

Abstract
Bile acids are generated in the liver and are traditionally recognized for their regulatory role in multiple metabolic processes including bile acid homeostasis, nutrient absorption, and cholesterol homeostasis. Recently, bile acids emerged as signaling molecules that, as ligands for the bile acid receptors farnesoid X receptor (FXR) and TGR5, activate and integrate multiple complex signaling pathways involved in lipid and glucose metabolism. Bile acid sequestrants are pharmacologic molecules that bind to bile acids in the intestine resulting in the interruption of bile acid homeostasis and, consequently, reduction in low-density lipoprotein cholesterol levels in hypercholesterolemia. Bile acid sequestrants also reduce glucose levels and improve glycemic control in persons with type 2 diabetes mellitus (T2DM). This article examines the mechanisms by which bile acid–mediated activation of FXR and TGR5 signaling pathways regulate lipid and glucose metabolism and the potential implications for bile acid sequestrant–mediated regulation of lipid and glucose levels in T2DM.
Keywords: Bile acid receptor, Bile acid sequestrant, Cholesterol, Glucose control, Lipid control, Type 2 diabetes mellitus
Introduction
Bile acid synthesis is the major pathway of cholesterol catabolism in the liver; approximately 500 mg of cholesterol is converted into bile acids daily in the adult human liver [1].
The first and rate-limiting step in the primary (classic or neutral) pathway of conversion of cholesterol to bile acids is catalyzed by cytochrome P450 enzyme cholesterol 7 α-hydroxylase (CYP7A1) [1]. Traditionally recognized for their role in regulating lipid absorption, bile acids are now known to be involved in the regulation of multiple metabolic processes including lipid and glucose metabolism and energy homeostasis through the activation of multiple signaling pathways. This article examines the effects of interrupting bile acid enterohepatic recirculation using bile acid sequestrants not only on lipid but also on glycemic control and the potential mechanism(s) of action involved in the bile acid–mediated regulation of these processes.
Clinical Effects of Bile Acid Sequestrants
Bile acid sequestrants (cholestyramine, colestipol, colestimide, and colesevelam) are positively charged nondigestible resins that bind to bile acids in the intestine to form an insoluble complex that is excreted in the feces. This process reduces bile acid levels in the liver, promoting the increased synthesis of bile acids from cholesterol and reducing hepatic cholesterol levels [2].
The clinical lipid-lowering utility of bile acid sequestrants was demonstrated more than two decades ago during the Lipid Research Clinics Coronary Primary Prevention Trials, in which long-term administration of cholestyramine in men with hypercholesterolemia resulted in overall reductions in total cholesterol and low-density lipoprotein cholesterol (LDLC) of 13.4% and 20.3%, respectively, compared with placebo (4.9% and 7.7%, respectively). These reductions were accompanied by a 19% reduction in the incidence of coronary heart disease [3, 4]. Similarly, treatment with colestipol (2 or 8 g twice daily) reduced LDLC levels in patients with mild hypercholesterolemia by 12% or 24%, respectively (P ≤ 0.05) [5]. Monotherapy with colesevelam resulted in mean reductions in LDLC of 15% to 19% in adults with hypercholesterolemia [6, 7]. Furthermore, the addition of colesevelam to therapy with atorvastatin (10 mg) or simvastatin (10 mg) reduced LDLC levels by 48% or 42%, respectively, an increase over colesevelam (12%) or atorvastatin (38%) alone or colesevelam (16%) or simvastatin (26%) alone, respectively [8, 9].
Bile acid sequestrants have also been shown to improve glycemic control in patients with type 2 diabetes mellitus (T2DM). In initial studies in patients with T2DM and dyslipidemia, cholestyramine therapy decreased LDLC levels by 28% and resulted in improved glycemic control, as defined by a 13% reduction in mean plasma glucose levels, a median reduction of 0.22 g/d in urinary glucose excretion, and a tendency toward a lower concentration of glycosylated hemoglobin A1c (HbA1c) [10]. In three 26-week, double-blind, placebo-controlled studies, the addition of colesevelam to the antidiabetes regimen of patients with T2DM who were inadequately controlled on their current metformin, sulfonylurea, or insulin therapies alone or in combination with other oral antidiabetes agents resulted in additional mean reductions in HbA1c of 0.54% (metformin and sulfonylurea) and 0.50% (insulin). These patients also experienced mean reductions in their LDLC levels of 15.9%, 16.7%, and 12.8% (P < 0.001 for all) for those receiving colesevelam added to ongoing metformin, sulfonylurea, and insulin therapy, respectively [11–13]. Colesevelam is the only bile acid sequestrant approved for glycemic control in patients with T2DM who are uncontrolled on their current treatment regimen.
