

Abstract
Mastitis is a prevalent disease in dairy cows. Gram-negative bacteria, which express the pro-inflammatory molecule lipopolysaccharide (LPS), are responsible for the majority of acute clinical cases of mastitis. Previous studies have identified differential susceptibility of human and bovine endothelial cells (EC) to the pro-inflammatory and injury-inducing effects of LPS. The Toll-like receptor (TLR)-4 signaling pathway, which is activated by LPS, has been well studied in humans, but not in ruminants. Human myeloid differentiation-factor 88 (MyD88) and TIR-domain containing adaptor protein (TIRAP) are critical proteins in the LPS-induced NF-κB and apoptotic signaling pathways. To assess the role of the bovine orthologs of these proteins in bovine TLR-4 signaling, dominant-negative constructs were expressed in bovine EC, and LPS-induced NF-κB activation and apoptosis evaluated. The results from this study indicate that bovine MyD88 and TIRAP play functional roles in transducing LPS signaling from TLR-4 to downstream effector molecules involved in NF-κB activation, and that TIRAP promotes apoptotic signaling.
Keywords: Apoptosis, Bovine, Endothelial cell, Endotoxin, Inflammation, Innate immunity, Lipopolysaccharide, Toll-like receptor
1. Introduction
In humans, septic shock is one of the most common causes of death in intensive care units and is a major cause of morbidity and mortality worldwide [1,2]. In dairy cows, mastitis remains among one of the most prevalent diseases [3]. Mastitis is also one of the most costly diseases of food production animals with worldwide economic losses associated with this disease estimated to approach $35 billion annually [4]. Systemic complications associated with mastitis include the development of septic shock. Gram-negative bacteria are responsible for ≥50% of the cases of septic shock in humans [5,6] and the majority of the acute clinical cases of mastitis in cows, which are correspondingly the most likely cases of mastitis to develop into septic shock [7,8].
A highly pro-inflammatory component expressed by all Gram-negative bacteria, lipopolysaccharide (LPS), has been implicated in the pathogenesis of septic shock and the deleterious inflammatory responses associated with acute clinical mastitis [7–9]. The vascular endothelium is a key host target of LPS and the pathogenesis of certain complications associated with septic shock, including the development of vascular leak syndromes, has been attributed to LPS-induced endothelial dysfunction and/or injury [10,11].
Endothelial recognition of and responses to LPS are mediated, in part, by LPS binding protein (LBP) and CD-14, which facilitate the presentation of the lipid A portion of LPS to MD-2 and the leucine-rich repeat domains of Toll-like receptor (TLR)-4 [12–14]. TLR-4 signaling is promulgated by the recruitment of adaptor molecules to its Toll-IL-1 receptor (TIR) domain. Two known adaptor molecules, myeloid differentiation factor 88 (MyD88) [15] and TIR domain-containing adaptor protein (TIRAP), also known as MyD88-adaptor-like (Mal) [16–18], facilitate TLR-4-induced NF-κB activation. TLR-4 recruitment of TIRAP and MyD88 leads to the recruitment and activation of IL-1 receptor activated kinase (IRAK)-4, a serine-threonine kinase that trans-phosphorylates IRAK-1 [19–21]. IRAK-1 then undergoes autophosphorylation, which enables TNF-α receptor activated factor-6 (TRAF-6) recruitment to the receptor complex, subsequent oligomerization and polyubiquination of TRAF-6, and downstream activation of the IκB kinase (IKK) complex. The IKK complex, which is made up of IKK α, β, and γ is then capable of phosphorylating IκB, which causes it to be ubiquitinated and undergo proteosomal degradation. Following IκB degradation, NF-κBis able to translocate from the cytoplasm to the nucleus and promote the induction of cytokines and adhesion molecules, including IL-6, IL-8, TNF-α, and E-selectin [22].
In addition to their role in NF-κB activation, TIRAP and MyD88 have established roles in mediating LPS-induced apoptotic signaling in human EC [23,24]. Another protein, FAS-associated death domain protein (FADD), has been demonstrated to promote LPS-induced apoptotic signaling in both human and bovine EC [25,26]. FADD is an adaptor molecule that is able to recruit pro-caspase-8 through homotypic death effector domain binding. Once pro-caspase-8 is bound, it is able to undergo self-cleavage, which initiates the activation of caspases, resulting in apoptosis [27,28]. At the present time, the nature of the interaction of TIRAP, MyD88 and FADD in promoting caspase activation in response to LPS remains to be clearly defined.
