mesothelial cells were Muc4-negative in all specimens. Muc4 expression in ovarian carcinoma cells in effusions was significantly higher than in solid tumors; however, there is a lack of association between Muc4 expression and other clinicopathologic parameters, including survival [54]. Molecularly, inhibition of Muc4 expression suppresses pancreatic tumor cell growth and metastasis [55,56]. Like in pancreatic cancer cells, Muc4 directly interacts with and stabilizes HER2 in ovarian cancer cells, and it has been demonstrated that Muc4 activates ERRB2 (Her2) signaling and enhances the motility of human ovarian cancer cells in vitro [57], probably through phosphorylation of focal adhesion kinase (FAK), Akt and ERK, downstream effectors of Her2.
Mesothelin
Mesothelin has been known as a cell-surface marker for cancer diagnosis and target-based therapy. Besides pancreatic carcinoma and malignant mesothelioma, ovarian cancer is one of the most common types of carcinoma that overexpress mesothelin [58–63]. Upregulation of mesothelin can be important in the formation of tumor ascites in ovarian cancer patients, as mesothelin has been demonstrated to promote anchorage-independent growth and prevents anoikis via the extracellular signal-regulated kinase signaling pathway (Figure 3) [64]. Mesothelin immunoreactivity (>5% of tumor cells) was present in 55% of ovarian serous carcinomas [63]. Kaplan–Meier analysis demonstrated that a diffuse mesothelin staining (>50% of tumor cells) in primary high-grade ovarian carcinomas significantly correlated with prolonged survival in patients who had advanced stage disease and had received optimal debulking surgery followed by chemotherapy [63]. It is of great interest to note that a longer survival only occurred in tumors with diffuse mesothelin expression (>50%), but not in those with focal expression. This finding implies that a tumor containing greater numbers of mesothelin-positive tumor cells was more likely to elicit a response from the immune system, whereas a tumor with focal mesothelin-positive cells would continue to progress unchecked as the majority of tumor cells fail to express the tumor antigen. This view is supported by the following evidence. A humoral response to mesothelin has been found in patients who suffered from ovarian cancer and mesothelioma [65]. Elevated levels of mesothelin-specific antibodies were detected in the sera of 41.7% of patients with ovarian cancer and the immunogenicity of mesothelin is associated with high mesothelin expression in tumor cells. Besides the humoral response, a cell-mediated immune response may also play an important role. It has been reported that vaccination with a GM-CSF-transduced allogenic pancreatic tumor vaccine induced human mesothelin-specific CD8+ T cells in pancreatic cancer patients [66]. Among those patients who received the vaccine, three of 14 patients had prolonged survival. It is also noted that among seven pancreatic tumor genes identified by serial analysis of gene expression as overexpressed on pancreatic cancer, only mesothelin was recognized by T cells from the three patients, but not by T cells from other patients. Therefore, the correlation between the generation of vaccine-dependent T-cell responses to mesothelin and long-term survival argues that it is a promising candidate for mesothelin-specific immunotherapy [67]. Alternatively, the cell–cell adhesion mediated by mesothelin and mucin 16 (CA125 molecules) [68] may lead to cohesive tumor growth in ovarian carcinoma, which expresses mesothelin and prevents cancer cells from dissemination or metastasis in the peritoneal cavity. Recently, a fully humanized antimesothelin monoclonal antibody, M912, has been developed, and the antibody can specifically attack cancer cells engineered to express mesothelin in the presence of peripheral blood mononuclear cells isolated from healthy donors, probably through antibody-dependent cellular cytotoxicity. Future clinical trials may determine if M912 has a potential for cancer treatment in patients with pancreatic and ovarian cancer [69].
Apolipoprotein E
Besides its well-known role in lipid transportation, apolipoprotein (apo) E may also act as a potential tumor-associated marker in ovarian cancer. In a previous study [70], apoE expression was frequently detected in ovarian serous carcinomas, but was not detected in serous borderline tumors and normal ovarian surface epithelium. Inhibition of apoE expression using an apoE-specific siRNA led to G2 cell-cycle arrest and apoptosis in apoE-expressing ovarian cancer cells, but not in apoE-negative cell lines. Clinically, expression of apoE in nuclei was significantly associated with a better survival in patients who presented with peritoneal effusions at the time of diagnosis [70]. This finding indicates that apoE-expressing tumor cells are more sensitive to chemotherapeutic agents. One of the possible mechanisms underlying a new role of apoE in cancer cell proliferation and survival is that endogenous apoE molecules enhance the lipid transport into tumor cells through the low-density lipoprotein (LDL) receptor family members that are expressed in ovarian cancer cells. Besides, apoE mediates signal transduction upon binding to the LDL receptor family members. This autocrine loop results in activation of the downstream survival signals. This view is supported by a number of studies showing that apoE, upon binding to LDL receptor family members, initiates cell signaling and exerts a variety of biological effects [71–73]. It can be speculated that during tumor progression, some ovarian carcinomas overexpress apoE and become molecularly dependent on the strategy by overexpressing to maintain cell survival through the autocrine mechanism of ligand-receptor binding. Therefore, inhibition of apoE expression inactivates the apoE-related pathway(s), resulting in cell-cycle arrest and apoptosis. In addition to lipid transport and signal transduction as described above, apoE may modify the tumor microenvironment to maintain tumor proliferation and survival, as a previous study has demonstrated that apoE induces expression of subendothelial heparan sulfate proteoglycan [74], which may be critical for apoE in preventing programmed cell death [75]. Studies on apoE knockout mice [76–78] have further suggested that macrophage-derived apoE interacted with the extracellular matrix, and that such interactions result in the retention of lipoproteins in the vessel wall and affect the bioavailability of cytokines and growth factors sequestered in the extracellular matrix [79]. Future studies are necessary to assess if a similar paracrine mechanism exists in ovarian carcinomas.
Kallikreins
Kallikreins are serine proteases that are involved in a variety of physiological processes from tissue remodeling to pathogenesis of neoplastic diseases [80]. Numerous studies have reported specific types of kallikreins that are overexpressed in the major types of carcinomas [81]. For example, kallikreins 4, 5, 6, 7, 9, 10, 13 and 14 have been reported to be biomarkers associated with ovarian, breast, prostate and testicular cancer [82–84]. Recent studies further suggest that kallikreins directly contribute to the development of cancer by participating in extracellular matrix degradation, cellular invasion and metastasis [81,85–87]. Cancer cells may utilize kallikreins to transform the tissue microenvironment, peritoneal walls for example, and facilitate tumor implantation and dissemination. All kallikreins are secreted proteins and can be detected in body fluids, suggesting a role of kallikreins in cancer diagnosis [82,88–90]. Indeed, kallikrein 3, also known as prostate specific antigen (PSA), is one the most useful tumor markers for cancer screening, diagnosis, prognosis and monitoring.
In ovarian cancer, kallikrein 4 is expressed in the majority of ovarian carcinoma cells, and its expression level could be associated with paclitaxel resistance [91]. However, expression of kallikrein 4 in carcinoma cells of effusions or solid tumors from ovarian cancer patients does not significantly correlate with survival [92]. Based on ELISA, profiling of secreted kallikreins in the supernatants of cancer effusions has shown that kallikrein 6, 7, 8 and 10 are the major secreted kallikreins associated with ovarian cancer effusions [93,94]. This is supported by the higher gene expression of these kallikreins in serous ovarian/peritoneal carcinoma compared with malignant peritoneal mesothelioma based on gene-expression profiling [95]. Furthermore, kallikreins 6, 7, 8 and 10 showed the highest statistical power in distinguishing ovarian cancer from benign controls and other cancer groups and these kallikreins could diagnose false-negative cases based on cytology [93]. The mechanisms of increased levels of kallikreins in ovarian cancer body fluids remain to be determined. Elevated kallikrein expression is probably a molecular genetic event or a result of promoter activation by the estrogen receptor signaling pathway [96]. It could be envisioned that kallikreins in tumor effusions may contribute to the formation of effusion because kallikreins may enhance the permeability and/or destroy the angiolymphatic vessels on the peritoneal wall. Further studies are required to determine the molecular mechanism by which kallikreins promote ovarian cancer growth, and assess if ovarian cancer-specific kallikreins can be used in target-based therapy and in prevention of ovarian cancer effusions. Upregulation in several kallikreins, including kallikrein 4, 5, 6 and 10, has been shown to associate with a worse clinical outcome (shorter disease-free interval and overall survival) in ovarian cancer patients, although conflicting results have also been reported [92,97].
