

Abstract
Phrenic long term facilitation (pLTF) is a form of respiratory plasticity induced by acute intermittent hypoxia. pLTF requires spinal serotonin receptor activation, new BDNF synthesis and TrkB receptor activation. Spinal adenosine 2A (A2A) receptor activation also elicits phrenic motor facilitation, but by a distinct mechanism involving new TrkB synthesis. Because extracellular adenosine increases during hypoxia, we hypothesized that A2A receptor activation contributes to acute intermittent hypoxia (AIH)-induced pLTF. A selective A2A receptor antagonist (MSX-3, 8 μg kg−1, 12 μl) was administered intrathecally (C4) to anaesthetized, vagotomized and ventilated male Sprague–Dawley rats before AIH (three 5 min episodes, 11% O2). Contrary to our hypothesis, pLTF was greater in MSX-3 versus vehicle (aCSF) treated rats (97 ± 6%vs. 49 ± 4% at 60 min post-AIH, respectively; P < 0.05). MSX-3 and aCSF treated rats did not exhibit facilitation without AIH (time controls; 7 ± 5% and 9 ± 9%, respectively; P > 0.05). A second A2A receptor antagonist (ZM2412385, 7 μg kg−1, 7 μl) enhanced pLTF (85 ± 11%, P < 0.05), but an adenosine A1 receptor antagonist (DPCPX, 3 μg kg−1, 10 μl) had no effect (51%± 8%, P > 0.05), indicating specific A2A receptor effects. Intrathecal methysergide (306 μg kg−1, 15μl) blocked AIH-induced pLTF in both MSX-3 and aCSF treated rats, confirming that enhanced pLTF is serotonin dependent. Intravenous MSX-3 (140 μg kg−1, 1 ml) enhanced both phrenic (104 ± 7%vs. 57 ± 5%, P < 0.05) and hypoglossal LTF (46 ± 13%vs. 28 ± 10%; P < 0.05). In conclusion, A2A receptors constrain the expression of serotonin-dependent phrenic and hypoglossal LTF following AIH. A2A receptor antagonists (such as caffeine) may exert beneficial therapeutic effects by enhancing the capacity for AIH-induced respiratory plasticity.
Introduction
Plasticity is a fundamental property of the respiratory control system (Mitchell & Johnson, 2003). One frequently studied model of respiratory plasticity is phrenic long-term facilitation (pLTF) following acute intermittent hypoxia (AIH; Bach & Mitchell, 1996; Baker & Mitchell, 2000). Although considerable recent progress has been made towards an understanding of cellular/synaptic mechanisms giving rise to pLTF (Feldman et al. 2003; Mitchell & Johnson, 2003), our understanding remains incomplete.
Current evidence supports a mechanism whereby spinal serotonin receptor activation initiates (Baker-Herman & Mitchell, 2002), but does not maintain, AIH-induced pLTF (Fuller et al. 2001b). Specifically, Gq protein-coupled metabotropic serotonin 2 (5-HT2) receptor activation on phrenic motor neurons is believed to activate downstream kinases (e.g. PKC), leading to new protein synthesis, including new synthesis of brain derived neurotrophic factor (BDNF) (Baker-Herman & Mitchell, 2002; Feldman et al. 2003; Baker-Herman et al. 2004; Feldman et al. 2005). Spinal BDNF subsequently elicits pLTF by activation of its high affinity receptor, TrkB (Baker-Herman et al. 2004).
Although serotonergic signalling initiates cellular events underlying pLTF, we recently reported an alternative mechanism of long-lasting phrenic motor facilitation that involves TrkB trans-activation (Golder et al. 2008). Specifically, activation of spinal Gs protein-coupled metabotropic adenosine 2A (A2A) receptors stimulates new synthesis and phosphorylation of an immature TrkB isoform within phrenic motor neurons, thereby inducing phrenic motor facilitation (Golder et al. 2008).
Since moderate hypoxia increases extracellular adenosine levels via active transport and ATP degradation (Winn et al. 1981; Gourine et al. 2002), we hypothesized that adenosine release (and subsequent A2A receptor activation) contributes to AIH-induced pLTF expression. To test this hypothesis, we delivered intrathecal, cervical injections of adenosine receptor antagonists to localize effective drug concentrations to the vicinity of phrenic motor nuclei. A similar approach has been used extensively to study spinal mechanisms of pain perception (Yaksh & Rudy, 1977) and AIH-induced pLTF (Baker-Herman & Mitchell, 2002; Macfarlane & Mitchell, 2007; Wilkerson et al. 2008). An experimental advantage of this approach is the ability to bathe both phrenic motor nuclei (which span four spinal segments; Goshgarian & Rafols, 1981) with drug, while preventing effective drug concentrations elsewhere in the central nervous system. Simultaneous recordings of phrenic and hypoglossal nerve activity provide an internal control for unintended drug distribution (Baker-Herman & Mitchell, 2002).
Contrary to our hypothesis, we discovered a novel mechanism whereby spinal adenosine A2A receptors constrain (versus contribute to) AIH-induced pLTF. A2A receptor modulation of pLTF may provide unique insights concerning fundamental cellular/synaptic mechanisms regulating AIH-induced respiratory plasticity.
Methods
Animals
All experiments were performed on adult (3–6 months) male Sprague–Dawley rats (303–390 g, colonies 236/217/218a; Harlan Inc., Indianapolis, IN, USA). The University of Wisconsin Animal Care and Use committee approved all experimental protocols performed in this study.
In the first series of experiments we tested the hypothesis that spinal adenosine A2A receptors contributed to AIH-induced pLTF by using spinal application of a selective receptor antagonist. The second series was designed to confirm that A2A receptor inhibition per se was responsible for enhanced pLTF. Therefore, a distinct A2A receptor antagonist was used to confirm our results, and contrasted with spinal administration of an A1 receptor antagonist. The third experimental series tested whether spinal A2A receptor antagonist-enhanced pLTF required spinal serotonin receptor activity, confirming that the mechanism is similar to pLTF in normal animals. In the final experimental series, we tested whether systemic (intravenous) A2A receptor antagonist administration enhanced both phrenic and hypoglossal LTF.
Surgical preparation
Anaesthesia was induced with isoflurane (2.5–3.5% in 50% O2, balance N2), and isoflurane was continued during surgical preparations. Once surgical procedures (described below) had been completed, rats were converted to urethane anaesthesia over 15 min via intravenous injection (1.75 g kg−1). Adequate anaesthetic depth was tested by lack of any pressor or respiratory neural response to toe pinch with a haemostat. After conversion to urethane anaesthesia, a continuous intravenous infusion (4–6.5 ml kg−1 h−1) of a 1: 4 mixture of 6% hetastarch and lactated Ringer solution was implemented to maintain fluid balance and acid–base status. A tracheal cannula was placed in the neck to enable artificial ventilation (Rodent Respirator, model 683, Harvard Apparatus, Holliston, MA, USA; tidal volume = 2.5 ml). A rapidly responding flow-through carbon dioxide analyser (Capnogard, Novametrix, Wallingford, CT, USA) was placed on the expired limb of a Y-tube connected to the tracheal cannula to enable measurements of end-tidal CO2 partial pressures 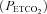 . The vagus nerves were cut in the mid-cervical region to prevent entrainment of respiratory neural activity with the ventilator. During ventilation, rats were subjected to neuromuscular blockade with pancuronium bromide (2.5 mg kg−1, i.v.). A polyethylene catheter (PE50, Intramedic) was placed in the right femoral artery and blood pressure was monitored with a pressure transducer (Gould, P23ID). A three-way stop-cock, attached to the arterial catheter, was used to withdraw blood samples (0.2–0.4 ml) for blood gas analysis (ABL-500, Radiometer; Copenhagen, Denmark); during an experiment, blood gas measurements were made during baseline, during the first hypoxic episode, and at 15, 30 and 60 min post-intermittent hypoxia. Body temperature was monitored with a rectal thermometer (Fisher Scientific, Pittsburg, PA, USA) and maintained (37.5 ± 1°C) with a heated surgical table.