Cows are exquisitely sensitive to LPS [29]. Interestingly, bovine EC are more sensitive to the pro-inflammatory and injurious effects of LPS than human EC. In response to LPS, injury-independent permeability is induced to a greater extent in bovine versus human endothelial monolayers [30]. Further, bovine EC are directly sensitive to the apoptotic-inducing effects of LPS, whereas human EC are resistant [31–33]. This latter finding has been attributed to differential expression of the anti-apoptotic protein, FLICE-like inhibitory protein (FLIP) [34,35]. Finally, human FADD has been demonstrated to inhibit LPS-induced NF-κB activation, whereas the bovine ortholog lacks this downregulatory effect [25,36,37]. Thus, differences in both the responses and signaling mediators involved in promoting these responses have been identified between human and bovine EC.
Recently, bovine TIRAP and MyD88 were cloned and sequenced [38]. Unlike their human orthologs, the functional roles of these proteins in mediating LPS responses in bovine EC have yet to be ascertained. Because differences between bovine and human EC response to LPS have been reported, and human, but not bovine FADD, is able to downregulate NF-κB activation, it cannot be simply assumed that the functions of TIRAP and MyD88 are conserved across species. Thus, the objective of the current study was to use dominant-negative constructs of these proteins to functionally characterize the role of bovine TIRAP and MyD88 in mediating bovine TLR-4-induced NF-κB and apoptotic signaling.
2. Materials and methods
2.1. Materials
Highly purified LPS extracted from Escherichia coli serotype 0111:B4 was purchased from List Biological Laboratories, Inc., (Campbell, CA). Recombinant human TNF-α and staurosporine were obtained from R&D Systems, Inc., (Minneapolis, MN) and Sigma Chemical Co., (St. Louis, MO), respectively.
2.2. Cell culture
Bovine aortic (BAEC) and pulmonary artery (BPAEC) endothelial cells (generous gifts of Dr. L.M. Sordillo, Michigan State University, East Lansing, MI and Dr. C.J. Czuprynski, University of Wisconsin, Madison, WI) were cultured in Ham's F12K medium (Irvine Scientific Sales Co., Santa Ana, CA) supplemented with 10% fetal bovine serum (FBS) (Hyclone Laboratories, Logan, UT), heparin (100 μg/ml), insulin (10 μg/ml), transferrin (5 μg/ml) (Sigma Chemical Co.), HEPES (20 mM), L-glutamine (2 mM), penicillin (100 U/ml) and streptomycin (100 μg/ml) (Cambrex BioScience Walkersville, Inc., Walkersville, MD). The Phoenix amphotropic packaging cell line (ATCC, Manassas, VA) was cultured in Dulbecco's Modified Eagle's Medium (Sigma Chemical Co.) supplemented with 10% heat inactivated FBS, non-essential amino acids (1×)(Cambrex BioScience Walkersville, Inc.), L-glutamine (2 mM), penicillin (100 U/ml) and streptomycin (100 μg/ml). All cells were cultured at 37 °C in the presence of 5% CO2.
2.3. Cloning and generation of bovine TIRAP and MyD88 dominant-negative (D/N) constructs
Primers were designed to amplify sequence encoding amino acids 152–296 of bovine MyD88 (NM_001014382), which corresponds only to the TIR domain of the full-length protein. The forward primer (5′CCGGAATTCGACCCCCTAGGGCAAAAGCCCGAG3′) and the reverse primer (5′CCGCTCGAGCTATCAGGGCATGGACAGGGCCTTGGC3′) also included sequence encoding 5′ EcoRI (gaa ttc) and 3′ XhoI (ctc gag) restriction sites to enable subsequent insertion of the construct into the pCR3.V64 Met FLAG vector (gift from Dr. Jurg Tschopp, Institute of Biochemistry of the University of Lausanne, Switzerland). PCR amplification was performed using 2 μl of first-strand cDNA, which was generated from bovine mammary epithelial cell RNA, iQ Supermix (Bio-Rad Laboratories, Hercules, CA), and the primers described above. The cycling conditions were 95 °C for 3 min, followed by 35 cycles of the following: 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min.
To generate the TIRAP D/N construct, primers were designed to amplify the entire coding region of bovine TIRAP (NM_001039962). The forward primer (5′CCGGAATTCATGGCATCATCAACCTCC3′) and the reverse primer (5′CCGCTCGAGCTATCAGCCAAGGGTCTGCAG3′) also included sequence encoding 5′ EcoRI and 3′ XhoI restriction sites. PCR amplification was performed with these primers as described above. Subsequently, P136 in the construct was mutated to H136 using the Quikchange Site-Directed Mutagenesis Kit (Stratagene, Inc., La Jolla, CA) using the following primers: 5′CTTCGCGACGCCACCCATGGTGGCGCCATCGTG3′ and 5′CACGATGGCGCCACCATGGGTGGCGTCGCGAAG3′.