Claudin
Members of the claudin family function as tight junction proteins, which are present at epithelial and endothelial cell interface. Upregulation of some claudin members has been found in several malignancies. In ovarian cancer, claudin overexpression was first identified by Morin et al. using serial analysis of gene expression to profile the transcriptome in ovarian serous carcinomas [59,98], and this initial finding was subsequently validated by several reports [61,95,99–101] (please see a review article [102]). These profiling studies demonstrated that claudin-3, -4 and -7 are the most abundant and consistently expressed claudins in ovarian cancer. Indeed, transcriptome analysis has shown that claudin-3 and claudin-4, and the kallikrein family of proteins, were among the most highly overexpressed genes in ovarian carcinoma when compared with human ovarian surface epithelial cells [101]. Further study has demonstrated that claudin overexpression may promote ovarian tumorigenesis and metastasis through increased invasion and survival of tumor cells through a matrix metalloproteinase-2 mediated pathway [103]. Quantitative proteomic analysis, together with mRNA expression levels, demonstrated that claudin-4 was associated with cisplatin resistance in ovarian cancer cells [104]. Claudin-3 and claudin-7 expression in effusions have been found to be independent predictors for poor survival in patients with ovarian cancer effusions [105]. As claudin-3 and -4 have been reported as the low- and high-affinity receptors, respectively, for the cytotoxic Clostridium perfringens enterotoxin (CPE), investigators have tested the hypothesis that both claudins are potential therapeutic targets in ovarian cancer, especially for those developing chemoresistance. The initial data has shown that CPE-based therapy has shown efficacy in inhibiting xenograft tumor growth in a mouse model of chemotherapy-resistant freshly explanted human ovarian cancer [106]. As claudin-3 and -4 are elevated and secreted in many ovarian carcinomas, they may represent useful biomarkers for detection and prognosis, in addition to serving as ideal targets for therapy.
HLA-G
Upregulation of human leukocyte antigen-G (HLA-G) in many human cancers exemplifies similar strategies used by the feto–placental unit and cancer to evade immune response. Like other cancer types, ovarian tumor cells take advantage of a similar molecular strategy to escape from immune recognition in response to the expression of a variety of tumor-associated antigens. The molecular mechanisms shared by feto–placental and cancerous tissues to counteract immune responses remains enigmatic, but it has been shown that the expression of nonclassical major histocompatibility (MHC) antigens plays an important role [107]. HLA-G, a nonclassical MHC molecule, has been well established as a molecule involved in immune regulation, especially in immune tolerance. Several studies have provided cogent evidence of HLA-G expression in human ovarian tumor tissues, and the potential roles of HLA-G as a novel biomarker in cancer detection, diagnosis and prediction of clinical outcome in ovarian cancer patients [107]. HLA-G expression has been shown to facilitate tumor cell evasion from the immune system, as both membrane-bound and secreted forms of HLA-G (sHLA-G) protect malignant cells from being attacked by immune cells [107]. Besides, sHLA-G may suppress angiogenesis through an apoptotic pathway and by direct binding to CD160 receptor expressed by endothelial cells [108].
Several studies have attempted to correlate HLA-G expression with a patient's clinical outcome, including overall survival and the risk of developing metastatic diseases. It turns out that HLA-G expression can be associated with either a favorable or an unfavorable clinical outcome, depending on cancer types. In ovarian carcinomas, HLA-G expression in effusions is associated with tumor susceptibility to chemotherapy [109]. In that study, the immunoreactivity of HLA-G expression in ovarian cancer cells was significantly lower in effusions obtained during or after chemotherapy than that without chemotherapy. Moreover, the percentage of HLA-G-positive tumor cells obtained prior to chemotherapy significantly correlated with better overall survival. The above finding appears paradoxical to the expected role of HLA-G in propelling tumor progression. It is possible that the HLA-G-positive cells may become more sensitive to chemotherapeutic agents or other treatment modalities, but this awaits further studies for confirmation.
Singer et al. applied a sensitive ELISA to determine sHLA-G levels in the supernatant of ascites from ovarian and breast carcinoma and benign controls [110]. They found that malignant ascites contained higher levels of sHLA-G than benign ascites. The investigators based on receiver operating characteristic (ROC) curves, and demonstrated an excellent performance of HLA-G ELISA as a diagnostic tool to detect ovarian and breast cancer. The presence of sHLA in ovarian carcinoma tissues strongly suggests that sHLA-G can be used as a tumor marker in blood for cancer detection. In fact, the serum levels of sHLA-G were elevated in patients with a variety of neoplastic diseases including ovarian cancer [111]. Although this study was preliminary, it provides another promising biomarker for future clinical trials to determine its potential clinical applications, especially for its role in detecting early-stage and curable diseases. Further studies are required to assess whether a combination of selected secreted biomarkers, including sHLA-G, may provide a better strategy in increasing both sensitivity and specificity than single markers alone for cancer detection in body fluids.
Folate receptor-α
Folate receptor (FR) mediates receptor-dependent cellular uptake of folate (vitamin B9), which is involved in one-carbon transfer reactions that are required for RNA and DNA synthesis. Among different FRs, FR-α has been extensively studied in human cancer. In normal tissues, FR-α has a very limited tissue distribution, while in neoplastic tissues, FR-α can be detected in carcinomas arising from different organs including nonmucinous ovarian carcinoma, endometrial carcinoma and cervix carcinoma, as well as in a variety of nongenital tract tumors. In ovarian cancer, over 90% of nonmucinous types of ovarian cancer overexpress FR-α, and the higher level of FR-α expression correlates with the higher grade of malignancy [112]. The high expression of FR-α in cancer has led to extensive research regarding its potential role as a target for molecular therapy and as a diagnostic marker. It has been demonstrated that FR immunoreactivity may be useful in differential diagnosis of ovarian carcinoma from malignant mesothelioma and metastatic breast cancer, morphologic mimickers of ovarian cancer. However, FR expression in ovarian cancer effusions does not predict drug response or survival [113]. A recent study has demonstrated that ovarian cancer patients have elevated levels of functional intact FR-α in their blood samples, a finding supporting the potential use of circulating FR-α as a biomarker of early ovarian cancer [114]. The high expression of FR in ovarian cancer nevertheless supports efforts to investigate their validity as molecular therapeutic targets in this disease. To this end, a humanized monoclonal antibody directed to FR-α, MORAb-003, has been developed [115], and an early-phase clinical trial has demonstrated good tolerability and promising clinical benefit in ovarian cancer patients. Besides the antibody approach, BGC 945, a thymidylate synthase inhibitor preferentially transported into FR-α-overexpressing tumors, may have selected antitumor effects in FR-α-positive tumors, of which its clinical promise awaits further studies [116].
NF-κB transcription factor
The binding of NF-κB transcription factors to κB elements in promoters and enhancers can either induce or repress gene expression, affecting processes such as cell proliferation, inflammation, immune response and programmed cell death (reviewed in [117]). The five NF-κB family members RelA (p65), RelB, c-Rel, p50/p105 (NF-κB1) and p52/p100 (NF-κB2) form homodimers or heterodiomers, and are typically retained in the cytoplasm in complex with inhibitors of NF-κB (IκBs). Upon cell stimulation, the IκBs are phosphorylated in a site-specific manner by activated IκB kinase (IKK) complexes, the most common consisting of IKKα and IKKβ and of a regulatory IKKγ subunit, also named NF-κB essential modulator (NEMO) [117].