Bile Acid Receptors
In addition to facilitating the dietary absorption of lipids, bile acids act as ligands for the nuclear receptor farnesoid X receptor (FXR) and the G-protein-coupled receptor (GPCR) TGR5 in the liver and intestine. However, in addition to their role as receptor-mediated signaling molecules, certain bile acids also exert non–receptor-mediated effects on cellular responses such as improvement of the endoplasmic reticulum (ER) stress response. Obesity-induced ER stress can lead to the development of insulin resistance and T2DM. However, administration of the bile acid taurine-conjugated ursodeoxycholic acid (TUDCA) alleviated ER stress in cells and whole animals [14]. In addition, administration of TUDCA to obese and diabetic mice resulted in lowered glucose levels, improved insulin sensitivity, and resolution of fatty liver disease [14]. Evaluation of the mechanisms regulating these non–receptor-mediated effects of bile acids and their contribution to the lipid- and glucose-lowering effects of bile acid sequestrants is beyond the scope of this article.
FXR and Bile Acid Synthesis
FXR is a member of the nuclear receptor superfamily of ligand-activated transcription factors. FXR is expressed at high levels in the liver and intestine and is most potently activated by the primary bile acid chenodeoxycholic acid [15]. In the liver, as the bile acid pool increases in size, bile acid activation of FXR upregulates expression of the gene encoding the inhibitory nuclear receptor small heterodimer partner (SHP) [15]. SHP represses activation of several transcription factors including liver X receptor, liver receptor homologue-1 (LRH-1), and hepatocyte nuclear factor-4α (HNF-4α), subsequently suppressing, in humans, the LRH-1–mediated activation of CYP7A1, thereby inhibiting the first step in cholesterol catabolism (Fig. 1). Bile acid–mediated repression of HNF-4α also inhibits transcription of CYP7A1 [16].
Fig. 1.
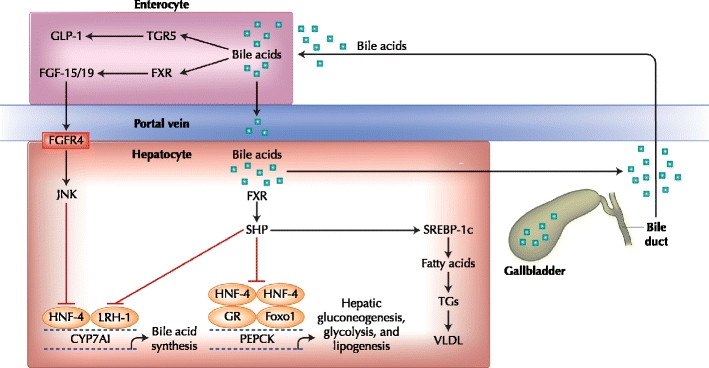
FXR- and TGR5-mediated regulation of bile acid synthesis and lipid and glucose metabolism in the liver and intestine. Bile acid synthesis and lipid and glucose metabolism are regulated in the liver and intestine via pathways involving the bile acid receptors FXR and TGR5. In the liver, bile acids activate FXR resulting in upregulation of SHP, an inhibitor of bile acid synthesis, gluconeogenesis, and fatty acid synthesis. In the intestine, bile acid activation of FXR upregulates FGF-15/19 and ultimately inhibits bile acid synthesis. CYP7A1—cytochrome P450 enzyme cholesterol 7 α-hydroxylase; FGF-15/19—fibroblast growth factor 15/19; FGFR4—fibroblast growth factor receptor 4; FXR—farnesoid X receptor; GLP-1—glucagon-like peptide-1; GR—glucocorticoid receptor; HNF-4—hepatocyte nuclear factor-4; JNK—c-jun N-terminal kinase; LRH-1—liver receptor homologue-1; PEPCK—phosphoenolpyruvate carboxykinase; SHP—small heterodimer partner; SREBP-1c—sterol regulatory element-binding protein-1c; TGs—triglycerides; VLDL—very low density lipoprotein. (Adapted from Staels [49] and Thomas et al. [50].)
A second pathway of bile acid–mediated repression of its own synthesis involves FXR-mediated induction of fibroblast growth factor-19 (FGF-19; [FGF-15 in mice]) in the intestine. In the intestine, transintestinal transport of bile acids after a meal induces activation of intestinal FXR, resulting in expression of FGF-19 [17]. Binding of FGF-19 to surface hepatocyte fibroblast growth factor receptor 4 results in a c-Jun N-terminal kinase–mediated repression of CYP7A1 transcription, a potent SHP-independent alternative pathway of CYP7A1 repression (Fig. 1) [17].
TGR5 and Bile Acid Synthesis
Bile acids can also bind to and activate TGR5 (also known as GPBAR1, M-BAR, and BG37), a member of the rhodopsin-like superfamily of transmembrane GPCRs [18, 19]. Expression of TGR5 is detected in multiple tissues, with the highest level of expression detected in the gallbladder, ileum, and colon, and with lower levels of expression in brown adipose tissue, liver, and intestine; TGR5 expression has not been detected in murine or human hepatocytes [19].
TGR5 is activated by multiple bile acids, with lithocholic acid being the most potent natural agonist (EC50 of 530 nM) [18]. Bile acid–mediated activation of TGR5 results in internalization of the receptor, activation of extracellular-regulated kinase, the mitogen-activated protein-kinase pathway, and ultimately stimulation of cyclic adenosine monophosphate (cAMP) synthesis. This, in turn, results in the activation of protein kinase A, phosphorylation of the cAMP-response element-binding protein, and transactivation of target gene expression [20]. Activation of TGR5 may play a role in energy and glucose homeostasis.