. The vagus nerves were cut in the mid-cervical region to prevent entrainment of respiratory neural activity with the ventilator. During ventilation, rats were subjected to neuromuscular blockade with pancuronium bromide (2.5 mg kg−1, i.v.). A polyethylene catheter (PE50, Intramedic) was placed in the right femoral artery and blood pressure was monitored with a pressure transducer (Gould, P23ID). A three-way stop-cock, attached to the arterial catheter, was used to withdraw blood samples (0.2–0.4 ml) for blood gas analysis (ABL-500, Radiometer; Copenhagen, Denmark); during an experiment, blood gas measurements were made during baseline, during the first hypoxic episode, and at 15, 30 and 60 min post-intermittent hypoxia. Body temperature was monitored with a rectal thermometer (Fisher Scientific, Pittsburg, PA, USA) and maintained (37.5 ± 1°C) with a heated surgical table.
The left phrenic and hypoglossal nerves were isolated using a dorsal approach, cut distally, desheathed and placed on bipolar silver electrodes to record respiratory neural activity. Phrenic and XII nerve signals were amplified (100 000×), band-pass filtered (300–10 000 Hz, Model 1800, A-M Systems, Carlsborg, WA, USA), rectified and integrated (Paynter filter; time constant, 50 ms, MA-821, CWE Inc., Ardmore, PA, USA). The resulting integrated nerve bursts were digitized (8 kHz) and analysed using a WINDAQ data acquisition system (DATAQ Instruments, Akron, OH, USA).
In some rats, the spinal column was exposed dorsally, followed by laminectomy at cervical level 2 (C2), where a small incision was made in dura. A soft silicone catheter (2 French; Access Technologies, Skokie, IL, USA) was inserted caudally through the incision until tip was located at approximately C4. The catheter was attached to a 50 μl Hamilton syringe filled with adenosinergic drug solutions or vehicle to allow localized drug injections into the cervical spinal cord. In experiments using a serotonin receptor antagonist, a second catheter was placed to allow drug administration at different times. The final surgical step was placement of the intrathecal catheter. Rats were then converted to urethane anaesthesia and allowed a minimum of 1 h to stabilize before beginning experimental protocol. Upon completion of experimental protocols, all animals were killed by urethane overdose.
Experimental protocol
The experimental phase began at least 1 h after conversion to urethane anaesthesia when apnoeic and recruitment thresholds were determined by lowering 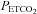 and increasing ventilation until rhythmic nerve bursts could no longer be detected (apnoeic threshold). After approximately 1 min, the ventilator rate was slowly decreased or inspired CO2 was slowly increased until rhythmic nerve bursts resumed (i.e. recruitment threshold). Baseline conditions were then established by holding
and increasing ventilation until rhythmic nerve bursts could no longer be detected (apnoeic threshold). After approximately 1 min, the ventilator rate was slowly decreased or inspired CO2 was slowly increased until rhythmic nerve bursts resumed (i.e. recruitment threshold). Baseline conditions were then established by holding 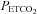 2 mmHg above the recruitment threshold and allowing for stable neural activity (≥15 min). At this point an arterial blood sample was taken to document baseline blood gas levels. Throughout the protocol, arterial
2 mmHg above the recruitment threshold and allowing for stable neural activity (≥15 min). At this point an arterial blood sample was taken to document baseline blood gas levels. Throughout the protocol, arterial 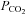 was maintained within ±1.5 mmHg of baseline levels by manipulation of inspired CO2 and/or ventilation rate. Subsets of rats (see Spinal treatment groups for details of n values) received intrathecal injections of vehicle solution (artificial cerebral spinal fluid (aCSF; mm): 120 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 23 NaHCO3, 10 glucose, pH 7.3–7.45), 10% saline or drug (see below) slowly injected over 1 min, 5 min prior to first hypoxic episode (or equivalent time for control groups). Acute hypoxic exposures consisted of three 5 min episodes of isocapnic (±1.5 mmHg) hypoxia (11% inspired O2,
was maintained within ±1.5 mmHg of baseline levels by manipulation of inspired CO2 and/or ventilation rate. Subsets of rats (see Spinal treatment groups for details of n values) received intrathecal injections of vehicle solution (artificial cerebral spinal fluid (aCSF; mm): 120 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 23 NaHCO3, 10 glucose, pH 7.3–7.45), 10% saline or drug (see below) slowly injected over 1 min, 5 min prior to first hypoxic episode (or equivalent time for control groups). Acute hypoxic exposures consisted of three 5 min episodes of isocapnic (±1.5 mmHg) hypoxia (11% inspired O2, 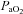 = 35–45 mmHg), separated by 5 min intervals of baseline oxygen levels (51% inspired O2,
= 35–45 mmHg), separated by 5 min intervals of baseline oxygen levels (51% inspired O2, 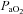 ≥ 150 mmHg). After the third hypoxic episode, rats were returned to baseline inspired oxygen levels and maintained for the duration of an experiment. To test the hypothesis that spinal A2A receptors contributed to AIH-induced pLTF, a selective A2A receptor antagonist (MSX-3, Sigma-Aldrich) was injected via an intrathecal catheter over the cervical spinal cord (C3–5). In this series of experiments, MSX-3 was dissolved in aCSF (10% saline) to an effective concentration determined from preliminary experiments (12 μl, 490 μm; 8 μg kg−1), and delivered 5 min prior to AIH. To provide additional support for A2A receptor inhibition enhancing phrenic LTF, a distinct A2A receptor antagonist (ZM241385, 7 μl, 1 mm; 10% DMSO/aCSF, 7 μg kg−1; Tocris) and an adenosine A1 receptor antagonist (8-cyclopentyl-1,3-dipropylxanthine (DPCPX), 10 μl, 330 μm, 10% DMSO/aCSF, 3 μg kg−1; Tocris Bioscience, Ellisville, MO, USA) were administered identically to MSX-3 and/or vehicle (aCSF). To test whether serotonin receptor activity was required for enhanced pLTF, a subset of rats were exposed to AIH following pretreatment with the broad-spectrum serotonin receptor antagonist methysergide maleate (15 μl, 20 mm, 300 μg kg−1; Sigma-Aldrich), dissolved in aCSF (Baker-Herman & Mitchell, 2002); methysergide was administered 20 min prior to MSX-3 or vehicle injections. Control rats received either vehicle (aCSF, 12 μl) or drug (MSX-3) 5 min prior to initiating an experimental protocol without AIH. The final series of experiments tested whether systemic MSX-3 injections could augment both phrenic and hypoglossal (XII) LTF; thus, rats received MSX-3 dissolved in saline (140 μg kg−1, 1 ml) infused intravenously over 1 min 5 min prior to hypoxic exposure. Again, control rats received MSX-3, in the absence of hypoxia, 5 min prior to initiating a protocol. The following treatment groups were studied.