In the ‘canonical’ NF-κB pathway, activated by TNF-α, IL-1 and other stimuli, IκBα is phosphorylated at ser32 and ser36 predominantly by IKKβ, and is subjected thereafter to ubiquitination and degradation in the 26S proteosome. This releases the NF-κB p65/p50 heterodimer to translocate into the nucleus. Additional NF-κB activation pathways have been described, and their diversity, in addition to the existence of hundreds of NF-κB activators and target genes [118], further contributes to the myriad of cellular functions and effects of NF-κB.
Post-translational modifications regulate the activity of the IKK complex, IκB proteins and the NF-κB subunits themselves, among them phosphorylation of NF-κB p65 at Ser536, which results in enhanced transactivation potential and nuclear translocation of NF-κB p65 [119]. NF-κB activation depends on the cellular setting and can result in both cell survival and apoptosis/necrosis. Pro-survival NF-κB signaling involves increased proliferation via cyclin D1 activation, suppression of the JNK cascade and induction of angiogenesis, invasion and metastasis by upregulating the expression of molecules such as VEGF, VCAM-1 and MMPs [120]. NF-κB is activated by many oncogenes, for example, HER-2 and HRAS, and the PI3K/AKT pathway has been implicated in this process. NF-κB additionally confers resistance to cell death through upregulation of the expression of antiapoptotic molecules, including the inhibitor of caspase activation cFLIP, inhibitor of apoptosis (IAP) family members (cIAP1, cIAP2 and XIAP), and Bcl-2 family members (A1/Bfl1 and Bcl-XL) [121,122]. Data available from in vitro and in vivo ovarian carcinoma models suggest that inhibition of NF-κB enhances the efficacy of cisplatin and paclitaxel in the treatment of this tumor [123,124].
NF-κB activation has been known to be associated with tumor progression in ovarian cancer. More recently, Klienberg et al. have studied NF-κB p65 and IκBα protein expression in ovarian carcinoma effusions and corresponding primary carcinomas and solid metastases [125]. Nuclear NF-κB p65 expression, indicating NF-κB activation, was detected in the majority of tumors at all anatomic sites, with less frequent cytoplasmic NF-κB p65 and IκBα expression. Comparative analysis of matched effusions, primary carcinomas and solid metastases demonstrated a higher percentage of cells expressing nuclear NF-κB p65, cytoplasmic NF-κB p65 and IκBα in the solid lesions compared with effusions. NF-κB p65 phosphorylation at Ser536 was found in 94% of the effusions using western blotting. In effusions, high (>25% of cells) nuclear NF-κB p65 expression was significantly associated with a larger volume of residual disease and poor response to chemotherapy at disease recurrence. Nuclear NF-κB p65 expression correlated with poor progression-free survival in univariate survival analysis. Nuclear NF-κB p65 expression was an independent predictor of shorter progression-free survival in Cox analysis. These data suggest that NF-κB p65 is frequently expressed and activated in advanced-stage ovarian carcinoma. The observation that nuclear NF-κB p65 expression in effusions is associated with poor progression-free survival supports its involvement in ovarian cancer cell survival.
Osteopontin
Osteopontin (OPN) is a cell surface and secretory glycoprotein containing an arginine–glycine–aspartate (RGD) motif. Osteopontin plays a critical role in a variety of physiological process from embryonic development, and wound healing, to tumor development. Osteopontin is important for cellular proliferation, metastasis and the prevention of apoptosis. Osteopontin is frequently expressed in ovarian cancer tissues, and Mok et al. first demonstrated osteopontin as a potential serum biomarker in ovarian cancer patients [126]. It has also been reported that osteopontin, together with other markers, may complement CA125 expression and serves as a new diagnostic marker panel in those cases with low or undetectable levels of CA125 [127,128]. The molecular mechanisms underlying the oncogenic role of osteopontin remain largely unknown. However, osteopontin has been demonstrated to promote cancer metastasis by enhancing cell survival and invasion through Akt-mediated HIF-1α upregulation and MMP9 activation [129,130]. Osteopontin has been know as a survival signal by activation of the PI3K/Akt pathway, with subsequent upregulation of Bcl-xL and activation of NF-κB, and may enhance angiogenesis to further promote tumor growth [131]. Recently, it has been shown that osteopontin is a substrate of caspase-8, and proteolytic cleavage by casepase-8 and its cleavage product subsequently induces cell death via the p53 pathway [132]. Like other cell-surface proteins, osteopontin appears as a potential target for the antibody-based therapies in human cancer. Indeed, an early but exciting study has suggested that the humanized antiosteopontin antibody, hu1A12, may be a promising therapeutic agent for the treatment of human breast cancer, and perhaps other types of cancer including ovarian cancer, with osteopontin upregulation [133].
Major challenges in ovarian cancer research
With the advent of a large amount of data derived from genome-wide-based analysis, especially from TCGA, ovarian cancer research has been entering an era replete with tumor-associated genes and protein markers. How to efficiently characterize those ovarian cancer genes in a biological context and, more importantly, translate the results from gene discovery to clinical applications remains a daunting task that will continue to challenge scientists and physicians in the decades to come. The overexpressed genes as exemplified above clearly illustrate their promise as new diagnostic markers and therapeutic targets. Nevertheless, substantiating their clinical applications depends on our further understanding of the mechanisms behind how those gene products promote tumor progression. There are at least three major issues concerning the current and future studies of molecular etiology in ovarian cancer from the perspective of ovarian cancer-associated genes. First, as discussed above, it has now been established that ovarian carcinomas are a heterogeneous group of diseases, each with a distinct molecular pathogenesis (Figure 1) [1]. Unfortunately, many previous studies have not taken the heterogeneity of ovarian cancer into serious account, as they have combined different histologic subtypes in their molecular analysis. As a consequence, the results may not be reproducible and the biological and clinical significance of genes of interest remains unclear. Second, despite numerous studies that have focused on scrutinizing the ovaries for precursor lesions, the origin of ovarian cancer has perplexed investigators for decades. Recent morphologic and molecular genetic studies have suggested the fallopian tube epithelium rather than the long-thought ovarian surface epithelium as the possible origin of some serous carcinomas. The implication of such findings challenges many of the previous reports demonstrating the ‘overexpressed’ ovarian cancer-associated genes in which their expression levels in carcinoma are always compared with their ‘normal’ counterparts, ovarian surface epithelium. As the gene-expression profiles in ovarian surface epithelium, which is of the mesothelial origin, are distinct from fallopian tube epithelium, which is of the Müllerian origin, whether the overexpressed genes previously reported are indeed upregulated when they are compared with a more likely origin of ovarian serous carcinoma, that is, fallopian tube epithelium, should be rigorously revisited [134]. Third, the majority of previous studies on ovarian cancer-associated genes have focused on primary tumors. However, the mortality and morbidity of most ovarian carcinomas come from recurrent chemoresistant diseases, rather than the primary tumors that are usually excised. Thus, understanding and targeting the molecular underpinnings of recurrent diseases should represent another main focus in studying ovarian cancer associated genes in order to gain more therapeutic options for chemoresistant recurrent tumors. Given the fact that the main focus of TCGA is on the primary ovarian serous carcinoma, future efforts in studying the pathogenesis and molecular targets should be devoted to analyze their recurrent counterparts also.
Executive summary.
Recent advances in the pathogenesis of ovarian cancer
A number of new ovarian cancer-associated genes and pathways have been identified, and they provide new insights into how ovarian cancer develops.
Several overexpressed proteins in ovarian cancer are secreted and are promising serum biomarkers for early detection when used in combination.