Bile acid sequestrants cause alterations in the bile acid pool, the size of which is tightly regulated in the liver and the intestine by a negative feedback mechanism that prevents cytotoxic accumulation of bile acids [15]. Early studies into the effects of bile acid sequestrants showed that they interrupted the enterohepatic circulation of bile acids and increased fecal excretion of bile acids, resulting in decreased hepatic bile acid levels. As bile acid levels decreased, the FXR-mediated repression of CYP7A1 was reduced, upregulating expression of CYP7A1 and resulting in an increase in bile acid synthesis and reduced hepatic cholesterol levels. Reduced hepatic cholesterol levels promote activation of the sterol regulatory element-binding protein (SREBP)-2, ultimately resulting in increased expression of LDL receptors and increased clearance of LDLC from the blood. In parallel, increased secretion of very low-density lipoprotein (VLDL) particles and a transient rise in triglyceride levels are often observed [2, 21].
Bile acid sequestrants also increase high-density lipoprotein cholesterol (HDLC) levels. In murine studies, as bile acid levels decrease, the FXR-mediated activation of scavenger receptor BI (SR-BI) expression is also decreased. This receptor is responsible for the hepatic uptake of HDLC. It can be hypothesized that the diminished expression of SR-BI and resultant decrease in uptake of HDLC may contribute to the increase in HDLC levels seen after administration of bile acid sequestrants in humans [15, 22]. In addition, bile acid–induced downregulation of apolipoprotein (apo) A-I expression may be counteracted by bile acid sequestrants [23], resulting in increased apo A-I plasma levels. These effects may be mediated in part via the bile acid receptors FXR and TGR5, which regulate genes involved in lipid and glucose metabolism.
FXR and Lipid Metabolism
Studies have shown that activation of FXR alters the transcription of several genes involved in triglyceride synthesis and lipid metabolism. Mice deficient in FXR (Fxr -/-) have increased plasma non-HDLC and triglyceride levels, increased synthesis of apo B–containing lipoproteins, mainly VLDL, and increased absorption of intestinal cholesterol. Fxr -/- mice also have increased levels of plasma HDLC coincident with a reduced rate of plasma HDLC ester clearance [22]. Accordingly, mice deficient in the FXR target gene SHP (Shp -/-) have increased levels of the transcription factor SREBP-1c, which controls the expression of genes involved in lipogenesis (Table 1) [24].
Table 1.
Positive and negative FXR- and TGR5-mediated modulation of genes involved in lipid, glucose, and energy metabolism
| Study | Function | Gene | Regulation |
|---|---|---|---|
| FXR | |||
| Goodwin et al. [16] | Bile acid metabolism | CYP7A1 | Repressed (through SHP induction) |
| Lambert et al. [22], Watanabe et al. [24] | Lipogenesis | SREBP-1c | Repressed (through SHP induction) |
| Lambert et al. [22] | TG metabolism | Apo B | Repressed |
| Kast et al. [31] | TG metabolism | Apo C-II | Induced |
| Kast et al. [31] | TG metabolism | Apo C-III | Repressed |
| Cariou and Staels [52] | TG metabolism | VLDLR | Induced |
| Claudel et al. [23] | HDL metabolism | Apo A-1 | Repressed |
| Ma et al. [25] | Glucose metabolism | PEPCK | Induced and repressed |
| Ma et al. [25] | Glucose metabolism | G6Pase | Repressed |
| TGR5 | |||
| Watanabe et al. [44] | Energy metabolism | Iodothyronine deiodinase type 2 (D2) | Induced |
Apo—apolipoprotein; CYP7A1—cytochrome P450 enzyme cholesterol 7 α-hydroxylase; FXR—farnesoid X receptor; G6Pase—glucose-6 phosphatase; HDL—high-density lipoprotein; PEPCK—phosphoenolpyruvate carboxykinase; SHP—small heterodimer partner; SREBP-1c—sterol regulatory element-binding protein-1c; TG—triglyceride; VLDLR—very low-density lipoprotein receptor. (Adapted from Cariou and Staels [52].)
In addition, Fxr -/- mice have approximately twofold higher levels of circulating free fatty acids (FFAs), compared with wild-type mice, and accumulate fat in the liver [25]. Elevated levels of FFAs and excessive accumulation of fat in the liver can lead to the development of nonalcoholic fatty liver disease and hepatic steatosis. Interestingly, studies have shown that activation of FGF-19 can increase fatty acid oxidation and inhibit fatty acid synthesis via activation of SHP, inhibition of SREBP-1c, and changes in signal transducer and activator of transcription 3 and peroxisome proliferator-activated receptor (PPAR) coactivator-1β signaling pathways in murine models of diet-induced diabetes and obesity [26, 27]. These studies suggest a potential role for FGF-19–mediated signaling effects in fatty liver disease and in the resolution of some effects associated with the metabolic syndrome. The mechanism(s), if any, that integrates the effects of FGF-19 on obesity and diabetes and the lipid- and glucose-lowering effects of bile acid sequestrants remain to be investigated.