The PCR products for both the MyD88 and TIRAP D/N constructs were digested with EcoRI and XhoI, and ligated into the pCR3.V64 Met FLAG vector. This vector contains a 5′ BamHI site followed by a coding region for an initiator methionine, a FLAG tag (DYKDDDDK), and a spacer of two amino acids (EF) immediately before the EcoRI–XhoI PCR product insertion site. Bacteria were transformed with the ligation reactions and clones from bacteria containing appropriately sized inserts were fully sequenced. Correct clones were then subcloned into the BamHI and XhoI restriction sites of the pBMN-IRES-PURO retroviral expression vector (kindly provided by Dr. Gary Nolan, Stanford University, Stanford, CA and subsequently modified by Dr. Kyle Garton, University of Washington, Seattle, WA).
2.4. Stable expression of TIRAP and MyD88 D/N constructs
High-titer retrovirus was prepared from the Phoenix amphotropic packaging cell line as previously described [35]. Briefly, 24 μg of endotoxin free-expression plasmid were diluted with ddH2O in a final volume of 1.314 ml in a 5 ml polystyrene tube. After a gentle vortex, 186 μl of 2 M CaCl2 were added to each tube, followed by another gentle vortex. Using a pasteur pipette, 1.5 ml of 2× HEPES-buffered saline (HBS) were added to the tube followed by aggressive bubbling for 10 s. One hundred millimeter culture plates containing Phoenix cells at ~75% confluence were spotted with 10 μl of chloroquine (1000×) and the plasmid/CaCl2/HBS solution gently added. The medium in the plates was changed at 10 and 24 h post-transfection. At 48 h post-transfection, the media supernatants containing viral particles were collected and filtered through 0.45 μm Millex-HV filters (Millipore Corp., Bedford, MA), aliquotted into 15 ml polypropylene tubes, and stored at −80 °C.
For infection of BAEC and BPAEC, cells were seeded into 6-well plates and grown until reaching ~80% confluence. The growth medium was replaced with 2.5 ml of retroviral supernatant supplemented with 8 μg/ml of polybrene and 10 mM of HEPES, and the plate centrifuged for 2 h at 1430 × g. An additional 2.5 ml of growth media were added to each well and the cells were incubated for 10 h (5% CO2,37 °C), after which the retroviral-containing supernatant was replaced with normal growth medium. Twenty-four hours post-infection, growth media was supplemented with 2 μmg/ml puromycin (Sigma Chemical Co.) to select for cells stably expressing the construct of interest. To confirm expression, cDNA, which was generated from RNA isolated from stably infected EC using the RNeasy Mini Kit (Qiagen Inc., Valencia, CA), was amplified using a FLAG-specific forward primer (5′TGGATTACAAAGACGATGACG3′) and the construct-specific reverse primers described above. The resulting product was sequenced in both directions using the CEQ8000 automated DNA sequencer and DTCS Quickstart chemistry (Beckman Coulter, Fullerton, CA). Western blotting was performed as previously described with 1:2000 diluted anti-FLAG antibody conjugated to horseradish peroxidase [35] to further confirm expression of the constructs in the EC.
2.5. NF-κB luciferase assay
Sequence from the pNiFty-Luc2 vector (InvivoGen Corp., San Diego, CA), which encodes five NF-κB sites, the proximal E-selectin promoter, and luciferase, was subcloned into a shuttle vector. This composite, engineered promoter is NF-κB-specific and lacks the AP-1/CREB site found in the endogenous E-selectin promoter. The entire expression cassette encoding the reporter construct was transferred into an adenovirus genome vector and virus expressing the construct was generated commercially by Vector Biolabs, Inc. (Philadelphia, PA).
To assay for NF-κB-dependent luciferase expression, EC were seeded into 96-well black, clear bottom plates and grown to confluence. EC were then exposed for 24 h to 5 × 106 PFU/ml of adenovirus diluted in EC culture medium. The adenovirus was aspirated from each well, and EC were subsequently exposed to media alone (Ham's F12K medium supplemented with 5% heat-inactivated FBS, 20 mM HEPES, and 0.5% BSA), or media containing LPS or TNF-α. Luciferase activity was measured using the Bright-Glo Luciferase Assay System (Promega Corp., San Luis Obispo, CA) and a Veritas microplate luminometer (Turner Biosystems Inc., Sunnyvale, CA).
2.6. E-selectin enzyme-linked immunosorbent assay (ELISA)
EC were seeded into 96-well plates and cultured until confluent. Following treatment, cells were washed twice with RPMI-1640 medium (Cambrex BioScience Walkersville, Inc.,) supplemented with 2.5% bovine calf serum and fixed for 10 min with 0.5% glutaraldehyde in PBS at room temperature. Monolayers were washed and then incubated for 1 h at 37 °C with an anti-E-selectin (CD-62E) rabbit polyclonal antibody (NeoMarkers, Inc., Fremont, CA) diluted 1:1000 in wash buffer. The cells were washed twice and incubated as above with horseradish peroxidase-conjugated goat anti-rabbit antibodies (BD Biosciences Corp., San Jose, CA) diluted 1:2000 in wash buffer. The wells were then washed five times with PBS and 100 μl of 3,3′,5,5′-tetramethylbenzidine substrate solution (Kirkegaard and Perry Laboratories Inc., Gaithersburg, MD) was added to each well. The reaction was stopped by the addition of 100 μl of 2 M H2SO4 and the absorbance read at 450 nm on a microplate reader (Bio-Tec Instruments, Inc., Winooski, VT). A background correction reading at 565 nm was subtracted from the 450 nm absorbance readings.