Exciting progress has suggested many ovarian cancer overexpressed genes are potentially targetable and their inhibitors, including neutralizing antibodies and enzyme inhibitors, may serve as a new wave of new cancer drugs in some ovarian cancers.
Challenges in current & future studies in ovarian cancer research
Ovarian cancer is unique among human cancer, as it is composed of several histological subtypes developing along distinct molecular pathways. It is essential to separate them in all clinical and basic studies.
Recurrent, chemoresistant tumors, rather than their primary counterparts, are the main cause of mortality. These tumors should be included in molecular studies of ovarian cancer.
New evidence has suggested the fallopian tube is the precursor of ‘ovarian’ cancer, and challenges the long-standing paradigm that ‘ovarian’ cancer arises from the ovary. Whether the overexpressed genes previously reported are indeed upregulated when they are compared with a more likely origin of ovarian serous carcinoma, that is, fallopian tube epithelium, should be rigorously revisited.
Footnotes
Financial & competing interests disclosure: The authors have received the following grants: NIH/NCI RO1CA129080 and RO1CA103937. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Contributor Information
Ie-Ming Shih, Departments of Pathology and Oncology, Johns Hopkins University School of Medicine, 1550 Orleans Street, CRB-2, Baltimore, MD 21212, USA, Tel: +1 410 502 7774, Fax: +1 410 502 7943.
Ben Davidson, Division of Pathology, Norwegian Radium Hospital, Rikshospitalet University Hospital, Oslo, Norway and Faculty Division Radiumhospitalet, the Medical Faculty, University of Oslo, Oslo, Norway.
Bibliography
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.Cho KR, Shih IM. Ovarian cancer. Annu Rev Pathol Mech Dis. 2009;4:287–313. doi: 10.1146/annurev.pathol.4.110807.092246. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪ A comprehenisve review of recent advances in the pathogenesis of ovarian cancer research.
- 2.Wang TL, Maierhofer C, Speicher MR, et al. Digital karyotyping. Proc Natl Acad Sci USA. 2002;99:16156–16161. doi: 10.1073/pnas.202610899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakayama K, Nakayama N, Jinawath N, et al. Amplicon profiles in ovarian serous carcinomas. Int J Cancer. 2007;120:2613–2617. doi: 10.1002/ijc.22609. [DOI] [PubMed] [Google Scholar]; ▪ Describes the genome-wide DNA copy number changes in serous ovarian cancer using purified tumor samples.
- 4.Sturgeon CM, Duffy MJ, Stenman UH, et al. National Academy of Clinical Biochemistry laboratory medicine practice guidelines for use of tumor markers in testicular, prostate, colorectal, breast, and ovarian cancers. Clin Chem. 2008;54:E11–79. doi: 10.1373/clinchem.2008.105601. [DOI] [PubMed] [Google Scholar]; ▪▪ A comprehensive overview of protein biomarkers in ovarian cancer.
- 5.Shih IM, Sokoll LJ, Chan DW. Tumor markers in ovarian cancer. In: Diamandis EP, Fritsche HA, Lilja H, Chan DW, Schwartz MK, editors. Tumor markers physiology, pathobiology, technology and clinical applications. AACC Press; PA, USA: 2002. pp. 239–252. [Google Scholar]
- 6.Collinson F, Jayson G. New therapeutic agents in ovarian cancer. Curr Opin Obstet Gynecol. 2009;21:44–53. doi: 10.1097/GCO.0b013e32831ffe71. [DOI] [PubMed] [Google Scholar]
- 7.Yap TA, Carden CP, Kaye SB. Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer. 2009;9:167–181. doi: 10.1038/nrc2583. [DOI] [PubMed] [Google Scholar]
- 8.Shih IM, Wang TL. Notch signaling, γ-secretase inhibitors, and cancer therapy. Cancer Res. 2007;67:1879–1882. doi: 10.1158/0008-5472.CAN-06-3958. [DOI] [PubMed] [Google Scholar]; ▪▪ A succinct review to summarize the potential use of anti-Notch3 therapy in future clinical application.
- 9.Nickoloff BJ, Osborne BA, Miele L. Notch signaling as a therapeutic target in cancer: a new approach to the development of cell fate modifying agents. Oncogene. 2003;22:6598–6608. doi: 10.1038/sj.onc.1206758. [DOI] [PubMed] [Google Scholar]
- 10.Park JT, Li M, Nakayama N, et al. Notch-3 gene amplification in ovarian cancer. Cancer Res. 2006;66:6312–6318. doi: 10.1158/0008-5472.CAN-05-3610. [DOI] [PubMed] [Google Scholar]; ▪ The first report to demonstrate Notch3 gene amplification in ovarian cancer.
- 11.Shih Ie M, Sheu JJ, Santillan A, et al. Amplification of a chromatin remodeling gene, Rsf-1/HBXAP, in ovarian carcinoma. Proc Natl Acad Sci USA. 2005;102:14004–14009. doi: 10.1073/pnas.0504195102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi JH, Sheu JJ, Guan B, et al. Functional analysis of 11q13.5 amplicon identifies Rsf-1 (HBXAP) as a gene involved in paclitaxel resistance in ovarian cancer. Cancer Res. 2009;69:1407–1415. doi: 10.1158/0008-5472.CAN-08-3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shamay M, Barak O, Shaul Y. HBXAP, a novel PHD-finger protein, possesses transcription repression activity. Genomics. 2002;79:523–529. doi: 10.1006/geno.2002.6717. [DOI] [PubMed] [Google Scholar]
- 14.Shamay M, Barak O, Doitsh G, Ben-Dor I, Shaul Y. Hepatitis B virus pX interacts with HBXAP, a PHD finger protein to coactivate transcription. J Biol Chem. 2002;277:9982–9988. doi: 10.1074/jbc.M111354200. [DOI] [PubMed] [Google Scholar]
- 15.Loyola A, Huang JY, LeRoy G, et al. Functional analysis of the subunits of the chromatin assembly factor RSF. Mol Cell Biol. 2003;23:6759–6768. doi: 10.1128/MCB.23.19.6759-6768.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LeRoy G, Loyola A, Lane WS, Reinberg D. Purification and characterization of a human factor that assembles and remodels chromatin. J Biol Chem. 2000;275:14787–14790. doi: 10.1074/jbc.C000093200. [DOI] [PubMed] [Google Scholar]
- 17.Aihara T, Miyoshi Y, Koyama K, et al. Cloning and mapping of SMARCA5 encoding hSNF2H, a novel human homologue of Drosophila ISWI. Cytogenet Cell Genet. 1998;81:191–193. doi: 10.1159/000015027. [DOI] [PubMed] [Google Scholar]
- 18.Vignali M, Hassan AH, Neely KE, Workman JL. ATP-dependent chromatin-remodeling complexes. Mol Cell Biol. 2000;20:1899–1910. doi: 10.1128/mcb.20.6.1899-1910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flanagan JF, Peterson CL. A role for the yeast SWI/SNF complex in DNA replication. Nucleic Acids Res. 1999;27:2022–2028. doi: 10.1093/nar/27.9.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cosma MP, Tanaka T, Nasmyth K. Ordered recruitment of transcription and chromatin remodeling factors to a cell cycle- and developmentally regulated promoter. Cell. 1999;97:299–311. doi: 10.1016/s0092-8674(00)80740-0. [DOI] [PubMed] [Google Scholar]
- 21.Abhiman S, Iyer LM, Aravind L. BEN: a novel domain in chromatin factors and DNA viral proteins. Bioinformatics. 2008;24:458–461. doi: 10.1093/bioinformatics/btn007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cha XY, Pierce RC, Kalivas PW, Mackler SA. NAC-1, a rat brain mRNA, is increased in the nucleus accumbens three weeks after chronic cocaine self-administration. J Neurosci. 1997;17:6864–6871. doi: 10.1523/JNEUROSCI.17-18-06864.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roitman MF, Wheeler RA, Carelli RM. Nucleus accumbens neurons are innately tuned for rewarding and aversive taste stimuli, encode their predictors, and are linked to motor output. Neuron. 2005;45:587–597. doi: 10.1016/j.neuron.2004.12.055. [DOI] [PubMed] [Google Scholar]
- 24.Koob GF. Drug addiction: the yin and yang of hedonic homeostasis. Neuron. 1996;16:893–896. doi: 10.1016/s0896-6273(00)80109-9. [DOI] [PubMed] [Google Scholar]
- 25.Mackler SA, Korutla L, Cha XY, et al. NAC-1 is a brain POZ/BTB protein that can prevent cocaine-induced sensitization in the rat. J Neurosci. 2000;20:6210–6217. doi: 10.1523/JNEUROSCI.20-16-06210.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalivas PW, Duffy P, Mackler SA. Interrupted expression of NAC-1 augments the behavioral responses to cocaine. Synapse. 1999;33:153–159. doi: 10.1002/(SICI)1098-2396(199908)33:2<153::AID-SYN5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 27.Ishibashi M, Nakayama K, Yeasmin S, et al. A BTB/POZ gene, NAC-1, a tumor recurrence-associated gene, as a potential target for taxol resistance in ovarian cancer. Clin Cancer Res. 2008;14:3149–3155. doi: 10.1158/1078-0432.CCR-07-4358. [DOI] [PubMed] [Google Scholar]
- 28.Nakayama K, Nakayama N, Davidson B, et al. A BTB/POZ protein, NAC-1, is related to tumor recurrence and is essential for tumor growth and survival. Proc Natl Acad Sci USA. 2006;103:18739–18744. doi: 10.1073/pnas.0604083103. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪ The first report to demonstrate the biological role of NAC1 in ovarian carcinoma.