≥ 150 mmHg). After the third hypoxic episode, rats were returned to baseline inspired oxygen levels and maintained for the duration of an experiment. To test the hypothesis that spinal A2A receptors contributed to AIH-induced pLTF, a selective A2A receptor antagonist (MSX-3, Sigma-Aldrich) was injected via an intrathecal catheter over the cervical spinal cord (C3–5). In this series of experiments, MSX-3 was dissolved in aCSF (10% saline) to an effective concentration determined from preliminary experiments (12 μl, 490 μm; 8 μg kg−1), and delivered 5 min prior to AIH. To provide additional support for A2A receptor inhibition enhancing phrenic LTF, a distinct A2A receptor antagonist (ZM241385, 7 μl, 1 mm; 10% DMSO/aCSF, 7 μg kg−1; Tocris) and an adenosine A1 receptor antagonist (8-cyclopentyl-1,3-dipropylxanthine (DPCPX), 10 μl, 330 μm, 10% DMSO/aCSF, 3 μg kg−1; Tocris Bioscience, Ellisville, MO, USA) were administered identically to MSX-3 and/or vehicle (aCSF). To test whether serotonin receptor activity was required for enhanced pLTF, a subset of rats were exposed to AIH following pretreatment with the broad-spectrum serotonin receptor antagonist methysergide maleate (15 μl, 20 mm, 300 μg kg−1; Sigma-Aldrich), dissolved in aCSF (Baker-Herman & Mitchell, 2002); methysergide was administered 20 min prior to MSX-3 or vehicle injections. Control rats received either vehicle (aCSF, 12 μl) or drug (MSX-3) 5 min prior to initiating an experimental protocol without AIH. The final series of experiments tested whether systemic MSX-3 injections could augment both phrenic and hypoglossal (XII) LTF; thus, rats received MSX-3 dissolved in saline (140 μg kg−1, 1 ml) infused intravenously over 1 min 5 min prior to hypoxic exposure. Again, control rats received MSX-3, in the absence of hypoxia, 5 min prior to initiating a protocol. The following treatment groups were studied.
Spinal treatment groups: (1) vehicle (aCSF) + AIH (n= 7); (2) vehicle, no AIH (n= 8); (3) MSX-3 + AIH (n= 8); (4) MSX-3, no AIH (n= 7); (5) ZM241385 + AIH (n= 5); (6) DPCPX + AIH (n= 5). A second intrathecal catheter was placed in rats pretreated with methysergide; thus, a subset of control rats also received dual-catheter placement: (1) vehicle/MSX-3 + AIH (n= 3); (2) methysergide/MSX-3 + AIH (n= 5); (3) vehicle/vehicle + AIH (n= 5); (4) methysergide/vehicle + AIH (n= 5); (5) methysergide/MSX-3, no AIH (n= 6).
Systemic treatment groups: (1) vehicle (saline) + AIH (n= 9); (2) MSX-3 + AIH (n= 12); (3) MSX-3, no AIH (n= 9).
Data analysis
Integrated phrenic and hypoglossal nerve burst amplitudes and burst frequencies were averaged over 1 min bins at each experimental time point (baseline, 15, 30 and 60 min). Nerve burst amplitudes were normalized by subtracting from the baseline value, then dividing by the baseline and reported as percentage change from baseline. Burst frequencies were also normalized to baseline, but expressed as an absolute difference in bursts per minute. All statistical comparisons between treatment groups for nerve amplitude (15, 30 and 60 min time points) were made using a two-way ANOVA with a repeated measures design. Since no differences were detected between hypoxic exposures within groups (data not shown), comparisons were made using two-way ANOVA of phrenic burst amplitude during the fifth minute of hypoxic episodes averaged from all three episodes. Nerve bursts from rats treated with methysergide (serotonin receptor antagonist), ZM241385 (A2A antagonist) and DPCPX (A1 antagonist) were compared at the 60 min post-AIH time point using a two-way ANOVA. Comparisons for mean arterial pressures, 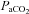 and
and 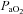 were made at baseline, hypoxia 1 and 60 min post-AIH using two-way ANOVA. All individual comparisons were made using the Student–Neuman–Keuls (SNK) post hoc test (Sigma-Stat version 2.03; Systat Software Inc., San Jose, CA, USA). Differences between groups were considered significant if P < 0.05. All values are expressed as means ±s.e.m.
were made at baseline, hypoxia 1 and 60 min post-AIH using two-way ANOVA. All individual comparisons were made using the Student–Neuman–Keuls (SNK) post hoc test (Sigma-Stat version 2.03; Systat Software Inc., San Jose, CA, USA). Differences between groups were considered significant if P < 0.05. All values are expressed as means ±s.e.m.
Results
Blood gases and mean arterial pressures
Measurements of arterial 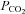 and
and 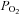 during baseline conditions were similar in all experimental groups (see Table 1). Furthermore, in all groups, arterial
during baseline conditions were similar in all experimental groups (see Table 1). Furthermore, in all groups, arterial 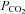 was regulated successfully within 1.5 mmHg of its baseline value and arterial
was regulated successfully within 1.5 mmHg of its baseline value and arterial 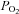 remained above 150 mmHg (Table 1). Mean arterial pressure (MAP) was similar between all treatment groups at baseline, during hypoxia and 60 min post-AIH (Table 1). Thus
remained above 150 mmHg (Table 1). Mean arterial pressure (MAP) was similar between all treatment groups at baseline, during hypoxia and 60 min post-AIH (Table 1). Thus 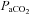 ,
, 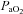 and MAP were stable throughout the experimental protocol in all groups.
and MAP were stable throughout the experimental protocol in all groups.
Table 1.
Measurements of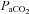 ,
, 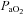 and mean arterial pressure (MAP) during baseline, hypoxia and 60 min post-hypoxia
and mean arterial pressure (MAP) during baseline, hypoxia and 60 min post-hypoxia
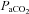 (mmHg) (mmHg) |
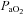 (mmHg) (mmHg) |
MAP (mmHg) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| baseline | hypoxia | 60 min | baseline | hypoxia | 60 min | baseline | hypoxia | 60 min | |
| Spinal Groups | |||||||||
| vehicle | 41.4 ± 1.0 | 43.1 ± 0.8 | 41.2 ± 1.1 | 260 ± 7 | 35 ± 2†‡ | 234 ± 15 | 118 ± 7 | 97 ± 8 | 106 ± 8 |
| vehicle (–AIH) | 44.6 ± 1.2 | 45.5 ± 1.2 | 44.9 ± 1.1 | 274 ± 9 | 270 ± 7 | 272 ± 7 | 95 ± 4 | 101 ± 3 | 103 ± 4 |
| MSX-3 | 41.9 ± 0.9 | 42.0 ± 1.0 | 41.5 ± 0.8 | 255 ± 8 | 38 ± 2†‡ | 247 ± 9 | 117 ± 7 | 80 ± 13† | 103 ± 8 |
| MSX3 (–AIH) | 44.0 ± 1.6 | 42.8 ± 1.5 | 43.8 ± 1.3 | 260 ± 14 | 254 ± 15 | 260 ± 10 | 112 ± 5 | 103 ± 6 | 99 ± 6 |
| DPCPX | 43.8 ± 1.4 | 43.7 ± 1.1 | 44.1 ± 1.3 | 269 ± 2 | 40 ± 0†‡ | 266 ± 2 | 137 ± 8‡ | 108 ± 11† | 118 ± 7 |
| ZM241385 | 43.1 ± 1.5 | 43.3 ± 2.2 | 43.2 ± 1.5 | 264 ± 1 | 37 ± 0†‡ | 244 ± 2 | 108 ± 8 | 83 ± 19 | 93 ± 7 |
| vehicle+vehicle | 43.6 ± 1.2 | 44.2 ± 1.2 | 43.5 ± 1.0 | 272 ± 5 | 37 ± 2† | 272 ± 9 | 116 ± 3 | 85 ± 10 | 101 ± 4 |
| methy+vehicle | 42.5 ± 1.1 | 45.3 ± 0.7 | 42.8 ± 1.2 | 279 ± 6 | 37 ± 1† | 273 ± 7 | 124 ± 9 | 109 ± 10 | 118 ± 8 |
| MSX-3+vehicle | 44.5 ± 1.6 | 45.1 ± 1.6 | 44.7 ± 1.3 | 287 ± 8 | 37 ± 3 | 273 ± 6 | 108 ± 4 | 96 ± 10 | 92 ± 9 |
| MSX-3+methy | 42.1 ± 1.3 | 42.1 ± 1.5 | 43.4 ± 1.3 | 279 ± 12 | 43 ± 2† | 266 ± 7 | 126 ± 9 | 110 ± 15 | 119 ± 9 |
| Systemic Groups | |||||||||
| vehicle | 43.0 ± 0.6 | 42.9 ± 1.0 | 43.4 ± 0.6 | 270 ± 5 | 36 ± 1†‡ | 256 ± 7 | 113 ± 6 | 110 ± 5 | 107 ± 8 |
| MSX-3 | 43.8 ± 0.5 | 42.5 ± 0.9 | 44.2 ± 0.5 | 258 ± 7 | 35 ± 1†‡ | 242 ± 9 | 120 ± 4 | 98 ± 8† | 103 ± 6† |
| vehicle (–AIH) | 43.4 ± 0.5 | 42.2 ± 1.2 | 43.4 ± 0.5 | 257 ± 12 | 272 ± 7 | 265 ± 15 | 117 ± 5 | 113 ± 4 | 117 ± 5 |
Values are means ±s.e.m. MAP, mean arterial pressure.