2.7. Caspase assay
EC were seeded into 96-well plates and cultured until confluent. Following treatment, caspase activity was measured with a fluorimetric homogenous caspases assay according to the manufacturer's instructions (Roche Molecular Biochemicals, Indianapolis, IN). The plates were analyzed on a fluorescence plate reader (Bio-Tec Instruments, Inc.) at a 485 nm excitation and a 530 nm emission, and caspase activity expressed relative to the medium control.
2.8. Statistical methods
A t-test was used to compare the mean responses between an experimental group and its respective control. All statistical analyzes were performed using GraphPad Prism version 4.02 for Windows (GraphPad Software, Inc., San Diego, CA). A P-value of <0.05 was considered significant.
3. Results and discussion
3.1. Expression of TIRAP and MyD88 D/N constructs
By virtue of its direct recruitment to TLR-4, TIRAP is one of the most proximal intracellular signaling molecules involved in LPS signaling [39]. TLR-4 interaction with TIRAP is mediated through reciprocal binding of TIR domains contained within each protein. A proline to histidine mutation at amino acid 125 (P125H) in the TIR domain of human TIRAP has been reported to impair its ability to bind to the TIR domain of TLR-4 [18]. Correspondingly, expression of human TIRAP constructs containing this mutation function to inhibit signaling mediated by endogenous TIRAP in a D/N manner and impair the ability of LPS to induce NF-κB activation and apoptosis [16,24]. MyD88 is another proximal LPS signaling molecule that is recruited to the TLR-4 intracellular signaling complex via its interaction with TIRAP. MyD88 contains two distinct protein binding domains, a TIR domain and a death domain. Expression of the human TIR domain of MyD88 has been demonstrated to block TLR-4-mediated NF-κB activation and apoptosis in a D/N fashion [15,23].
Bovine TIRAP and MyD88 possess the predicted protein binding domains necessary for their participation in TLR-4-mediated signaling and demonstrate 81% and 86% conservation, respectively, with their human orthologs at the amino acid level [38]. To determine whether bovine TIRAP and MyD88 mediate LPS-induced signaling in bovine EC, a retroviral infection system was used to stably transfect BPAEC and BAEC with vector, or vector containing cDNA encoding either TIRAP with a P136H mutation, or the TIR domain of MyD88 alone. The P136H mutation in bovine TIRAP corresponds with the P125H mutation of human TIRAP that has been reported to ablate its function. RT-PCR (data not shown) and Western blot analysis confirmed expression of the FLAG-tagged D/N constructs in both cell types (Fig. 1).
Fig. 1.
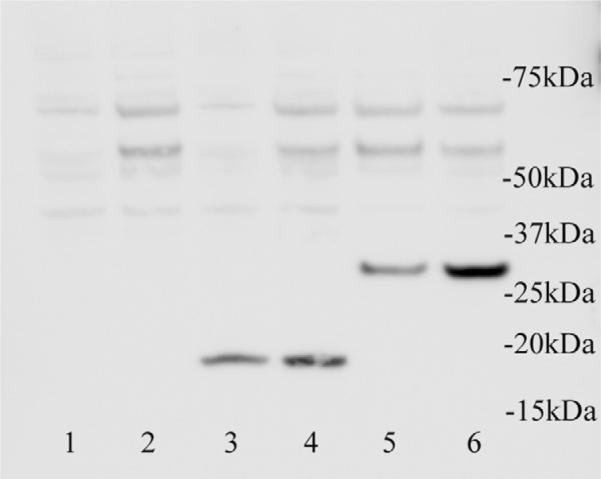
Expression of TIRAP and MyD88 dominant-negative (D/N) constructs in bovine aortic (BAEC) and pulmonary artery (BPAEC) endothelial cells. BAEC (lanes 1, 3 and 5) and BPAEC (lanes 2, 4 and 6) were stably transfected with either vector alone (lanes 1 and 2), or vector containing cDNA encoding either the TIR domain of MyD88 (lanes 3 and 4) or full-length TIRAP containing P136 mutated to H136 (lanes 5 and 6). An anti-FLAG antibody was used to detect the expression of the FLAG-tagged constructs. Molecular mass (in kDa) is indicated.