- 29.Nakayama K, Nakayama N, Wang TL, Shih IM. NAC-1 Controls cell growth and survival by repressing transcription of Gadd45GIP1, a candidate tumor suppressor. Cancer Res. 2007;67:8058–8064. doi: 10.1158/0008-5472.CAN-07-1357. [DOI] [PubMed] [Google Scholar]
- 30.Jinawath N, Vasoontara C, Yap KL, et al. NAC-1, a potential stem cell pluripotency factor, contributes to paclitaxel resistance in ovarian cancer through inactivating Gadd45 pathway. Oncogene. 2009;28:1941–1948. doi: 10.1038/onc.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davidson B, Berner A, Trope CG, Wang TL, Shih IeM. Expression and clinical role of the bric-a-brac tramtrack broad complex/poxvirus and zinc protein NAC-1 in ovarian carcinoma effusions. Hum Pathol. 2007;38:1030–1036. doi: 10.1016/j.humpath.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 32.Kuhajda FP. Fatty acid synthase and cancer: new application of an old pathway. Cancer Res. 2006;66:5977–5980. doi: 10.1158/0008-5472.CAN-05-4673. [DOI] [PubMed] [Google Scholar]
- 33.Kuhajda FP, Jenner K, Wood FD, et al. Fatty acid synthesis: a potential selective target for antineoplastic therapy. Proc Natl Acad Sci USA. 1994;91:6379–6383. doi: 10.1073/pnas.91.14.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪ Describes the evidence for fatty acid synthase-based target therapy for human cancer.
- 34.Gansler TS, Hardman W, 3rd, Hunt DA, Schaffel S, Hennigar RA. Increased expression of fatty acid synthase (OA-519) in ovarian neoplasms predicts shorter survival. Hum Pathol. 1997;28:686–692. doi: 10.1016/s0046-8177(97)90177-5. [DOI] [PubMed] [Google Scholar]
- 35.Epstein JI, Carmichael M, Partin AW. OA-519 (fatty acid synthase) as an independent predictor of pathologic state in adenocarcinoma of the prostate. Urology. 1995;45:81–86. doi: 10.1016/s0090-4295(95)96904-7. [DOI] [PubMed] [Google Scholar]
- 36.Alo PL, Visca P, Marci A, et al. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer. 1996;77:474–482. doi: 10.1002/(SICI)1097-0142(19960201)77:3<474::AID-CNCR8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 37.Rashid A, Pizer ES, Moga M, et al. Elevated expression of fatty acid synthase and fatty acid synthetic activity in colorectal neoplasia. Am J Pathol. 1997;150:201–208. [PMC free article] [PubMed] [Google Scholar]
- 38.Milgraum LZ, Witters LA, Pasternack GR, Kuhajda FP. Enzymes of the fatty acid synthesis pathway are highly expressed in in situ breast carcinoma. Clin Cancer Res. 1997;3:2115–2120. [PubMed] [Google Scholar]
- 39.Pizer ES, Wood FD, Heine HS, et al. Inhibition of fatty acid synthesis delays disease progression in a xenograft model of ovarian cancer. Cancer Res. 1996;56:1189–1193. [PubMed] [Google Scholar]
- 40.Orita H, Coulter J, Tully E, Kuhajda FP, Gabrielson E. Inhibiting fatty acid synthase for chemoprevention of chemically induced lung tumors. Clin Cancer Res. 2008;14:2458–2464. doi: 10.1158/1078-0432.CCR-07-4177. [DOI] [PubMed] [Google Scholar]
- 41.Jensen V, Ladekarl M, Holm-Nielsen P, Melsen F, Soerensen FB. The prognostic value of oncogenic antigen 519 (OA-519) expression and proliferative activity detected by antibody MIB-1 in node-negative breast cancer. J Pathol. 1995;176:343–352. doi: 10.1002/path.1711760405. [DOI] [PubMed] [Google Scholar]
- 42.Shurbaji MS, Kalbfleisch JH, Thurmond TS. Immunohistochemical detection of a fatty acid synthase (OA-519) as a predictor of progression of prostate cancer. Hum Pathol. 1996;27:917–921. doi: 10.1016/s0046-8177(96)90218-x. [DOI] [PubMed] [Google Scholar]
- 43.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]; ▪▪ Review article that summarizes the recent advances in fatty acid synthase expression in cancer.
- 44.Alo PL, Visca P, Framarino ML, et al. Immunohistochemical study of fatty acid synthase in ovarian neoplasms. Oncol Rep. 2000;7:1383–1388. doi: 10.3892/or.7.6.1383. [DOI] [PubMed] [Google Scholar]
- 45.Ross J, Najjar AM, Sankaranarayanapillai M, et al. Fatty acid synthase inhibition results in a magnetic resonance-detectable drop in phosphocholine. Mol Cancer Ther. 2008;7:2556–2565. doi: 10.1158/1535-7163.MCT-08-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Menendez JA, Vellon L, Mehmi I, et al. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc Natl Acad Sci USA. 2004;101:10715–10720. doi: 10.1073/pnas.0403390101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang HQ, Altomare DA, Skele KL, et al. Positive feedback regulation between AKT activation and fatty acid synthase expression in ovarian carcinoma cells. Oncogene. 2005;24:3574–3582. doi: 10.1038/sj.onc.1208463. [DOI] [PubMed] [Google Scholar]
- 48.Zhou W, Han WF, Landree LE, et al. Fatty acid synthase inhibition activates AMP-activated protein kinase in SKOV3 human ovarian cancer cells. Cancer Res. 2007;67:2964–2971. doi: 10.1158/0008-5472.CAN-06-3439. [DOI] [PubMed] [Google Scholar]
- 49.Grunt TW, Wagner R, Grusch M, et al. Interaction between fatty acid synthase- and ErbB-systems in ovarian cancer cells. Biochem Biophys Res Commun. 2009;385:454–459. doi: 10.1016/j.bbrc.2009.05.085. [DOI] [PubMed] [Google Scholar]
- 50.Westgaard A, Schjolberg AR, Cvancarova M, et al. Differentiation markers in pancreatic head adenocarcinomas: MUC1 and MUC4 expression indicates poor prognosis in pancreatobiliary differentiated tumours. Histopathology. 2009;54:337–347. doi: 10.1111/j.1365-2559.2009.03227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bafna S, Singh AP, Moniaux N, et al. MUC4, a multifunctional transmembrane glycoprotein, induces oncogenic transformation of NIH3T3 mouse fibroblast cells. Cancer Res. 2008;68:9231–9238. doi: 10.1158/0008-5472.CAN-08-3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Theodoropoulos G, Carraway CA, Carraway KL. MUC4 involvement in ErbB2/ErbB3 phosphorylation and signaling in response to airway cell mechanical injury. J Cell Biochem. 2009;107:112–122. doi: 10.1002/jcb.22106. [DOI] [PubMed] [Google Scholar]
- 53.Workman HC, Sweeney C, Carraway KL., 3rd The membrane mucin Muc4 inhibits apoptosis induced by multiple insults via ErbB2-dependent and ErbB2-independent mechanisms. Cancer Res. 2009;69:2845–2852. doi: 10.1158/0008-5472.CAN-08-2089. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪ Very recent study demonstrating how Muc4 participates in the pathogenesis of cancer.