Significant different than baseline;
significant different than vehicle (–AIH).
Short-term hypoxic responses
Spinally treated rats
Spinal injection of an A2A receptor antagonist (MSX-3, 12 μl) had no effect on baseline frequency or amplitude of peak integrated inspiratory phrenic or hypoglossal nerve bursts (data not shown), thus assuring that pLTF comparisons relative to baseline were appropriate. Typical phrenic neurograms during experimental protocols for each treatment group are illustrated in Fig. 1. Hypoxia elicited a rapid increase in phrenic and hypoglossal burst amplitude in vehicle (133 ± 10%, n= 7; 180 ± 48%, n= 5; respectively; P < 0.001) and MSX-3 treated rats (184 ± 12%, n= 9; 176%± 16%, n= 6; respectively; P < 0.001; Fig. 2). Phrenic burst amplitude from MSX-3 treated rats was significantly greater than in vehicle-treated rats (184 ± 12%versus 133 ± 10%, respectively; P= 0.007); however, there was no difference in hypoglossal burst amplitude (180 ± 28%versus 176 ± 16%, respectively, P= 0.816; Fig. 2).
Figure 1. Representative phrenic neurograms depicting experimental protocols in rats from different treatment groups.

A, intrathecal vehicle (aCSF) prior to AIH. B, intrathecal A2A receptor antagonist (MSX-3) prior to AIH. C, time control receiving intrathecal MSX-3 without AIH. D, time control receiving intrathecal vehicle without AIH. MSX-3 pretreatment enhanced pLTF relative to AIH exposed rats given control, vehicle injections. Neither time control experiment exhibited pLTF.
Figure 2. Hypoxic phrenic and hypoglossal responses during hypoxic episodes (i.e. short-term hypoxic response).

Changes in phrenic and hypoglossal (XII) burst amplitudes during final minute of hypoxic exposures (average of 3) from spinal (▪) vehicle (aCSF; phr, n= 7; XII, n= 5) and A2A receptor antagonist (MSX-3; phr, n= 8; XII, n= 6); systemic (□) vehicle (saline; phr, n= 9; XII, n= 5) and MSX-3 (Phr, n= 12; XII, n= 4) treated rats. Phrenic burst amplitudes from spinal MSX-3 treated rats were increased versus vehicle treated rats, but not in rats with systemic MSX-3 administration. Values are means ±s.e.m. #Different from baseline; †different from vehicle, ANOVA, P < 0.05.
Systemically treated rats
Exposure to hypoxia elicited a rapid increase in phrenic and hypoglossal burst amplitudes in rats receiving intravenous vehicle (saline: 157 ± 17%, n= 9; 151 ± 20%, n= 5; respectively; P < 0.001) and MSX-3 (177 ± 23%, n= 12; 270 ± 80%, n= 4; respectively; P < 0.001). However, in contrast to intrathecal drug administration, short-term hypoxic responses were not different between experimental groups for either nerve (P > 0.15; Fig. 2).
Spinal A2A, but not A1 receptors constrain AIH-induced pLTF
AIH-induced pLTF in vehicle treated rats (artificial CSF, 12 μl; Figs 2 and 3A) is expressed as an increase in phrenic burst amplitude (49 ± 4%, 60 min post-AIH; P < 0.001; n= 7). Phrenic burst amplitude was also significantly greater in vehicle treated rats receiving AIH versus those not exposed to AIH (i.e. time controls: 9 ± 5%, P < 0.001, n= 8; Fig. 3A) at 60 min post-AIH.
Figure 3. Time course of phrenic and hypoglossal (XII) burst amplitude changes following spinal drug (or vehicle) injections with or without (ctrl) AIH.

Changes in phrenic (A) and XII (B) burst amplitudes (percentage change from baseline) in rats receiving vehicle + AIH (♦, n= 7), MSX-3 + AIH (▪, n= 8), vehicle – AIH (♦n= 8) and MSX-3 – AIH (□, n= 7). Spinal MSX-3 augmented phrenic burst amplitude responses following AIH, indicative of enhanced pLTF. However, similar enhancement of XII LTF was not observed, suggesting that drug distribution was restricted to the spinal cord. Values are means ±s.e.m. #Different from baseline; †different from vehicle at the same time point; ‡different from MSX-3 ctrl at the same time point; *different from vehicle ctrl at the same time point; RMANOVA; P < 0.05.
To test whether spinal Gs protein-coupled A2A receptors contribute to pLTF, two distinct antagonists, MSX-3 (12 μl, 8 μg kg−1) and ZM241385 (7 μl, 7 μg kg−1), were administered to the intrathecal space of the cervical spinal cord (C3–5). Contrary to our original hypothesis, phrenic burst amplitude was increased above baseline levels at 60 min post-AIH (MSX-3: 97 ± 10%, n= 8; Fig. 3A; ZM241385: 85 ± 15%, n= 5, P < 0.001; Fig. 4A) and were significantly greater than vehicle-treated rats receiving AIH (49 ± 4%, P < 0.001; Fig. 3A) or controls not exposed to AIH (vehicle: 9 ± 5%, n= 8; P < 0.001; MSX-3: 10 ± 6%, n= 7; P < 0.001; Fig. 3A). However, no difference in phrenic burst amplitude was detected between MSX-3 and ZM241385 treatment at the 60 min time point (P= 0.458, Fig. 4A).
Figure 4. Effect of spinal adenosine A1, A2A and serotonin receptor antagonists on pLTF.

A, changes in phrenic burst amplitudes (% baseline) at 60 min post-AIH are depicted in rats receiving intrathecal injections of vehicle (aCSF), spinal adenosine A1 receptor antagonist (DPCPX, n= 6) and two A2A receptor antagonists (MSX-3, n= 8; and ZM241385, n= 5). †Different from vehicle; ‡different from DPCPX. B, with dual catheter placement, vehicle + vehicle with AIH (n= 4), methyergide + vehicle with AIH (n= 5), MSX-3 + vehicle with AIH (n= 4) and methysegide + MSX-3 with AIH. pLTF enhancement is not observed following spinal A1 receptor inhibition, in contrasted to A2A receptor antagonists. Enhanced pLTF following MSX-3 is effectively blocked by methysergide. Values are means ±s.e.m. †Different from vehicle + vehicle; ‡different from methysegide + vehicle; #different from methysegide + MSX-3; ANOVA; P < 0.05.
To address possible concerns that spinal A2A receptor inhibition alone induces pLTF, rats were treated with intrathecal MSX-3 but without AIH (n= 7); in these rats, no significant increase in phrenic burst amplitude was observed at any time post-AIH (15 min: 13 ± 8%, P= 0.353; 30 min: 13 ± 8%, P= 0.242; 60 min: 10 ± 6%, P= 0.24; Fig. 3A).