3.2. Expression of TIRAP and MyD88 D/N constructs inhibit LPS-induced NF-κB activation
To evaluate the functional roles of TIRAP and MyD88 in promoting NF-κB signaling, BPAEC expressing either vector alone or vector encoding TIRAP or MyD88 D/N constructs were treated with media or LPS (100 ng/ml) for 6 h and assayed for NF-κB activity (Fig. 2). Expression of the TIRAP D/N completely inhibited LPS-induced NF-κB activation (Fig. 2A), whereas the MyD88 D/N construct inhibited ~18% of this response (Fig. 2B). To rule out the possibility that expression of either the MyD88 or TIRAP D/N construct non-specifically inhibited NF-κB activation, BPAEC expressing these constructs were also stimulated for 6 h with TNF-α (100 ng/ml) (Fig. 2). TNF-α is well-described activator of NF-κB in EC [40,41], however, distinct transmembrane receptors and associated proximal intracellular signaling molecules mediate TNF-α- and LPS-induced NF-κB activation. Consistent with the predicted roles of bovine TIRAP and MyD88 in mediating signaling elicited by LPS, but not TNF-α, expression of either D/N construct demonstrated no effect on TNF-α-induced NF-κB activation.
Fig. 2.

Effect of the expression of bovine TIRAP or MyD88 dominant-negative (D/N) constructs on LPS-induced NF-κB activation in bovine pulmonary artery endothelial cells (BPAEC). BPAEC stably transfected with either vector (A and B), or vector encoding for TIRAP (A) or MyD88 (B) D/N constructs were treated with media, LPS (100 ng/ml), or TNF-α (100 ng/ml) for 6 h, and NF-κB activity was assayed. Vertical bars represent mean (± S.E.) NF-κB activity relative to media controls. **Significantly (P < 0.01) decreased compared to BPAEC expressing vector alone and exposed to identical treatment.
3.3. Expression of TIRAP and MyD88 D/N constructs inhibit LPS-induced expression of E-selectin
The assay used to measure NF-κB activity (Fig. 2) relied on adenoviral transfection of EC with a reporter construct encoding the luciferase gene under the control of a pure NF-κB promoter. The advantage of this assay is that it allows for the sole measurement of NF-κB-dependent signaling. However, this assay does not preclude the possibility that bovine TIRAP and MyD88 were restricted in their participation in LPS-induced NF-κB-dependent gene expression under the artificial conditions of expression of an exogenously induced reporter construct. To exclude this possibility, the effects of expression of the D/N constructs on expression of endogenous E-selectin, which is regulated by NF-κB [22], were studied (Fig. 3). BPAEC expressing either vector alone or vector encoding TIRAP or MyD88 D/N constructs were treated with media, LPS (100 ng/ml), or TNF-α (100 ng/ml) for 8 h and assayed for E-selectin expression by ELISA (Fig. 3). The TIRAP D/N construct completely inhibited LPS-induced E-selectin expression (Fig. 3A), whereas, the MyD88 D/N construct inhibited this response by ~20% (Fig. 3B). Neither D/N construct had any demonstrable effect on TNF-α-induced E-selectin expression. These experiments investigating the NF-κB-dependent expression of an endogenous protein (Fig. 3) are in agreement with those investigating NF-κB-dependent expression of an exogenously introduced reporter construct (Fig. 2).
Fig. 3.

Effect of the expression of bovine TIRAP or MyD88 dominant-negative (D/N) constructs on LPS-induced expression of E-selectin in bovine pulmonary artery endothelial cells (BPAEC). BPAEC stably transfected with either vector (A and B), or vector encoding for TIRAP (A) or MyD88 (B) D/N constructs were treated with media, LPS (100 ng/ml), or TNF-α (100 ng/ml) for 8 h, and assayed by ELISA for E-selectin expression. Vertical bars represent mean (± S.E.) E-selectin expression relative to media controls. **Significantly (P < 0.01) decreased compared to BPAEC expressing vector alone and exposed to identical treatment.
EC are notoriously difficult to transfect and retroviral-mediated transduction systems have been successfully used to express constructs of interest in this cell type. We have established with this system and subsequent selection with puromycin that ~99% of the selected EC express the construct of interest [35]. To preclude that the observed effects of the D/N constructs were the result of where they stably integrated into the cell genome rather than a result of their D/N function when expressed, several wells of infected EC were selected for and then pooled. Thus, the pooled EC from the different transductions contained constructs of interest inserted in different sites of the genome. To further rule out any site-specific integration effects and to evaluate whether these D/N constructs exert their effect on bovine EC derived from a different anatomical source, the retroviral infection system was used to stably transfect BAEC with vector, or vector containing cDNA encoding either the TIRAP or MyD88 D/N constructs (Fig. 1). These EC were then treated with media, LPS (100 ng/ml), or TNF-α (100 ng/ml) for 8 h and assayed for E-selectin expression by ELISA (Fig. 4). Similar to the results with the BPAEC, the TIRAP D/N construct completely inhibited LPS-induced E-selectin expression, whereas, the MyD88 D/N construct only partially blocked this response. TNF-α-induced E-selectin expression was unaffected by expression of either D/N construct.