- 54.Davidson B, Baekelandt M, Shih IeM. MUC4 is upregulated in ovarian carcinoma effusions and differentiates carcinoma cells from mesothelial cells. Diagn Cytopathol. 2007;35:756–760. doi: 10.1002/dc.20771. [DOI] [PubMed] [Google Scholar]
- 55.Singh AP, Chaturvedi P, Batra SK. Emerging roles of MUC4 in cancer: a novel target for diagnosis and therapy. Cancer Res. 2007;67:433–436. doi: 10.1158/0008-5472.CAN-06-3114. [DOI] [PubMed] [Google Scholar]
- 56.Singh AP, Moniaux N, Chauhan SC, Meza JL, Batra SK. Inhibition of MUC4 expression suppresses pancreatic tumor cell growth and metastasis. Cancer Res. 2004;64:622–630. doi: 10.1158/0008-5472.can-03-2636. [DOI] [PubMed] [Google Scholar]
- 57.Ponnusamy MP, Singh AP, Jain M, et al. MUC4 activates HER2 signalling and enhances the motility of human ovarian cancer cells. Br J Cancer. 2008;99:520–526. doi: 10.1038/sj.bjc.6604517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang K, Pastan I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci USA. 1996;93:136–140. doi: 10.1073/pnas.93.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪▪ Original paper to report the cloning of mesothelin and provide new evidence that mesothelin is a novel marker in ovarian cancer.
- 59.Hough CD, Sherman-Baust CA, Pizer ES, et al. Large-scale serial analysis of gene expression reveals genes differentially expressed in ovarian cancer. Cancer Res. 2000;60:6281–6287. [PubMed] [Google Scholar]
- 60.Ordonez NG. Application of mesothelin immunostaining in tumor diagnosis. Am J Surg Pathol. 2003;27:1418–1428. doi: 10.1097/00000478-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 61.Lu KH, Patterson AP, Wang L, et al. Selection of potential markers for epithelial ovarian cancer with gene expression arrays and recursive descent partition analysis. Clin Cancer Res. 2004;10:3291–3300. doi: 10.1158/1078-0432.CCR-03-0409. [DOI] [PubMed] [Google Scholar]
- 62.Schaner ME, Ross DT, Ciaravino G, et al. Gene expression patterns in ovarian carcinomas. Mol Biol Cell. 2003;14:4376–4386. doi: 10.1091/mbc.E03-05-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yen MJ, Hsu CY, Mao TL, et al. Diffuse mesothelin expression correlates with prolonged patient survival in ovarian serous carcinoma. Clin Cancer Res. 2006;12:827–831. doi: 10.1158/1078-0432.CCR-05-1397. [DOI] [PubMed] [Google Scholar]
- 64.Uehara N, Matsuoka Y, Tsubura A. Mesothelin promotes anchorage-independent growth and prevents anoikis via extracellular signal-regulated kinase signaling pathway in human breast cancer cells. Mol Cancer Res. 2008;6:186–193. doi: 10.1158/1541-7786.MCR-07-0254. [DOI] [PubMed] [Google Scholar]
- 65.Ho M, Hassan R, Zhang J, et al. Humoral immune response to mesothelin in mesothelioma and ovarian cancer patients. Clin Cancer Res. 2005;11:3814–3820. doi: 10.1158/1078-0432.CCR-04-2304. [DOI] [PubMed] [Google Scholar]
- 66.Thomas AM, Santarsiero LM, Lutz ER, et al. Mesothelin-specific CD8(+) T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients. J Exp Med. 2004;200:297–306. doi: 10.1084/jem.20031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hassan R, Bera T, Pastan I. Mesothelin: a new target for immunotherapy. Clin Cancer Res. 2004;10:3937–3942. doi: 10.1158/1078-0432.CCR-03-0801. [DOI] [PubMed] [Google Scholar]
- 68.Rump A, Morikawa Y, Tanaka M, et al. Binding of ovarian cancer antigen CA125/MUC16 to mesothelin mediates cell adhesion. J Biol Chem. 2004;279:9190–9198. doi: 10.1074/jbc.M312372200. [DOI] [PubMed] [Google Scholar]
- 69.Feng Y, Xiao X, Zhu Z, et al. A novel human monoclonal antibody that binds with high affinity to mesothelin-expressing cells and kills them by antibody-dependent cell-mediated cytotoxicity. Mol Cancer Ther. 2009 doi: 10.1158/1535-7163.MCT-08-0945. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen YC, Pohl G, Wang TL, et al. Apolipoprotein E is required for cell proliferation and survival in ovarian cancer. Cancer Res. 2005;65:331–337. [PubMed] [Google Scholar]; ▪ Original report demonstrating the new role of apolipoprotein E in ovarian cancer.
- 71.Ho YY, Deckelbaum RJ, Chen Y, Vogel T, Talmage DA. Apolipoprotein E inhibits serum-stimulated cell proliferation and enhances serum-independent cell proliferation. J Biol Chem. 2001;276:43455–43462. doi: 10.1074/jbc.M105325200. [DOI] [PubMed] [Google Scholar]
- 72.Mahley RW, Rall SC., Jr Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 73.Gliemann J. Receptors of the low density lipoprotein (LDL) receptor family in man. Multiple functions of the large family members via interaction with complex ligands. Biol Chem. 1998;379:951–964. [PubMed] [Google Scholar]
- 74.Pillarisetti S. Lipoprotein modulation of subendothelial heparan sulfate proteoglycans (perlecan) and atherogenicity. Trends Cardiovasc Med. 2000;10:60–65. doi: 10.1016/s1050-1738(00)00048-7. [DOI] [PubMed] [Google Scholar]
- 75.Chen G, Paka L, Kako Y, et al. A protective role for kidney apolipoprotein E. Regulation of mesangial cell proliferation and matrix expansion. J Biol Chem. 2001;276:49142–49147. doi: 10.1074/jbc.M104879200. [DOI] [PubMed] [Google Scholar]
- 76.Linton MF, Atkinson JB, Fazio S. Prevention of atherosclerosis in apolipoprotein E-deficient mice by bone marrow transplantation. Science. 1995;267:1034–1037. doi: 10.1126/science.7863332. [DOI] [PubMed] [Google Scholar]
- 77.Boisvert WA, Spangenberg J, Curtiss LK. Treatment of severe hypercholesterolemia in apolipoprotein E-deficient mice by bone marrow transplantation. J Clin Invest. 1995;96:1118–1124. doi: 10.1172/JCI118098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boisvert WA, Curtiss LK. Elimination of macrophage-specific apolipoprotein E reduces diet-induced atherosclerosis in C57BL/6J male mice. J Lipid Res. 1999;40:806–813. [PubMed] [Google Scholar]
- 79.Mazzone T. Apolipoprotein E secretion by macrophages: its potential physiological functions. Curr Opin Lipidol. 1996;7:303–307. doi: 10.1097/00041433-199610000-00008. [DOI] [PubMed] [Google Scholar]
- 80.Emami N, Diamandis EP. Utility of kallikrein-related peptidases (KLKs) as cancer biomarkers. Clin Chem. 2008;54:1600–1607. doi: 10.1373/clinchem.2008.105189. [DOI] [PubMed] [Google Scholar]
- 81.Borgono CA, Diamandis EP. The emerging roles of human tissue kallikreins in cancer. Nat Rev Cancer. 2004;4:876–890. doi: 10.1038/nrc1474. [DOI] [PubMed] [Google Scholar]
- 82.Obiezu CV, Diamandis EP. Human tissue kallikrein gene family: applications in cancer. Cancer Lett. 2005;224:1–22. doi: 10.1016/j.canlet.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 83.Yousef GM, Polymeris ME, Grass L, et al. Human kallikrein 5: a potential novel serum biomarker for breast and ovarian cancer. Cancer Res. 2003;63:3958–3965. [PubMed] [Google Scholar]
- 84.Yousef GM, Polymeris ME, Yacoub GM, et al. Parallel overexpression of seven kallikrein genes in ovarian cancer. Cancer Res. 2003;63:2223–2227. [PubMed] [Google Scholar]; ▪▪ Original study to demonstrate the kallikreins in ovarian cancer tissues.