Since adenosinergic A1 receptor inhibition increases synaptic transmission, reportedly via presynaptic disinhibition of neurotransmitter release (Bellingham & Berger, 1994; Dong & Feldman, 1995), we tested whether unintended effects of MSX-3 on A1 receptors could account for enhanced pLTF. In rats pre-treated with an A1 receptor antagonist (DPCPX, 10 μl, 3 μg kg−1), phrenic burst amplitude was above baseline at 60 min post-AIH (51 ± 8%, n= 6; P= 0.005; Fig. 4A) and greater than in control rats receiving vehicle or MSX-3 without AIH (9 ± 5%, n= 8; 10 ± 6%, n= 7; respectively; P < 0.001). Phrenic burst amplitude after A1 receptor inhibition (DPCPX) was not different from vehicle (49 ± 4%, P= 0.820, n= 7) treatment at 60 min post-AIH, but was significantly lower than in A2A antagonist treated rats following AIH (MSX-3: 97 ± 6%, n= 8; and ZM241385: 85 ± 15%, n= 5; P < 0.001; Fig. 4A). Together, these results confirm that A2A, and not A1, receptors are the relevant receptor subtype for enhanced pLTF. Thus, spinal injections of A2A receptor antagonists enhance AIH-induced pLTF, suggesting that endogenous spinal A2A receptors actually constrain rather than contribute to AIH-induced pLTF.
Enhanced pLTF requires spinal serotonin receptor activation
Since activation of spinal serotonin receptors is necessary for AIH-induced pLTF in normal rats (Baker-Herman & Mitchell, 2002), we hypothesized that spinal MSX-3 enhanced pLTF results from the same, serotonin-dependent mechanism. To test this hypothesis, a serotonin receptor antagonist (methysergide-maleate, 15 μl, 300 μg kg−1) was applied prior to MSX-3 administration and exposure to AIH. Because methysergide injections required placement of a second catheter into the intrathecal space, we repeated a subset of experiments (see Methods: Spinal treatment groups) to confirm that placement of two intrathecal catheters did not influence our results. Combined intrathecal methysergide and MSX-3 in the absence of hypoxia had no significant effect on frequency or amplitude of phrenic and hypoglossal nerve bursts (data not shown), and thus pLTF comparisons were made relative to baseline. pLTF after artificial CSF in one catheter and MSX-3 in the other (i.e. dual catheter placement) was significantly greater than in dual-vehicle injections at 60 min post-AIH (97 ± 13%, n= 4; 34 ± 4%, n= 4; respectively; P < 0.001; Fig. 4B), demonstrating enhanced pLTF. When pretreated with methysergide, pLTF was abolished in vehicle (−6 ± 9%, P < 0.001, n= 5) and MSX-3 treated rats at 60 min post-AIH (−5 ± 7%, P < 0.001, n= 5; Fig. 4B), confirming that enhanced pLTF is serotonin-dependent.
Spinal MSX-3 does not affect hypoglossal LTF
Our goal was to localize the effects of MSX-3 to the spinal cord, avoiding effects on brainstem respiratory neurons. Thus, recordings of hypoglossal burst activity were made to assure that similar effects (MSX-3 enhancement) were not observed in these cranial motor neurons. Rats receiving either intrathecal vehicle or MSX-3 injections exhibited significant hypoglossal LTF at 60 min post-AIH (47 ± 15%, P= 0.001, n= 5; 45 ± 8%, P < 0.001, n= 6, respectively; Fig. 3B), and these effects did not differ from one another (P= 0.884; Fig. 3B). In control rats receiving spinal MSX-3 without hypoxia, hypoglossal burst amplitude was not different from baseline at any time point (15 min: 0 ± 7%, P= 0.982; 30 min: −10 ± 8%, P= 0.707; 60 min: −4 ± 10%, P= 0.928, n= 7; Fig. 3B). Therefore, although spinal A2A receptor inhibition augments pLTF, it has no effect on hypoglossal LTF, suggesting that drug distribution was restricted to the region of interest (i.e. cervical spinal cord).
Intravenous MSX-3 enhances hypoglossal and phrenic LTF
Although the primary purpose of this study was to test whether spinal A2A receptors contribute to AIH-induced pLTF, we also sought to know whether intravenous MSX-3, distributing to the entire CNS, would have similar effects on phrenic and hypoglossal LTF. Since MSX-3 is a water-soluble drug capable of crossing the blood–brain barrier (Schindler et al. 2005), we predicted that systemic MSX-3 would enhance both phrenic and hypoglossal LTF. In rats receiving intravenous vehicle (saline) injection, hypoglossal nerve burst amplitude was not significantly different from baseline at 60 min post-AIH (28 ± 10%, P= 0.349, n= 4; Fig. 5B). Thus, hypoglossal LTF is weak in the specific rats used in this experimental series, a finding previously reported in certain sub-strains of Sprague–Dawley rats (Fuller et al. 2001a). However, in rats pre-treated with MSX-3, robust hypoglossal LTF was observed 60 min post-AIH (46 ± 13%, P= 0.008, n= 4), an effect significantly greater than in rats receiving MSX-3 without AIH (1 ± 6%, P= 0.01; Fig. 5B).
Figure 5. Time course of phrenic and hypoglossal (XII) burst amplitude changes following systemic drug injections.

Changes in integrated phrenic (A) and hypoglossal (XII) (B) burst amplitudes from rats receiving intravenous vehicle with AIH (saline, ♦ phrenic n= 9, XII n= 6), MSX-3 with AIH (▪; phrenic n= 12, XII n= 5), and control (ctrl) MSX-3 without AIH (□; phrenic n= 9, XII n= 4). Systemic MSX-3 enhanced both phrenic and XII LTF. Values are means ±s.e.m. #Different from baseline; †different from vehicle at the same time point; ‡different from MSX-3 ctrl at the same time point; RMANOVA; P < 0.05.
Similar to spinally treated rats, phrenic burst amplitude from vehicle (saline) treated rats was above baseline at 60 min post-AIH (57 ± 6%, P < 0.001, n= 9). Systemic MSX-3 significantly enhanced pLTF 60 min post-AIH (107 ± 9%, P < 0.001, n= 12; Fig. 5A). Following intravenous MSX-3 administration without AIH, phrenic burst amplitude was significantly above baseline levels at the 60 min time point (19 ± 8%, P= 0.024, n= 9; Fig. 5A), suggesting that certain A2A receptor sites may influence respiratory motor activity following systemic, but not spinal, MSX-3 administration. Nevertheless, phrenic burst amplitude after vehicle (saline) and MSX-3 treatment was significantly greater than MSX-3 without AIH (P < 0.001, respectively; Fig. 5A). Collectively, these results demonstrate that systemic A2A antagonists enhance AIH-induced respiratory plasticity in both hypoglossal and phrenic motor output.
Respiratory frequency facilitation
AIH-induced frequency LTF in this experimental preparation is generally small and inconsistent (Baker-Herman & Mitchell, 2008). In spinally treated rats receiving AIH, phrenic burst frequency increased over baseline in vehicle (7 ± 3 bursts min−1, P= 0.03, n= 7) and MSX-3 treated rats at 60 min post-AIH (9 ± 2 bursts min−1, P= 0.002, n= 8; Fig. 6A). However, no burst frequency differences were detected between vehicle and MSX-3 treated rats (P= 0.20). Phrenic burst frequency following spinal MSX-3 alone was not different from baseline at any measured time point (15 min: 1 ± 1 bursts min−1, P= 0.996; 30 min: 1 ± 1 bursts min−1, P= 0.903; 60 min: 1 ± 2 bursts min−1, P= 0.982; n= 7), and was significantly less than in vehicle (P= 0.011) and MSX-3 treated rats (P < 0.001) 60 min post-AIH. Burst frequency responses in vehicle and MSX-3 treated rats without AIH not did not differ (P= 0.287; Fig. 6A).
Figure 6. Time course of changes in respiratory nerve burst frequency in experimental groups.