In murine models predisposed to develop hypertriglyceridemia (KK/Ay and ob/ob), administration of the primary bile acid cholic acid (CA) or the synthetic FXR agonist (GW4064) resulted in decreased plasma FFAs, decreased triglycerides (VLDL triglycerides), decreased cholesterol (HDLC), and increased LDLC levels [24]. The decreased cholesterol levels observed following FXR activation are due largely to a decrease in circulating HDLC levels. Results from several studies in murine models of hypercholesterolemia (ApoE -/-, ob/ob, and db/db) confirm that FXR activation resulted in a reduction in HDLC plasma levels [24, 28•].
Administration of bile acids also repressed expression of the transcription factor SREBP-1c and its lipogenic target genes in murine hepatocytes and liver in an SHP-dependent manner (Table 1) [24, 29]. Other mechanisms implicated in FXR-mediated hypotriglyceridemia include FXR-activated expression of PPAR-α and its target gene pyruvate dehydrogenase kinase 4, which promote fatty acid oxidation, increased expression of apo C-II (an activator of lipoprotein lipase activity), and decreased expression of apo C-III and angiopoietin-like protein 3, which are both lipoprotein lipase inhibitors (Table 1) [30–32]. The relative contribution of triglyceride production versus triglyceride clearance is unknown.
Hypothetical Glucose-lowering Mechanisms
FXR and Hepatic Glucose Metabolism
The liver plays a central role in the control of blood glucose homeostasis by maintaining a balance between glucose production and utilization (Fig. 1). Evidence for a role of bile acids in glucose metabolism was revealed by studies showing that bile acid composition and pool size are altered in animal and human models of diabetes [33, 34]. A link between FXR and glucose metabolism was provided by the observation that hepatic expression of the gene encoding FXR was decreased following quantitative analysis of hepatic FXR mRNA expression in a rat model of type 1 diabetes. Accordingly, this was accompanied by increased expression of CYP7A1, which likely contributes to the enlarged bile acid pool that is also characteristic of such diabetic animals [35]. As seen in the rat model of type 1 diabetes, hepatic expression of FXR also decreased with age in a rat model of T2DM [35]. Hepatocytes from nondiabetic rats were incubated with glucose and insulin to assess the effects of glucose and insulin on hepatic FXR expression. Incubation of rat hepatocytes with insulin repressed expression of FXR, an effect that is reduced by glucose. These results suggested that diabetes impairs the normal regulation of FXR expression [35].
A potential role for FXR in the regulation of glucose metabolism was provided by the observation that pharmacologic treatment with CA for 5 days resulted in decreased expression of phosphoenolpyruvate carboxykinase (PEPCK), glucose-6-phosphatase (G6Pase), and fructose 1,6-bisphosphatase, enzymes critically involved in the regulation of hepatic gluconeogenesis (Table 1), in nondiabetic wild-type but not Fxr -/- mice [25]. In contrast to the results obtained with CA, in vivo treatment of nondiabetic wild-type mice with the synthetic FXR agonist GW4064 induced expression of PEPCK, whereas activation of FXR with GW4064 in the diabetic db/db mouse model reduced expression of PEPCK and G6Pase. Together, these results suggest that bile acids may regulate hepatic glucose utilization and production [15, 36].
Fxr -/- mice also have decreased levels of glycogen in the liver and develop transient hypoglycemia upon fasting [37]. In keeping with a potential role for FXR in regulation of the kinetics of glucose metabolism, analysis of Fxr -/- mice subject to an overnight fast followed by re-feeding with a high-carbohydrate diet resulted in increased L-pyruvate kinase and lipogenic gene expression, potentially revealing an additional role for FXR in regulating hepatic carbohydrate metabolism [38]. Together, these data provide evidence of a critical role for bile acids and FXR in the dynamic regulation of glucose metabolism [15]. However, given the contradictory evidence (different animal models and tested ligands), a precise mechanism of FXR regulation of glucose metabolism remains unclear.
FXR and Insulin Sensitivity
Hyperinsulinemic-euglycemic clamp studies in Fxr -/- mice have led to the observation that FXR may also play a partial role in regulating insulin sensitivity [25, 39]. Fxr -/- mice exhibit peripheral insulin resistance and impaired insulin signaling in insulin-sensitive tissues such as skeletal muscle and white adipose tissue. Moreover, activation of FXR in db/db mice and treatment of db/db and ob/ob mice with GW4064 resulted in significant improvement in insulin sensitivity [36, 39]. However, thus far, no studies have been performed in genetically predisposed or diet-induced models of diabetes.
TGR5 and Glucose Metabolism
Mice deficient in TGR5 (Tgr5 -/-) have normal glucose levels and do not develop overt diabetes [40]. However, the observation that murine intestinal cells secrete the gut-derived incretin hormone glucagon-like peptide-1 (GLP-1) in a Tgr5-dependent manner following in vitro stimulation with the bile acids lithocholic acid and deoxycholic acid revealed a role for TGR5 in glucose homeostasis [41, 42•].