- 85.Michael IP, Sotiropoulou G, Pampalakis G, et al. Biochemical and enzymatic characterization of human kallikrein 5 (hK5), a novel serine protease potentially involved in cancer progression. J Biol Chem. 2005;280:14628–14635. doi: 10.1074/jbc.M408132200. [DOI] [PubMed] [Google Scholar]
- 86.Kapadia C, Ghosh MC, Grass L, Diamandis EP. Human kallikrein 13 involvement in extracellular matrix degradation. Biochem Biophys Res Commun. 2004;323:1084–1090. doi: 10.1016/j.bbrc.2004.08.206. [DOI] [PubMed] [Google Scholar]
- 87.Ghosh MC, Grass L, Soosaipillai A, Sotiropoulou G, Diamandis EP. Human kallikrein 6 degrades extracellular matrix proteins and may enhance the metastatic potential of tumour cells. Tumour Biol. 2004;25:193–199. doi: 10.1159/000081102. [DOI] [PubMed] [Google Scholar]
- 88.Luo LY, Katsaros D, Scorilas A, et al. The serum concentration of human kallikrein 10 represents a novel biomarker for ovarian cancer diagnosis and prognosis. Cancer Res. 2003;63:807–811. [PubMed] [Google Scholar]
- 89.Diamandis EP, Scorilas A, Fracchioli S, et al. Human kallikrein 6 (hK6): a new potential serum biomarker for diagnosis and prognosis of ovarian carcinoma. J Clin Oncol. 2003;21:1035–1043. doi: 10.1200/JCO.2003.02.022. [DOI] [PubMed] [Google Scholar]
- 90.Diamandis EP, Yousef GM, Soosaipillai AR, Bunting P. Human kallikrein 6 (zyme/protease M/neurosin): a new serum biomarker of ovarian carcinoma. Clin Biochem. 2000;33:579–583. doi: 10.1016/s0009-9120(00)00182-x. [DOI] [PubMed] [Google Scholar]
- 91.Xi Z, Kaern J, Davidson B, et al. Kallikrein 4 is associated with paclitaxel resistance in ovarian cancer. Gynecol Oncol. 2004;94:80–85. doi: 10.1016/j.ygyno.2004.03.044. [DOI] [PubMed] [Google Scholar]
- 92.Davidson B, Xi Z, Klokk TI, et al. Kallikrein 4 expression is up-regulated in epithelial ovarian carcinoma cells in effusions. Am J Clin Pathol. 2005;123:360–368. doi: 10.1309/PTBB-5BPC-KX8K-9V69. [DOI] [PubMed] [Google Scholar]
- 93.Shih IeM, Salani R, Fiegl M, et al. Ovarian cancer specific kallikrein profile in effusions. Gynecol Oncol. 2007;105:501–507. doi: 10.1016/j.ygyno.2007.01.018. [DOI] [PubMed] [Google Scholar]; ▪▪ Original report demonstrating the profiles of kallikreins in ovarian cancer effusions.
- 94.Kuk C, Kulasingam V, Gunawardana CG, et al. Mining the ovarian cancer ascites proteome for potential ovarian cancer biomarkers. Mol Cell Proteomics. 2009;8:661–669. doi: 10.1074/mcp.M800313-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Davidson B, Zhang Z, Kleinberg L, et al. Gene expression signatures differentiate ovarian/peritoneal serous carcinoma from diffuse malignant peritoneal mesothelioma. Clin Cancer Res. 2006;12:5944–5950. doi: 10.1158/1078-0432.CCR-06-1059. [DOI] [PubMed] [Google Scholar]
- 96.Yousef GM, Diamandis EP. The new human tissue kallikrein gene family: structure, function, and association to disease. Endocr Rev. 2001;22:184–204. doi: 10.1210/edrv.22.2.0424. [DOI] [PubMed] [Google Scholar]
- 97.Yousef GM, Diamandis EP. Kallikreins as cancer biomarkers. In: Diamandis EP, Fritsche HA, Lilja H, Chan DW, Schwartz S, editors. Tumor markers – physiology, pathobiology, technolgy and clinical applications. 1st. American Association for Clinical Chemistry; Washington DC, USA: 2002. pp. 465–469. [Google Scholar]
- 98.Hough CD, Cho KR, Zonderman AB, Schwartz DR, Morin PJ. Coordinately up-regulated genes in ovarian cancer. Cancer Res. 2001;61:3869–3876. [PubMed] [Google Scholar]; ▪ Original report demonstrating that claudin protein is one of the markers in ovarian cancer as compared with its benign counterpart.
- 99.Rangel LB, Agarwal R, D'Souza T, et al. Tight junction proteins claudin-3 and claudin-4 are frequently overexpressed in ovarian cancer but not in ovarian cystadenomas. Clin Cancer Res. 2003;9:2567–2575. [PubMed] [Google Scholar]
- 100.Hibbs K, Skubitz KM, Pambuccian SE, et al. Differential gene expression in ovarian carcinoma: identification of potential biomarkers. Am J Pathol. 2004;165:397–414. doi: 10.1016/S0002-9440(10)63306-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bignotti E, Tassi RA, Calza S, et al. Gene expression profile of ovarian serous papillary carcinomas: identification of metastasis-associated genes. Am J Obstet Gynecol. 2007;196:245, E241–E211. doi: 10.1016/j.ajog.2006.10.874. [DOI] [PubMed] [Google Scholar]
- 102.Morin PJ. Claudin proteins in human cancer: promising new targets for diagnosis and therapy. Cancer Res. 2005;65:9603–9606. doi: 10.1158/0008-5472.CAN-05-2782. [DOI] [PubMed] [Google Scholar]; ▪ Recent review article summarizing the translational role of claudin protein in ovarian cancer.
- 103.Agarwal R, D'Souza T, Morin PJ. Claudin-3 and claudin-4 expression in ovarian epithelial cells enhances invasion and is associated with increased matrix metalloproteinase-2 activity. Cancer Res. 2005;65:7378–7385. doi: 10.1158/0008-5472.CAN-05-1036. [DOI] [PubMed] [Google Scholar]; ▪ Original study showing the possible mechanism by which claudin proteins contribute to ovarian cancer progression.