Changes in phrenic nerve burst frequency from baseline (Δfrequency; bursts min−1) in rats receiving spinal (A) and intravenous (B) treatments: vehicle with AIH (♦ aCSF or saline, respectively), MSX-3 with AIH (▪), MSX-3 time-control without AIH (□) and vehicle control without AIH (♦). Phrenic burst frequency in rats receiving AIH increased from baseline at 60 min post-AIH, indicative of frequency LTF. However, this response was unaffected by MSX-3. #Different from baseline; ‡different from MSX-3 ctrl, *different from vehicle ctrl; RMANOVA; P < 0.05.
Phrenic burst frequency in rats receiving intravenous vehicle (saline) or MSX-3 was increased over baseline at 60 min post-AIH (11 ± 2 bursts min−1, P < 0.001, n= 12; 12 ± 1 bursts min−1, P < 0.001, n= 9; respectively; Fig. 6B), but were not different from one another. Phrenic burst frequency following spinal MSX-3 treatment alone was not different from baseline at the 60 min time point (−2 ± 2, P= 0.333; n= 9), but was significantly decreased from MSX-3 and vehicle (P < 0.001, respectively) treated rats 60 min post-AIH (Fig. 6B). Therefore neither spinal nor systemic MSX-3 appears to augment AIH-induced burst frequency facilitation.
Discussion
Here we demonstrate that spinal A2A receptor activation constrains AIH-induced pLTF in rats, refuting our original hypothesis that these receptors contribute to pLTF. This is the first evidence that A2A receptor signalling modulates respiratory plasticity following acute intermittent hypoxia, and is difficult to reconcile with our previous report that A2A receptor activation without hypoxia elicits long-lasting phrenic motor facilitation (Golder et al. 2008). We now postulate that AIH-induced pLTF (Gq protein-mediated) and A2A receptor induced phrenic motor facilitation (Gs protein-mediated) are distinct mechanisms that interact via cross-talk inhibition. In this context, we propose that both serotonin receptors associated with phrenic LTF and A2A receptors are activated during AIH, but that serotonin-dependent mechanisms predominate. Thus, instead of inducing phrenic motor facilitation, cross-talk inhibition from the A2A receptor-dependent pathway constrains serotonin-dependent pLTF during AIH.
Since systemic MSX-3 augments both hypoglossal and phrenic LTF, whereas spinal MSX-3 augments pLTF alone, we suggest that the A2A receptor constraint to AIH-induced LTF occurs within these respective motor nuclei. Elucidation of cellular/synaptic signalling events underlying MSX-3 enhanced pLTF may provide unique insights concerning regulatory mechanisms that govern respiratory LTF expression. Moreover, small molecules that cross the blood–brain barrier, targeting respiratory motor neurons, hold promise as a novel therapeutic strategy to treat respiratory insufficiency during lung, airway or neural disease (Mahamed & Mitchell, 2007; Mitchell, 2007).
Enhanced pLTF is serotonin dependent
pLTF is a form of respiratory plasticity induced by serotonin receptor activation (Feldman et al. 2003; Mitchell & Johnson, 2003). Our working model of AIH-induced pLTF is that episodic hypoxia activates raphe serotonergic neurons that project to phrenic motor nuclei. Spinal serotonin release during hypoxic episodes subsequently activates Gq protein-coupled 5-HT2 receptors on or near phrenic motor neurons, and initiates intracellular cascades that underlie pLTF.
A2A receptors in the nucleus tractus solitarii (NTS) augment electrically evoked serotonin release in superfused rat medullary slices via presynaptic mechanisms (Barraco et al. 1996). Additionally, selective A2A receptor agonists (e.g. PD125944, cgs21680) increase extracellular serotonin levels in the hippocampus, although only in the presence of A1 receptor antagonists (Okada et al. 1997). Hence, spinal A2A receptors may augment available serotonin via increased release and/or decreased re-uptake. This study provides no evidence for such an effect during AIH since, contrary to predictions, A2A receptor antagonists enhanced pLTF.
Other experimental interventions also induce serotonin receptor-dependent enhancement of pLTF, such as pre-conditioning with chronic intermittent hypoxia or chronic cervical dorsal rhizotomy (Kinkead et al. 1998; Ling et al. 2001). Ling and colleagues reported that chronic intermittent hypoxia enhanced the short-term hypoxic phrenic response and pLTF (Ling et al. 2001). The enhanced pLTF was methysergide sensitive, and of a similar magnitude to MSX-3 enhanced phrenic LTF (Ling et al. 2001). Cervical dorsal rhizotomy increases serotonergic innervation of the phrenic motor nucleus and enhances AIH-induced pLTF (Kinkead et al. 1998). Moreover cervical dorsal rhizotomy increases spinal BDNF and NT-3 levels in ventral cervical segments that encompass the phrenic motor nucleus (Johnson et al. 2000). MSX-3 enhanced pLTF may involve similar signalling molecules to these experimental treatments, but this hypothesis remains to be tested. Since both chronic intermittent hypoxia and cervical dorsal rhizotomy are lasting pre-conditions that elicit respiratory meta-plasticity (i.e. enhanced phrenic LTF; Mitchell & Johnson, 2003), MSX-3 is unique as an acute pharmacological intervention that produces the same experimental result. A more complete understanding of cellular mechanisms giving rise to A2A receptor antagonist enhancement of pLTF may also increase our understanding of signalling events that underlie ‘normal’ AIH-induced pLTF.
Potential cellular mechanism of MSX-3 enhanced pLTF
There is an abundance of literature demonstrating ‘cross-talk’ between Gq and Gs protein-coupled receptor pathways (Meszaros et al. 2000; Roy et al. 2006; Ryzhov et al. 2006). For example, PKC activation (e.g. 5-HT2 receptors) modulates Gs protein-coupled receptor (GsPCR) pathways (e.g. A2A), possibly through adenylyl cyclase or regulators of G-proteins (RGS; Zimmermann & Taussig, 1996; Lai et al. 1997; Cunningham et al. 2001; Roy et al. 2006). A2A receptor activation of adenylyl cyclase is inhibited by PKC phosphorylation, which prevents second messenger transduction through G-proteins (Zimmermann & Taussig, 1996; Lai et al. 1997). Moreover PKC is known to phosphorylate RGS proteins which, in turn, modulate activity of multiple G-proteins, including those involved in GsPCR signalling (e.g. Gαs; Cunningham et al. 2001). Although speculative, we propose that, during AIH, GqPCR mediated signalling (i.e. AIH-induced pLTF) predominates, and suppresses GsPCR mediated signalling via‘cross-talk’ inhibition.
Conversely, PKA may mediate inhibitory ‘cross-talk’ to GqPCR signalling via modulation of cellular reactive oxygen species (ROS) production. Hypoxia followed by re-oxygenation generates ROS by several cellular mechanisms (Zulueta et al. 1997; Li & Jackson, 2002; Abramov et al. 2007). Scavenging superoxide anions with a superoxide dismutase mimetic blocks AIH-induced pLTF, demonstrating that superoxide anions are necessary in its underlying mechanism (Macfarlane & Mitchell, 2007). Relevant ROS appear to be derived from spinal NADPH oxidase activity since spinal administration of the NADPH oxidase inhibitors apocynin and diphenyleneiodonium chloride (DP) abolish pLTF (Macfarlane et al. 2008; MacFarlane et al. 2009). In our working cellular/synaptic model of pLTF, ROS generated during AIH inhibit protein phosphatases that normally constrain the cellular cascade leading to pLTF (Macfarlane et al. 2008; Wilkerson et al. 2008). NADPH oxidase-derived ROS have been implicated in other forms of respiratory plasticity, including carotid sensory LTF in rats pre-treated with chronic intermittent hypoxia (Peng & Prabhakar, 2003; Peng et al. 2006). We suggest that modulatory influences of A2A receptors on pLTF (i.e. GqPCR-induced PMF) are indirectly mediated via regulation of NADPH oxidase activity as described above. Thus, MSX-3 may remove an endogenous adenosine A2A receptor/PKA-dependent constraint on transient ROS production necessary for AIH-induced pLTF. A2A receptor (and subsequent PKA) activation during AIH may inhibit NADPH-dependent ROS formation, maintaining suboptimal ROS levels for cellular mechanisms that normally gives rise to pLTF. Spinal A2A receptor antagonists may relieve this limitation, increasing ROS formation and enhancing pLTF.