Fig. 4.

Effect of the expression of bovine TIRAP or MyD88 dominant-negative (D/N) constructs on LPS-induced expression of E-selectin in bovine aortic endothelial cells (BAEC). BAEC stably transfected with either vector (A, B), or vector encoding for TIRAP (A) or MyD88 (B) D/N constructs were treated with media, LPS (100 ng/ml), or TNF-α (100 ng/ml) for 8 h, and assayed by ELISA for E-selectin expression. Vertical bars represent mean (± S.E.) E-selectin expression relative to media controls. **Significantly (P < 0.01) decreased compared to BAEC expressing vector alone and exposed to identical treatment.
3.4. Expression of the TIRAP D/N construct inhibits LPS-induced apoptosis
In addition to their roles in mediating NF-κB activity, both human TIRAP and MyD88 have been demonstrated to promote LPS-induced apoptotic signaling in human EC [23,24]. Differences in the mechanisms by which LPS induces apoptosis in human versus bovine EC have been implied by findings that demonstrate that bovine EC are directly sensitive to LPS-induced killing, whereas, human EC are only rendered sensitive to this effect when gene expression is inhibited [31]. This differential sensitivity has been attributed, in part, to differential expression of the FLICE-like inhibitory protein (FLIP), which inhibits LPS-induced caspase activation [35]. However, whether the bovine orthologs of the TLR-4 signaling molecules that promote pro-apoptotic signaling in human EC have a similar function in bovine EC has remained unknown.
To investigate whether bovine TIRAP and MyD88 are able to promote LPS-induced pro-apoptotic signaling, BPAEC and BAEC expressing vector alone or one of the D/N constructs were treated with media or LPS (100 ng/ml) for 12 h and assayed for caspase activity. The TIRAP D/N construct completely inhibited LPS-induced apoptosis in both cell types (Fig. 5), whereas the MyD88 D/N construct had no effect (data not shown). Expression of the TIRAP D/N construct had no effect on staurosporine-induced apoptosis, demonstrating the specificity of this construct to inhibit LPS-induced apoptotic signaling and ruling out that this construct globally inhibits apoptosis.
Fig. 5.

Effect of the expression of a bovine TIRAP dominant-negative (D/N) construct on LPS-induced caspase activation in bovine pulmonary artery (BPAEC) and aortic (BAEC) endothelial cells. BPAEC (A) and BAEC (B) stably transfected with either vector or vector encoding for a TIRAP D/N construct were treated with media, LPS (100 ng/ml), or staurosporine (1 μM) for 12 h, and assayed for caspase activity. Vertical bars represent mean (± S.E.) caspase activity relative to media controls. **Significantly (P < 0.01) decreased compared to cells expressing vector alone and exposed to identical treatment.
Because the activation of initiator caspases sets off an amplifying caspase cascade leading to apoptosis, it may not be surprising that the MyD88 D/N construct, which could only partially inhibit NF-κB activation, had no inhibitory effect on LPS-induced caspase activation. Although a detailed time course was not performed, it's possible that expression of the MyD88 D/N construct delayed the onset of initial caspase activation. It has also been reported that TIRAP contains a TRAF-6 binding site and that it may be capable of promoting LPS-induced signaling independently of MyD88 [42]. This may also explain why the MyD88 D/N construct only partially inhibited LPS-induced NF-κB activation, whereas the TIRAP D/N construct completely inhibited this response, as well as LPS-induced caspase activation.
In conclusion, the current study has established through the expression of D/N constructs that bovine TIRAP and MyD88 promote LPS-induced NF-κB activation in bovine EC. Further, this study has established a role for bovine TIRAP in promoting LPS-induced apoptotic signaling in bovine EC. To our knowledge, this is the first study to establish a functional role for bovine TIRAP in mediating responses to LPS in any bovine cell type. Thus, despite reported differences in the sensitivity of human and bovine EC to LPS, the function of TIRAP, which is the most proximal intracellular TLR-4 signaling molecule, is conserved across species. If the differential species-specific sensitivity to LPS is due to differences in the TLR-4 signaling pathway, the data presented here suggest that these differences occur downstream of TIRAP and MyD88.