Additional evidence implicating TGR5 in glucose homeostasis was provided by the observation that treatment of mice fed a high-fat diet with oleanolic acid (an extract of Olea Europaea [olive leaves] and a TGR5 agonist) protected against weight gain and resulted in reduced plasma glucose and insulin levels compared with controls [43].
Moreover, in vivo overexpression of TGR5 in a transgenic (TGR5-Tg) mouse model markedly improved glucose tolerance in mice fed a high-fat diet compared with controls. This improved glucose tolerance in TGR5-Tg mice was associated with robust secretion of GLP-1 and increased insulin release in response to an oral glucose load. Interestingly, there was an even greater postprandial effect on GLP-1 release and insulin secretion after a test meal than in response to the glucose challenge. The researchers hypothesized that this was because of an increased bile acid flux triggered by the test meal compared with the glucose challenge [42•]. These findings establish a role for TGR5 in bile acid–mediated regulation of glucose homeostasis.
TGR5 and Energy Homeostasis
Supplementation of high-fat-fed mice with CA decreased obesity and insulin resistance (through an increase in energy expenditure in brown adipose tissue) and increased the bile acid pool size [44]. These observations implicate bile acids in the regulation of the metabolic process through increases in energy expenditure via modulation of thermogenesis.
Evidence of a role for TGR5 in the regulation of energy expenditure was provided by observations that TGR5 is expressed in brown adipose tissue and that the bile acid–mediated increase in expression of the D2 gene that controls energy expenditure in brown adipose tissue is regulated primarily through the cAMP–protein kinase A signaling pathway [44]. TGR5 and D2 are also expressed in human skeletal muscle. Expression of D2 in skeletal muscle increased in a dose-dependent manner in response to incubation of skeletal muscle tissue with a TGR5 agonist but not with the FXR agonist GW4064, providing further confirmation for the TGR5-dependent regulation of energy expenditure [44]. However, despite the data supporting a role for a TGR5-cAMP–dependent pathway in regulation of energy homeostasis, the observation that Tgr5 -/- mice have normal triglyceride levels and do not gain weight when fed a regular diet suggests that more analyses are necessary to fully uncover the role of bile acids in the regulation of energy homeostasis [40].
Bile Acid Sequestrants in Lipid and Glycemic Control
Bile acid sequestrants are well known for their effects on lipid levels, particularly for reducing LDLC. Results from studies examining the effects of FXR on bile acid synthesis and lipid metabolism implicate FXR-dependent signaling pathways in the control of lipid metabolism by bile acid sequestrants [22, 24, 26, 27].
Less is known about the mechanisms involved in the glucose-lowering effects of bile acid sequestrants [10–13, 45•]. It can be hypothesized, given the results of studies examining the mechanisms of action involved in bile acid–mediated regulation of hepatic gluconeogenesis, that bile acid sequestrants may modulate FXR-dependent signaling pathways that regulate expression of PEPCK and other enzymes involved in hepatic gluconeogenesis [25, 33–35, 37].
Preliminary studies suggested that administration of the bile acid sequestrant colesevelam may act on glucose homeostasis by reducing insulin resistance and/or improving clearance of plasma glucose [46, 47]. In a rat model that develops insulin resistance and diet-induced obesity (DIO) when fed a high-energy diet, treatment with 2% colesevelam lowered plasma glucose levels following a 2-hour oral glucose tolerance test, whereas the insulin response was normalized (insulin peak in 15–30 min with return to baseline in 30–60 min) compared with DIO rats or DIO rats treated with the apical sodium codependent bile acid transport inhibitor SC-435 [47]. These results suggest that in this rat model of insulin resistance, colesevelam may reduce glucose levels by reducing insulin resistance and normalizing the first-phase insulin release [47].
In clinical studies, improved glycemic control in patients treated with colesevelam (3.75 g/d) compared with placebo was directly correlated with an increase in whole-body insulin sensitivity as measured by the Matsuda index, and not with an increase in peripheral insulin sensitivity as measured by the hyperinsulinemic-euglycemic clamp method [48]. In a separate study, stable isotope infusion analyses showed that significant reductions in fasting plasma glucose and HbA1c levels in patients receiving colesevelam compared with placebo were accompanied by an increase in plasma glucose clearance without the increase in endogenous glucose production seen in patients receiving placebo [46].
In addition, bile acid sequestrants may decrease glucose levels by effecting regulation of the TGR5-dependent secretion of the gut-derived incretin hormone GLP-1 [45•]. A study by Suzuki et al. [45•] showed that administration of colestimide (1500 mg, twice daily) resulted in decreased postprandial plasma glucose levels and increased secretion of GLP-1 in patients with T2DM and dyslipidemia. Although no comparisons were made with patients not receiving colestimide and the time to measurement of GLP-1 may have been too long to accurately reflect changes in GLP-1 levels, this study does provide some evidence to suggest that bile acid sequestrants may act in a TGR5-dependent manner to increase secretion of GLP-1 in patients with T2DM. Further studies are necessary to determine whether bile acid sequestrants act in an FXR- and/or TGR5-dependent manner to improve insulin sensitivity and insulin secretion in patients with T2DM.