- 104.Stewart JJ, White JT, Yan X, et al. Proteins associated with Cisplatin resistance in ovarian cancer cells identified by quantitative proteomic technology and integrated with mRNA expression levels. Mol Cell Proteomics. 2006;5:433–443. doi: 10.1074/mcp.M500140-MCP200. [DOI] [PubMed] [Google Scholar]
- 105.Kleinberg L, Holth A, Trope CG, Reich R, Davidson B. Claudin upregulation in ovarian carcinoma effusions is associated with poor survival. Hum Pathol. 2008;39:747–757. doi: 10.1016/j.humpath.2007.10.002. [DOI] [PubMed] [Google Scholar]; ▪ Report demonstrating the clinical significance of claudin overexpression in ovarian cancer.
- 106.Santin AD, Cane S, Bellone S, et al. Treatment of chemotherapy-resistant human ovarian cancer xenografts in C.B-17/SCID mice by intraperitoneal administration of Clostridium perfringens enterotoxin. Cancer Res. 2005;65:4334–4342. doi: 10.1158/0008-5472.CAN-04-3472. [DOI] [PubMed] [Google Scholar]
- 107.Sheu JJ, Shih IeM. Clinical and biological significance of HLA-G expression in ovarian cancer. Semin Cancer Biol. 2007;17:436–443. doi: 10.1016/j.semcancer.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fons P, Chabot S, Cartwright JE, et al. Soluble HLA-G1 inhibits angiogenesis through an apoptotic pathway and by direct binding to CD160 receptor expressed by endothelial cells. Blood. 2006;108:2608–2615. doi: 10.1182/blood-2005-12-019919. [DOI] [PubMed] [Google Scholar]
- 109.Davidson B, Elstrand MB, McMaster MT, et al. HLA-G expression in effusions is a possible marker of tumor susceptibility to chemotherapy in ovarian carcinoma. Gynecol Oncol. 2005;96:42–47. doi: 10.1016/j.ygyno.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 110.Singer G, Rebmann V, Chen YC, et al. HLA-G is a potential tumor marker in malignant ascites. Clin Cancer Res. 2003;9:4460–4464. [PubMed] [Google Scholar]
- 111.Rebmann V, Busemann A, Lindemann M, Grosse-Wilde H. Detection of HLA-G5 secreting cells. Hum Immunol. 2003;64:1017–1024. doi: 10.1016/j.humimm.2003.08.354. [DOI] [PubMed] [Google Scholar]; ▪ A study demonstrating the presence of secretory HLA-G in body fluid samples.
- 112.Kalli KR, Oberg AL, Keeney GL, et al. Folate receptor α as a tumor target in epithelial ovarian cancer. Gynecol Oncol. 2008;108:619–626. doi: 10.1016/j.ygyno.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yuan Y, Nymoen DA, Dong HP, et al. Expression of the folate receptor genes FOLR1 and FOLR3 differentiates ovarian carcinoma from breast carcinoma and malignant mesothelioma in serous effusions. Hum Pathol. 2009;40(10):1453–1460. doi: 10.1016/j.humpath.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 114.Basal E, Eghbali-Fatourechi GZ, Kalli KR, et al. Functional folate receptor α is elevated in the blood of ovarian cancer patients. PLoS ONE. 2009;4:E6292. doi: 10.1371/journal.pone.0006292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kalli KR. MORAb-003, a fully humanized monoclonal antibody against the folate receptor α, for the potential treatment of epithelial ovarian cancer. Curr Opin Investig Drugs. 2007;8:1067–1073. [PubMed] [Google Scholar]; ▪▪ Discussed the potential development of application of a humanized monoclonal antibody in treating cancer with FR-α upregulation.
- 116.Gibbs DD, Theti DS, Wood N, et al. BGC 945, a novel tumor-selective thymidylate synthase inhibitor targeted to α-folate receptor-overexpressing tumors. Cancer Res. 2005;65:11721–11728. doi: 10.1158/0008-5472.CAN-05-2034. [DOI] [PubMed] [Google Scholar]
- 117.Perkins ND. Integrating cell-signalling pathways with NF-κB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 118.Pahl HL. Activators and target genes of Rel/NF-κB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 119.Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor κB pathway. Oncogene. 2006;25:6717–6730. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- 120.Basseres DS, Baldwin AS. Nuclear factor-κB and inhibitor of κB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 121.Tang G, Minemoto Y, Dibling B, et al. Inhibition of JNK activation through NF-κB target genes. Nature. 2001;414:313–317. doi: 10.1038/35104568. [DOI] [PubMed] [Google Scholar]
- 122.Chen C, Edelstein LC, Gelinas C. The Rel/NF-κB family directly activates expression of the apoptosis inhibitor Bcl-x(L) Mol Cell Biol. 2000;20:2687–2695. doi: 10.1128/mcb.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mabuchi S, Ohmichi M, Nishio Y, et al. Inhibition of NFκB increases the efficacy of cisplatin in in vitro and in vivo ovarian cancer models. J Biol Chem. 2004;279:23477–23485. doi: 10.1074/jbc.M313709200. [DOI] [PubMed] [Google Scholar]
- 124.Mabuchi S, Ohmichi M, Nishio Y, et al. Inhibition of inhibitor of nuclear factor-κB phosphorylation increases the efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Clin Cancer Res. 2004;10:7645–7654. doi: 10.1158/1078-0432.CCR-04-0958. [DOI] [PubMed] [Google Scholar]
- 125.Kleinberg L, Dong HP, Holth A, et al. Cleaved caspase-3 and nuclear factor-κB p65 are prognostic factors in metastatic serous ovarian carcinoma. Hum Pathol. 2009;40:795–806. doi: 10.1016/j.humpath.2008.10.019. [DOI] [PubMed] [Google Scholar]; ▪ Recent article reporting the possible clinical correlation of p65 expression and clinical outcome in ovarian cancer.
- 126.Kim JH, Skates SJ, Uede T, et al. Osteopontin as a potential diagnostic biomarker for ovarian cancer. JAMA. 2002;287:1671–1679. doi: 10.1001/jama.287.13.1671. [DOI] [PubMed] [Google Scholar]
- 127.Visintin I, Feng Z, Longton G, et al. Diagnostic markers for early detection of ovarian cancer. Clin Cancer Res. 2008;14:1065–1072. doi: 10.1158/1078-0432.CCR-07-1569. [DOI] [PubMed] [Google Scholar]
- 128.Rosen DG, Wang L, Atkinson JN, et al. Potential markers that complement expression of CA125 in epithelial ovarian cancer. Gynecol Oncol. 2005;99:267–277. doi: 10.1016/j.ygyno.2005.06.040. [DOI] [PubMed] [Google Scholar]
- 129.Song G, Cai QF, Mao YB, et al. Osteopontin promotes ovarian cancer progression and cell survival and increases HIF-1α expression through the PI3-K/Akt pathway. Cancer Sci. 2008;99:1901–1907. doi: 10.1111/j.1349-7006.2008.00911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Song G, Ouyang G, Mao Y, et al. Osteopontin promotes gastric cancer metastasis by augmenting cell survival and invasion through Akt-mediated HIF-1α up-regulation and MMP9 activation. J Cell Mol Med. 2008 doi: 10.1111/j.1582-4934.2008.00540.x. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dai J, Peng L, Fan K, et al. Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells. Oncogene. 2009;28(38):3412–3422. doi: 10.1038/onc.2009.189. [DOI] [PubMed] [Google Scholar]
- 132.Kim HJ, Lee HJ, Jun JI, et al. Intracellular cleavage of osteopontin by caspase-8 modulates hypoxia/reoxygenation cell death through p53. Proc Natl Acad Sci USA. 2009;106(36):15326–15331. doi: 10.1073/pnas.0903704106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dai J, Li B, Shi J, et al. A humanized anti-osteopontin antibody inhibits breast cancer growth and metastasis in vivo. Cancer Immunol Immunother. 2009 doi: 10.1007/s00262-009-0754-z. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dubeau L. The cell of origin of ovarian epithelial tumours. Lancet Oncol. 2008;9:1191–1197. doi: 10.1016/S1470-2045(08)70308-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪ An extensive review on the possible origin of ovarian epithelial cancer.