Acknowledgements
The project was supported by the National Research Initiative of the U.S. Department of Agriculture Cooperative State Research, Education and Extension Service (grant#: 2005-35204-16028). Mention of trade names or commercial products is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
References
- [1].Parrillo JE, Parker MM, Natanson C, Suffredini AF, Danner RL, Cunnion RE, et al. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann Intern Med. 1990;113:227–42. doi: 10.7326/0003-4819-113-3-227. [DOI] [PubMed] [Google Scholar]
- [2].Pinner RW, Teutsch SM, Simonsen L, Klug LA, Graber JM, Clarke MJ, et al. Trends in infectious diseases mortality in the United States. JAMA. 1996;275:189–93. [PubMed] [Google Scholar]
- [3].Wells SJ, Ott SL, Seitzinger AH. Key health issues for dairy cattle—new and old. J Dairy Sci. 1998;81:3029–35. doi: 10.3168/jds.s0022-0302(98)75867-9. [DOI] [PubMed] [Google Scholar]
- [4].Wellenberg GJ, van der Poel WH, Van Oirschot JT. Viral infections and bovine mastitis: a review. Vet Microbiol. 2002;88:27–45. doi: 10.1016/s0378-1135(02)00098-6. [DOI] [PubMed] [Google Scholar]
- [5].Zanetti G, Baumgartner JD, Glauser MP. Sepsis and septic shock. Schweiz Med Wochenschr. 1997;127:489–99. [PubMed] [Google Scholar]
- [6].Dahmash NS, Chowdhury NH, Fayed DF. Septic shock in critically ill patients: aetiology, management and outcome. J Infect. 1993;26:159–70. doi: 10.1016/0163-4453(93)92815-e. [DOI] [PubMed] [Google Scholar]
- [7].Erskine RJ, Eberhart RJ. Post-milking teat dip use in dairy herds with high or low somatic cell counts. J Am Vet Med Assoc. 1991;199:1734–6. [PubMed] [Google Scholar]
- [8].Ziv G. Treatment of peracute and acute mastitis. Vet Clin North Am Food Anim Pract. 1992;8:1–15. doi: 10.1016/s0749-0720(15)30757-x. [DOI] [PubMed] [Google Scholar]
- [9].Opal SM. The host response to endotoxin, antilipopolysaccharide strategies, and the management of severe sepsis. Int J Med Microbiol. 2007;297:365–77. doi: 10.1016/j.ijmm.2007.03.006. [DOI] [PubMed] [Google Scholar]
- [10].Grandel U, Grimminger F. Endothelial responses to bacterial toxins in sepsis. Crit Rev Immunol. 2003;23:267–99. doi: 10.1615/critrevimmunol.v23.i4.20. [DOI] [PubMed] [Google Scholar]
- [11].Vallet B. Bench-to-bedside review: endothelial cell dysfunction in severe sepsis: a role in organ dysfunction? Crit Care. 2003;7:130–8. doi: 10.1186/cc1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab Invest. 2006;86:9–22. doi: 10.1038/labinvest.3700366. [DOI] [PubMed] [Google Scholar]
- [13].Dunzendorfer S, Lee HK, Soldau K, Tobias PS. Toll-like receptor 4 functions intracellularly in human coronary artery endothelial cells: roles of LBP and sCD14 in mediating LPS responses. FASEB J. 2004;18:1117–9. doi: 10.1096/fj.03-1263fje. [DOI] [PubMed] [Google Scholar]
- [14].Henneke P, Golenbock DT. Innate immune recognition of lipopolysaccharide by endothelial cells. Crit Care Med. 2002;30:S207–13. doi: 10.1097/00003246-200205001-00006. [DOI] [PubMed] [Google Scholar]
- [15].Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, et al. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–8. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- [16].Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413:78–83. doi: 10.1038/35092578. [DOI] [PubMed] [Google Scholar]
- [17].Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 2002;420:329–33. doi: 10.1038/nature01180. [DOI] [PubMed] [Google Scholar]
- [18].Horng T, Barton GM, Medzhitov R. TIRAP: an adapter molecule in the Toll signaling pathway. Nat Immunol. 2001;2:835–41. doi: 10.1038/ni0901-835. [DOI] [PubMed] [Google Scholar]
- [19].Fitzgerald KA, Rowe DC, Golenbock DT. Endotoxin recognition and signal transduction by the TLR4/MD2-complex. Microbes Infect. 2004;6:1361–7. doi: 10.1016/j.micinf.2004.08.015. [DOI] [PubMed] [Google Scholar]
- [20].Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- [21].Nishiya T, Kajita E, Horinouchi T, Nishimoto A, Miwa S. Distinct roles of TIR and non-TIR regions in the subcellular localization and signaling properties of MyD88. FEBS Lett. 2007;581:3223–9. doi: 10.1016/j.febslet.2007.06.008. [DOI] [PubMed] [Google Scholar]
- [22].Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bannerman DD, Tupper JC, Erwert RD, Winn RK, Harlan JM. Divergence of bacterial lipopolysaccharide pro-apoptotic signaling downstream of IRAK-1. J Biol Chem. 2002;277:8048–53. doi: 10.1074/jbc.M111249200. [DOI] [PubMed] [Google Scholar]