Unlike cholestyramine and colestipol, colesevelam has a high affinity for trihydroxy and dihydroxy bile acids in the intestine, leading to increased fecal excretion with colesevelam [49]. This differential bile acid–binding ability may allow colesevelam to have an alternative effect on the bile acid pool and on bile acid–mediated regulation of glucose metabolism. The metabolism of bile acids by the intestinal flora is altered in T2DM, and modification of the bile acid pool seen by bacteria by bile acids may influence secretion of GLP-1 by L cells. As such, differential bile acid–binding properties of distinct bile acid sequestrants may exist and may result in pathophysiologically relevant pharmacologic differences in action. However, definitive studies are necessary to examine the effect of alteration of the bile acid pool induced by bile acid sequestrants on activation of the bile acid receptors FXR and TGR5 and the potential effects on glycemic control in patients with T2DM.
Conclusions
Bile acid sequestrants cause alterations in the bile acid pool with resultant effects on lipid and glucose metabolism. Studies suggest that alterations of the enterohepatic circulation may regulate glucose homeostasis by modulating FXR- and TGR5-mediated pathways. Further studies examining the role of FXR- and TGR5-mediated signaling pathways on lipid and glucose metabolism in animal and human models of T2DM and obesity are needed to elucidate how these signals are integrated to mediate the lipid- and glucose-lowering effects of bile acid sequestrants.
Acknowledgments
Editorial assistance was provided by Luana Atherly, PhD, and funded by Daiichi Sankyo, Inc.
Disclosure
Dr. Bart Staels has received grant/research support from Daiichi Sankyo, Inc. Dr. Yehuda Handelsman has received grant/research support from Daiichi Sankyo, Inc., GlaxoSmithKline, Novo Nordisk, and Takeda Pharmaceuticals; has been a consultant for Bristol-Myers Squibb, Daiichi Sankyo, Inc., GlaxoSmithKline, Medtronic, Merck, Xoma, and Tethys; and has been on the speakers’ bureau for Daiichi Sankyo, Inc., GlaxoSmithKline, Merck, Bristol-Myers Squibb, and AstraZeneca. Dr. Vivian Fonseca has received research support (to Tulane)/grants from GlaxoSmithKline, Novartis, Novo Nordisk, Takeda Pharmaceuticals, AstraZeneca, Pfizer, sanofi-aventis, Eli Lilly, Daiichi Sankyo, Inc., Novartis, the US National Institutes of Health, and the American Diabetes Association. He has also received honoraria for consulting and lectures from GlaxoSmithKline, Novartis, Takeda Pharmaceuticals, Novo Nordisk, sanofi-aventis, Eli Lilly, and Daiichi Sankyo, Inc.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Contributor Information
Bart Staels, Email: Bart.Staels@pasteur-lille.fr.
Yehuda Handelsman, Email: yhandelsman@pacbell.net.
Vivian Fonseca, Email: vfonseca@tulane.edu.
References
Papers of particular interest, published recently, have been highlighted as: •Of importance
- 1.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 2.Insull W., Jr Clinical utility of bile acid sequestrants in the treatment of dyslipidemia: a scientific review. South Med J. 2006;99:257–273. doi: 10.1097/01.smj.0000208120.73327.db. [DOI] [PubMed] [Google Scholar]
- 3.The Lipid Research Clinics Coronary Primary Prevention Trial results. I. Reduction in incidence of coronary heart disease [no authors listed]. JAMA. 1984; 251:351–364. [DOI] [PubMed]
- 4.The Lipid Research Clinics Coronary Primary Prevention Trial results. II. The relationship of reduction in incidence of coronary heart disease to cholesterol lowering [no authors listed]. JAMA. 1984; 251:365–374. [PubMed]
- 5.Insull W, Jr, Davidson MH, Demke DM, et al. The effects of colestipol tablets compared with colestipol granules on plasma cholesterol and other lipids in moderately hypercholesterolemic patients. Atherosclerosis. 1995;112:223–235. doi: 10.1016/0021-9150(94)05418-I. [DOI] [PubMed] [Google Scholar]
- 6.Insull W, Jr, Toth P, Mullican W, et al. Effectiveness of colesevelam hydrochloride in decreasing LDL cholesterol in patients with primary hypercholesterolemia: a 24-week randomized controlled trial. Mayo Clin Proc. 2001;76:971–982. doi: 10.4065/76.10.971. [DOI] [PubMed] [Google Scholar]
- 7.Davidson MH, Dillon MA, Gordon B, et al. Colesevelam hydrochloride (cholestagel): a new, potent bile acid sequestrant associated with a low incidence of gastrointestinal side effects. Arch Intern Med. 1999;159:1893–1900. doi: 10.1001/archinte.159.16.1893. [DOI] [PubMed] [Google Scholar]
- 8.Hunninghake D, Insull W, Jr, Toth P, et al. Coadministration of colesevelam hydrochloride with atorvastatin lowers LDL cholesterol additively. Atherosclerosis. 2001;158:407–416. doi: 10.1016/S0021-9150(01)00437-3. [DOI] [PubMed] [Google Scholar]