Experimental Considerations
Drug localization
AIH-induced pLTF results from mechanisms on or near phrenic motor neurons (Mitchell et al. 2001). For example, pLTF is abolished after cervical spinal application of serotonin receptor antagonists or protein synthesis inhibitors (Baker-Herman & Mitchell, 2002), suggesting that pLTF results from plasticity in or near the phrenic motor nucleus. Contrary to our original hypothesis that spinal A2A receptors contribute to AIH-induced pLTF, A2A receptors appear to constrain these mechanisms.
MSX-3 applied to the cervical spinal cord augmented phrenic, but not hypoglossal, LTF. Since intravenous MSX-3 administration enhances both phrenic and hypoglossal LTF, hypoglossal motor output is capable of expressing enhanced LTF. Collectively, these results suggest that effective MSX-3 concentrations following intrathecal administration were restricted to spinal regions associated within the phrenic motor nucleus, and did not reach brainstem XII motor neurons.
In time-control rats receiving systemic MSX-3 without AIH, modest drift in phrenic burst amplitude was observed (Fig. 5A). A similar response was not observed in spinally treated rats (Fig. 3A). This effect may result from MSX-3 acting at the level of carotid chemoreceptors since A2A receptors are expressed by carotid body type I cells and inhibit their activity (Kobayashi et al. 2000; Conde et al. 2006). This hypothesis remains to be tested.
Significance
AIH induced pLTF is one of the most frequently studied models of respiratory plasticity (Mahamed & Mitchell, 2007). Our understanding of cellular/synaptic mechanisms giving rise to pLTF has increased substantially, yet critical details of the signalling mechanism are not yet known (Mahamed & Mitchell, 2007). Elucidation of cellular mechanisms whereby A2A receptors modulate pLTF expression may yield novel insights concerning fundamental mechanisms of hypoxia-induced respiratory plasticity. MSX-3 enhanced pLTF may ‘amplify’ existing signalling pathways to pLTF, possibly increasing our ability to discriminate these signalling events.
From a different perspective, multiple ventilatory control disorders share a common therapeutic goal: amplification of synaptic inputs onto respiratory motor neurons (Mitchell, 2007). A2A receptor antagonists represent an interesting class of small molecules that cross the blood–brain barrier (Sauer et al. 2000) and have the capacity to (indirectly) augment respiratory motor neuron function, at least in concert with AIH. Caffeine, an adenosine (A2A) receptor antagonist, is commonly used in neonatal intensive care units as a respiratory stimulant for premature newborn infants with respiratory instability (Finer et al. 2006). While, the localization of caffeine effects (central nervous system and/or peripheral chemoreceptors) is not known, current clinical use suggests that further development of selective adenosine receptor antagonists may be useful in augmenting respiratory motor output. Thus, A2A receptor antagonists may be of clinical interest in the treatment of respiratory insufficiency after spinal cord injury, during neurodegenerative diseases and, possibly, apnoea of prematurity and/or obstructive sleep apnoea (Mitchell, 2007).
Acknowledgments
This work was supported by grants NIH HL80209 and NRSA HL092785.
Glossary
Abbreviations
- A2A
adenosine 2A
- AIH
acute intermittent hypoxia
- PMF
phrenic motor facilitation
- ROS
reactive oxygen species
- pLTF
phrenic long-term facilitation
- XII
hypoglossal
Author contributions
M.H., F.G., S.M., and G.M., contributed to the conception, design, and analysis of experiments including interpretation of data and drafting/revising the final version of the article approved for publication. All experiments were carried out at University of Wisconsin, Madison, Wisconsin, USA.
Authors’ present addresses
F. J. Golder: Department of Clinical Studies – Philadelphia, University of Pennsylvania, United States 3900 Delancey Street, Philadelphia, PA 19104, USA
S. Mahamed: Department of Physiology, School of Medicine and Public Health, University of Wisconsin, 1300 University Avenue, Madison, WI 53706, USA
References
- Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci. 2002;22:6239–6246. doi: 10.1523/JNEUROSCI.22-14-06239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Determinants of frequency long-term facilitation following acute intermittent hypoxia in vagotomized rats. Respir Physiol Neurobiol. 2008;162:8–17. doi: 10.1016/j.resp.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TL, Mitchell GS. Episodic but not continuous hypoxia elicits long-term facilitation of phrenic motor output in rats. J Physiol. 2000;529:215–219. doi: 10.1111/j.1469-7793.2000.00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barraco RA, Helfman CC, Anderson GF. Augmented release of serotonin by adenosine A2a receptor activation and desensitization by CGS 21680 in the rat nucleus tractus solitarius. Brain Res. 1996;733:155–161. doi: 10.1016/0006-8993(96)00279-x. [DOI] [PubMed] [Google Scholar]
- Bellingham MC, Berger AJ. Adenosine suppresses excitatory glutamatergic inputs to rat hypoglossal motoneurons in vitro. Neurosci Lett. 1994;177:143–146. doi: 10.1016/0304-3940(94)90065-5. [DOI] [PubMed] [Google Scholar]
- Conde SV, Obeso A, Vicario I, Rigual R, Rocher A, Gonzalez C. Caffeine inhibition of rat carotid body chemoreceptors is mediated by A2A and A2B adenosine receptors. J Neurochem. 2006;98:616–628. doi: 10.1111/j.1471-4159.2006.03912.x. [DOI] [PubMed] [Google Scholar]
- Cunningham ML, Waldo GL, Hollinger S, Hepler JR, Harden TK. Protein kinase C phosphorylates RGS2 and modulates its capacity for negative regulation of Gα 11 signalling. J Biol Chem. 2001;276:5438–5444. doi: 10.1074/jbc.M007699200. [DOI] [PubMed] [Google Scholar]
- Dong XW, Feldman JL. Modulation of inspiratory drive to phrenic motoneurons by presynaptic adenosine A1 receptors. J Neurosci. 1995;15:3458–3467. doi: 10.1523/JNEUROSCI.15-05-03458.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Neverova NV, Saywell SA. Modulation of hypoglossal motoneuron excitability by intracellular signal transduction cascades. Respir Physiol Neurobiol. 2005;147:131–143. doi: 10.1016/j.resp.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Finer NN, Higgins R, Kattwinkel J, Martin RJ. Summary proceedings from the Apnea-of-Prematurity Group. Pediatrics. 2006;117:S47–51. doi: 10.1542/peds.2005-0620H. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Baker TL, Behan M, Mitchell GS. Expression of hypoglossal long-term facilitation differs between substrains of Sprague-Dawley rat. Physiol Genomics. 2001a;4:175–181. doi: 10.1152/physiolgenomics.2001.4.3.175. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Zabka AG, Baker TL, Mitchell GS. Phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. J Appl Physiol. 2001b;90:2000–2006. doi: 10.1152/jappl.2001.90.5.2001. [DOI] [PubMed] [Google Scholar]
- Golder FJ, Ranganathan L, Satriotomo I, Hoffman M, Lovett-Barr MR, Watters JJ, Baker-Herman TL, Mitchell GS. Spinal adenosine A2a receptor activation elicits long-lasting phrenic motor facilitation. J Neurosci. 2008;28:2033–2042. doi: 10.1523/JNEUROSCI.3570-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshgarian HG, Rafols JA. The phrenic nucleus of th albino rat: a correlative HRP and Golgi study. J Comp Neurol. 1981;201:441–456. doi: 10.1002/cne.902010309. [DOI] [PubMed] [Google Scholar]