- [24].Bannerman DD, Erwert RD, Winn RK, Harlan JM. TIRAP mediates endotoxin-induced NF-kappaB activation and apoptosis in endothelial cells. Biochem Biophys Res Commun. 2002;295:157–62. doi: 10.1016/s0006-291x(02)00638-1. [DOI] [PubMed] [Google Scholar]
- [25].Szperka ME, Connor EE, Paape MJ, Williams JL, Bannerman DD. Characterization of bovine FAS-associated death domain gene. Anim Genet. 2005;36:63–6. doi: 10.1111/j.1365-2052.2004.01207.x. [DOI] [PubMed] [Google Scholar]
- [26].Choi KB, Wong F, Harlan JM, Chaudhary PM, Hood L, Karsan A. Lipopolysaccharide mediates endothelial apoptosis by a FADD-dependent pathway. J Biol Chem. 1998;273:20185–8. doi: 10.1074/jbc.273.32.20185. [DOI] [PubMed] [Google Scholar]
- [27].Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;85:803–15. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- [28].Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326(Pt 1):1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Berczi I, Bertok L, Bereznai T. Comparative studies on the toxicity of Escherichia coli lipopolysaccharide endotoxin in various animal species. Can J Microbiol. 1966;12:1070–1. doi: 10.1139/m66-143. [DOI] [PubMed] [Google Scholar]
- [30].Bannerman DD, Goldblum SE. Direct effects of endotoxin on the endothelium: barrier function and injury. Lab Invest. 1999;79:1181–99. [PubMed] [Google Scholar]
- [31].Bannerman DD, Goldblum SE. Mechanisms of bacterial lipopolysaccharide-induced endothelial apoptosis. Am J Physiol Lung Cell Mol Physiol. 2003;284:L899–914. doi: 10.1152/ajplung.00338.2002. [DOI] [PubMed] [Google Scholar]
- [32].Harlan JM, Harker LA, Reidy MA, Gajdusek CM, Schwartz SM, Striker GE. Lipopolysaccharide-mediated bovine endothelial cell injury in vitro. Lab Invest. 1983;48:269–74. [PubMed] [Google Scholar]
- [33].Harlan JM, Harker LA, Striker GE, Weaver LJ. Effects of lipopolysaccharide on human endothelial cells in culture. Thromb Res. 1983;29:15–26. doi: 10.1016/0049-3848(83)90121-4. [DOI] [PubMed] [Google Scholar]
- [34].Bannerman DD, Eiting KT, Winn RK, Harlan JM. FLICE-like inhibitory protein (FLIP) protects against apoptosis and suppresses NF-kappaB activation induced by bacterial lipopolysaccharide. Am J Pathol. 2004;165:1423–31. doi: 10.1016/s0002-9440(10)63400-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Szperka ME, Connor EE, Paape MJ, Williams JL, Bannerman DD. Sequencing, chromosomal mapping, and functional characterization of bovine FLICE-like inhibitory protein (FLIP) Cytogenet Genome Res. 2006;112:90–7. doi: 10.1159/000087518. [DOI] [PubMed] [Google Scholar]
- [36].Zhande R, Dauphinee SM, Thomas JA, Yamamoto M, Akira S, Karsan A. FADD negatively regulates lipopolysaccharide signaling by impairing interleukin-1 receptor-associated kinase 1-MyD88 interaction. Mol Cell Biol. 2007;27:7394–404. doi: 10.1128/MCB.00600-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bannerman DD, Tupper JC, Kelly JD, Winn RK, Harlan JM. The Fas-associated death domain protein suppresses activation of NF-kappa B by LPS and IL-1 beta. J Clin Invest. 2002;109:419–25. doi: 10.1172/JCI14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Connor EE, Cates EA, Williams JL, Bannerman DD. Cloning and radiation hybrid mapping of bovine toll-like receptor-4 (TLR-4) signaling molecules. Vet Immunol Immunopathol. 2006;112:302–8. doi: 10.1016/j.vetimm.2006.03.003. [DOI] [PubMed] [Google Scholar]
- [39].West AP, Koblansky AA, Ghosh S. Recognition and signaling by toll-like receptors. Annu Rev Cell Dev Biol. 2006;22:409–37. doi: 10.1146/annurev.cellbio.21.122303.115827. [DOI] [PubMed] [Google Scholar]
- [40].Madge LA, Pober JS. TNF signaling in vascular endothelial cells. Exp Mol Pathol. 2001;70:317–25. doi: 10.1006/exmp.2001.2368. [DOI] [PubMed] [Google Scholar]
- [41].Beg AA, Finco TS, Nantermet PV, Baldwin AS., Jr Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of I kappa B alpha: a mechanism for NF-kappa B activation. Mol Cell Biol. 1993;13:3301–10. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mansell A, Brint E, Gould JA, O'Neill LA, Hertzog PJ. Mal interacts with tumor necrosis factor receptor-associated factor (TRAF)-6 to mediate NF-kappaB activation by toll-like receptor (TLR)-2 and TLR4. J Biol Chem. 2004;279:37227–30. doi: 10.1074/jbc.C400289200. [DOI] [PubMed] [Google Scholar]