- 9.Knapp HH, Schrott H, Ma P, et al. Efficacy and safety of combination simvastatin and colesevelam in patients with primary hypercholesterolemia. Am J Med. 2001;110:352–360. doi: 10.1016/S0002-9343(01)00638-6. [DOI] [PubMed] [Google Scholar]
- 10.Garg A, Grundy SM. Cholestyramine therapy for dyslipidemia in non-insulin-dependent diabetes mellitus. A short-term, double-blind, crossover trial. Ann Intern Med. 1994;121:416–422. doi: 10.7326/0003-4819-121-6-199409150-00004. [DOI] [PubMed] [Google Scholar]
- 11.Bays HE, Goldberg RB, Truitt KE, Jones MR. Colesevelam hydrochloride therapy in patients with type 2 diabetes mellitus treated with metformin: glucose and lipid effects. Arch Intern Med. 2008;168:1975–1983. doi: 10.1001/archinte.168.18.1975. [DOI] [PubMed] [Google Scholar]
- 12.Fonseca VA, Rosenstock J, Wang AC, et al. Colesevelam HCl improves glycemic control and reduces LDL cholesterol in patients with inadequately controlled type 2 diabetes on sulfonylurea-based therapy. Diabetes Care. 2008;31:1479–1484. doi: 10.2337/dc08-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldberg RB, Fonseca VA, Truitt KE, Jones MR. Efficacy and safety of colesevelam in patients with type 2 diabetes mellitus and inadequate glycemic control receiving insulin-based therapy. Arch Intern Med. 2008;168:1531–1540. doi: 10.1001/archinte.168.14.1531. [DOI] [PubMed] [Google Scholar]
- 14.Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lefebvre P, Cariou B, Lien F, et al. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 16.Goodwin B, Jones SA, Price RR, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. doi: 10.1016/S1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 17.Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 18.Kawamata Y, Fujii R, Hosoya M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 19.Maruyama T, Miyamoto Y, Nakamura T, et al. Identification of membrane-type receptor for bile acids (M-BAR) Biochem Biophys Res Commun. 2002;298:714–719. doi: 10.1016/S0006-291X(02)02550-0. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen A, Bouscarel B. Bile acids and signal transduction: role in glucose homeostasis. Cell Signal. 2008;20:2180–2197. doi: 10.1016/j.cellsig.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 21.Einarsson K, Ericsson S, Ewerth S, et al. Bile acid sequestrants: mechanisms of action on bile acid and cholesterol metabolism. Eur J Clin Pharmacol. 1991;40(Suppl 1):S53–S58. doi: 10.1007/BF01409410. [DOI] [PubMed] [Google Scholar]
- 22.Lambert G, Amar MJ, Guo G, et al. The farnesoid X-receptor is an essential regulator of cholesterol homeostasis. J Biol Chem. 2003;278:2563–2570. doi: 10.1074/jbc.M209525200. [DOI] [PubMed] [Google Scholar]
- 23.Claudel T, Sturm E, Duez H, et al. Bile acid-activated nuclear receptor FXR suppresses apolipoprotein A-I transcription via a negative FXR response element. J Clin Invest. 2002;109:961–971. doi: 10.1172/JCI14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watanabe M, Houten SM, Wang L, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113:1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116:1102–1109. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhatnagar S, Damron HA, Hillgartner FB. Fibroblast growth factor-19, a novel factor that inhibits hepatic fatty acid synthesis. J Biol Chem. 2009;284:10023–10033. doi: 10.1074/jbc.M808818200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomlinson E, Fu L, John L, et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology. 2002;143:1741–1747. doi: 10.1210/en.143.5.1741. [DOI] [PubMed] [Google Scholar]
- 28.• Hartman HB, Gardell SJ, Petucci CJ, et al. Activation of farnesoid X receptor prevents atherosclerotic lesion formation in LDLR-/- and apoE-/- mice. J Lipid Res. 2009; 50:1090–1100. This paper defines a role for FXR in atherosclerosis and the potential mechanisms involved by evaluating the in vivo effect of FXR activation on the formation of atherosclerotic lesions. [DOI] [PMC free article] [PubMed]
- 29.Zhang Y, Castellani LW, Sinal CJ, et al. Peroxisome proliferator-activated receptor-gamma coactivator 1alpha (PGC-1alpha) regulates triglyceride metabolism by activation of the nuclear receptor FXR. Genes Dev. 2004;18:157–169. doi: 10.1101/gad.1138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Claudel T, Inoue Y, Barbier O, et al. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology. 2003;125:544–555. doi: 10.1016/S0016-5085(03)00896-5. [DOI] [PubMed] [Google Scholar]