- Gourine AV, Llaudet E, Thomas T, Dale N, Spyer KM. Adenosine release in nucleus tractus solitarii does not appear to mediate hypoxia-induced respiratory depression in rats. J Physiol. 2002;544:161–170. doi: 10.1113/jphysiol.2002.024174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RA, Okragly AJ, Haak-Frendscho M, Mitchell GS. Cervical dorsal rhizotomy increases brain-derived neurotrophic factor and neurotrophin-3 expression in the ventral spinal cord. J Neurosci. 2000;20:RC77. doi: 10.1523/JNEUROSCI.20-10-j0005.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkead R, Zhan WZ, Prakash YS, Bach KB, Sieck GC, Mitchell GS. Cervical dorsal rhizotomy enhances serotonergic innervation of phrenic motoneurons and serotonin-dependent long-term facilitation of respiratory motor output in rats. J Neurosci. 1998;18:8436–8443. doi: 10.1523/JNEUROSCI.18-20-08436.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Conforti L, Millhorn DE. Gene expression and function of adenosine A2A receptor in the rat carotid body. Am J Physiol Lung Cell Mol Physiol. 2000;279:L273–282. doi: 10.1152/ajplung.2000.279.2.L273. [DOI] [PubMed] [Google Scholar]
- Lai HL, Yang TH, Messing RO, Ching YH, Lin SC, Chern Y. Protein kinase C inhibits adenylyl cyclase type VI activity during desensitization of the A2a-adenosine receptor-mediated cAMP response. J Biol Chem. 1997;272:4970–4977. doi: 10.1074/jbc.272.8.4970. [DOI] [PubMed] [Google Scholar]
- Li C, Jackson RM. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol Cell Physiol. 2002;282:C227–241. doi: 10.1152/ajpcell.00112.2001. [DOI] [PubMed] [Google Scholar]
- Ling L, Fuller DD, Bach KB, Kinkead R, Olson EB, Jr, Mitchell GS. Chronic intermittent hypoxia elicits serotonin-dependent plasticity in the central neural control of breathing. J Neurosci. 2001;21:5381–5388. doi: 10.1523/JNEUROSCI.21-14-05381.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane PM, Mitchell GS. Respiratory long-term facilitation following intermittent hypoxia requires reactive oxygen species formation. Neuroscience. 2007;35:1269–1272. doi: 10.1016/j.neuroscience.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Satriotomo I, Windelborn JA, Mitchell GS. NADPH oxidase activity is necessary for acute intermittent hypoxia-induced phrenic long-term facilitation. J Physiol. 2009;587:1931–1942. doi: 10.1113/jphysiol.2008.165597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane PM, Wilkerson JE, Lovett-Barr MR, Mitchell GS. Reactive oxygen species and respiratory plasticity following intermittent hypoxia. Respir Physiol Neurobiol. 2008;164:263–271. doi: 10.1016/j.resp.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamed S, Mitchell GS. Is there a link between intermittent hypoxia-induced respiratory plasticity and obstructive sleep apnoea? Exp Physiol. 2007;92:27–37. doi: 10.1113/expphysiol.2006.033720. [DOI] [PubMed] [Google Scholar]
- Meszaros JG, Gonzalez AM, Endo-Mochizuki Y, Villegas S, Villarreal F, Brunton LL. Identification of G protein-coupled signalling pathways in cardiac fibroblasts: cross talk between Gq and Gs. Am J Physiol Cell Physiol. 2000;278:C154–162. doi: 10.1152/ajpcell.2000.278.1.C154. [DOI] [PubMed] [Google Scholar]
- Mitchell GS. Respiratory plasticity following intermittent hypoxia: a guide for novel therapeutic approaches to ventilatory control disorders. In: Gaultier C, editor. Genetic Basis for Respiratory Control Disorders. 1st edn. New York: Springer; 2007. pp. 291–306. [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr Invited review: Intermittent hypoxia and respiratory plasticity. J Appl Physiol. 2001;90:2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Johnson SM. Neuroplasticity in respiratory motor control. J Appl Physiol. 2003;94:358–374. doi: 10.1152/japplphysiol.00523.2002. [DOI] [PubMed] [Google Scholar]
- Okada M, Kawata Y, Kiryu K, Mizuno K, Wada K, Tasaki H, Kaneko S. Effects of adenosine receptor subtypes on hippocampal extracellular serotonin level and serotonin reuptake activity. J Neurochem. 1997;69:2581–2588. doi: 10.1046/j.1471-4159.1997.69062581.x. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Reactive oxygen species in the plasticity of respiratory behaviour elicited by chronic intermittent hypoxia. J Appl Physiol. 2003;94:2342–2349. doi: 10.1152/japplphysiol.00613.2002. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Yuan G, Jacono FJ, Kumar GK, Prabhakar NR. 5-HT evokes sensory long term facilitation of the carotid body via activation of NADPH oxidase. J Physiol. 2006;576:289–295. doi: 10.1113/jphysiol.2006.116020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy AA, Nunn C, Ming H, Zou MX, Penninger J, Kirshenbaum LA, Dixon SJ, Chidiac P. Up-regulation of endogenous RGS2 mediates cross-desensitization between Gs and Gq signalling in osteoblasts. J Biol Chem. 2006;281:32684–32693. doi: 10.1074/jbc.M604416200. [DOI] [PubMed] [Google Scholar]
- Ryzhov S, Goldstein AE, Biaggioni I, Feoktistov I. Cross-talk between Gs- and Gq-coupled pathways in regulation of interleukin-4 by A2B adenosine receptors in human mast cells. Mol Pharmacol. 2006;70:727–735. doi: 10.1124/mol.106.022780. [DOI] [PubMed] [Google Scholar]
- Sauer R, Maurinsh J, Reith U, Fulle F, Klotz KN, Muller CE. Water-soluble phosphate prodrugs of 1-propargyl-8-styrylxanthine derivatives, A2A selective adenosine receptor antagonists. J Med Chem. 2000;43:440–448. doi: 10.1021/jm9911480. [DOI] [PubMed] [Google Scholar]
- Schindler CW, Karcz-Kubicha M, Thorndike EB, Muller CE, Tella SR, Ferre S, Goldberg SR. Role of central and peripheral adenosine receptors in the cardiovascular responses to intraperitoneal injections of adenosine A1 and A2A subtype receptor agonists. Br J Pharmacol. 2005;144:642–650. doi: 10.1038/sj.bjp.0706043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson JE, Satriotomo I, Baker-Herman TL, Watters JJ, Mitchell GS. Okadaic acid-sensitive protein phosphatases constrain phrenic long-term facilitation after sustained hypoxia. J Neurosci. 2008;28:2949–2958. doi: 10.1523/JNEUROSCI.5539-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn HR, Rubio R, Berne RM. Brain adenosine concentration during hypoxia in rats. Am J Physiol Heart Circ Physiol. 1981;241:H235–242. doi: 10.1152/ajpheart.1981.241.2.H235. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA. Studies on the direct spinal action of narcotics in the production of analgesia in the rat. J Pharmacol Exp Ther. 1977;202:411–428. [PubMed] [Google Scholar]
- Zimmermann G, Taussig R. Protein kinase C alters the responsiveness of adenylyl cyclases to G protein α and βγ subunits. J Biol Chem. 1996;271:27161–27166. doi: 10.1074/jbc.271.43.27161. [DOI] [PubMed] [Google Scholar]
- Zulueta JJ, Sawhney R, Yu FS, Cote CC, Hassoun PM. Intracellular generation of reactive oxygen species in endothelial cells exposed to anoxia-reoxygenation. Am J Physiol Lung Cell Mol Physiol. 1997;272:L897–902. doi: 10.1152/ajplung.1997.272.5.L897. [DOI] [PubMed] [Google Scholar]