- 31.Kast HR, Nguyen CM, Sinal CJ, et al. Farnesoid X-activated receptor induces apolipoprotein C-II transcription: a molecular mechanism linking plasma triglyceride levels to bile acids. Mol Endocrinol. 2001;15:1720–1728. doi: 10.1210/me.15.10.1720. [DOI] [PubMed] [Google Scholar]
- 32.Pineda Torra I, Claudel T, Duval C, et al. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol. 2003;17:259–272. doi: 10.1210/me.2002-0120. [DOI] [PubMed] [Google Scholar]
- 33.Andersen E, Karlaganis G, Sjovall J. Altered bile acid profiles in duodenal bile and urine in diabetic subjects. Eur J Clin Invest. 1988;18:166–172. doi: 10.1111/j.1365-2362.1988.tb01270.x. [DOI] [PubMed] [Google Scholar]
- 34.Uchida K, Makino S, Akiyoshi T. Altered bile acid metabolism in nonobese, spontaneously diabetic (NOD) mice. Diabetes. 1985;34:79–83. doi: 10.2337/diabetes.34.1.79. [DOI] [PubMed] [Google Scholar]
- 35.Duran-Sandoval D, Mautino G, Martin G, et al. Glucose regulates the expression of the farnesoid X receptor in liver. Diabetes. 2004;53:890–898. doi: 10.2337/diabetes.53.4.890. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Lee FY, Barrera G, et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci U S A. 2006;103:1006–1011. doi: 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cariou B, van Harmelen K, Duran-Sandoval D, et al. Transient impairment of the adaptive response to fasting in FXR-deficient mice. FEBS Lett. 2005;579:4076–4080. doi: 10.1016/j.febslet.2005.06.033. [DOI] [PubMed] [Google Scholar]
- 38.Duran-Sandoval D, Cariou B, Percevault F, et al. The farnesoid X receptor modulates hepatic carbohydrate metabolism during the fasting-refeeding transition. J Biol Chem. 2005;280:29971–29979. doi: 10.1074/jbc.M501931200. [DOI] [PubMed] [Google Scholar]
- 39.Cariou B, van Harmelen K, Duran-Sandoval D, et al. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem. 2006;281:11039–11049. doi: 10.1074/jbc.M510258200. [DOI] [PubMed] [Google Scholar]
- 40.Maruyama T, Tanaka K, Suzuki J, et al. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J Endocrinol. 2006;191:197–205. doi: 10.1677/joe.1.06546. [DOI] [PubMed] [Google Scholar]
- 41.Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun. 2005;329:386–390. doi: 10.1016/j.bbrc.2005.01.139. [DOI] [PubMed] [Google Scholar]
- 42.• Thomas C, Gioiello A, Noriega L, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009; 10:167–177. This paper evaluates the role of the TGR5 signaling pathway and a TGR5-specific agonist in the in vivo regulation of secretion of intestinal GLP-1. [DOI] [PMC free article] [PubMed]
- 43.Sato H, Genet C, Strehle A, et al. Anti-hyperglycemic activity of a TGR5 agonist isolated from Olea europaea. Biochem Biophys Res Commun. 2007;362:793–798. doi: 10.1016/j.bbrc.2007.06.130. [DOI] [PubMed] [Google Scholar]
- 44.Watanabe M, Houten SM, Mataki C, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 45.• Suzuki T, Oba K, Igari Y, et al. Colestimide lowers plasma glucose levels and increases plasma glucagon-like PEPTIDE-1 (7–36) levels in patients with type 2 diabetes mellitus complicated by hypercholesterolemia. J Nippon Med Sch. 2007; 74:338–343. This paper implicates TGR5-mediated secretion of GLP-1 in the mechanism governing the glucose-lowering effect of bile acid sequestrants. [DOI] [PubMed]
- 46.Beysen C, Murphy E, Deines K, et al. Colesevelam HCl Reduces Fasting Plasma Glucose Concentrations by Improving Plasma Glucose Clearance in Subjects With Type 2 Diabetes. Presented at the 69th Annual Meeting and Scientific Sessions of the American Diabetes Association. New Orleans, LA; June 5–9, 2009.
- 47.Shang Q, Saumoy M, Holst JJ, et al. Colesevelam improves insulin resistance in a diet-induced obesity (F-DIO) rat model by increasing the release of GLP-1. Am J Physiol Gastrointest Liver Physiol. 2009 Dec 31. [Epub ahead of print]. [DOI] [PubMed]
- 48.Schwartz SL, Lai YL, Xu J, et al. The effect of colesevelam hydrochloride on insulin sensitivity and secretion in patients with type 2 diabetes: a pilot study. Metab Syndr Relat Disord. 2010 Jan 8. [Epub ahead of print]. [DOI] [PubMed]
- 49.Staels B. A review of bile acid sequestrants: potential mechanism(s) for glucose-lowering effects in type 2 diabetes mellitus. Postgrad Med. 2009;121:25–30. doi: 10.3810/pgm.2009.05.suppl53.290. [DOI] [PubMed] [Google Scholar]
- 50.Thomas C, Pellicciari R, Pruzanski M, et al. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7:678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- 51.Thomas C, Pellicciari R, Pruzanski M, et al. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7:678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- 52.Cariou B, Staels B. FXR: a promising target for the metabolic syndrome? Trends Pharmacol Sci. 2007;28:236–243. doi: 10.1016/j.tips.2007.03.002. [DOI] [PubMed] [Google Scholar]