

Abstract
Coordinated and constructive physical activity is correlated with the maintenance of cognitive function in humans. Voluntary running also enhances neuroplasticity in adult and aging rodents, but the molecular pathways underlying these effects are still being elucidated. Considering the multifactorial nature of the biochemical links between physical activity and neurophysiology it is likely that there are many pharmacological mechanisms by which the beneficial actions of exercise can be effectively reproduced using chemical agents. Most studies to date have focused on brain-derived neurotrophic factor (BDNF) as a signaling target for the enhancement of neuronal function by exercise. The goal of the current review is to move beyond BDNF by exploring the diversity of molecular pathways regulated by physical activity in a variety of situations. We will discuss the availability and mechanism of action for several diverse physical activity pharmacomimetics. As physical activity enhances both neuroplasticity and cognition, understanding the molecular targets for these effects may lead to the development of protent new therapeutic interventions for age-related neurodegenerative conditions such as Alzheimer's disease.
Keywords: running, exercise, neurogenesis, serotonin, insulin-like growth factor, Wnt, nitric oxide, hippocampus
Introduction
Lifestyle factors such as exercise, diet, and nutrition are increasingly recognized as determinants of successful aging. Participation in recreational physical activity is correlated with reduced risk for dementia in elderly humans [1]. These changes occur in parallel with changes in the volume of brain regions associated with executive function [2] and spatial memory [3]. These correlations are upheld even after correcting for education and socioeconomic status – two additional determinants of age-related cognitive decline and exercise participation. Running also enhances learning and memory in healthy adults [4].
The cellular and molecular mechanisms underlying exercise-induced changes in brain function are currently being characterized in rodent models. Mice and rats will voluntarily run long distances of up to 10km/24hr in a running wheel [5, 6], and this activity paradigm has been used as a model for the effects of exercise in humans. By understanding the cellular and molecular consequences of wheel running in the rodent brain, it may be possible to develop novel therapeutic interventions that mimic the positive effects of exercise. In this review we will discuss the latest data and theories concerning the neurophysiological actions of exercise and how these are then chemically translated to improve cognitive function and synaptic activity. Our increased understanding of the multiple molecular events that are engendered by physical activity and how we can mimic them pharmacologically may help to facilitate the pharmacomimetic translation of other lifestyle alterations that have beneficial neurophysiological actions such as caloric restriction [7, 8]. To this date the most well-recognized chemical translator of the beneficial neurophysiological effects of exercise is brain-derived neurotrophic factor (BDNF). BDNF is a potent neuroprotective growth factor that enhances synaptic plasticity and memory through its actions on the TrkB receptor [9-11]. Physical exercise increases serum BDNF in humans [12] and central BDNF in rodents [6, 13]. Changes in BDNF are apparently necessary for the effects of exercise on cognition in rodents, as blocking BDNF signaling also prevents the enhancement of cognitive function following physical training [14]. The effects of exercise on synaptic plasticity are qualitatively similar to those of BDNF application [15], suggesting parallel actions in behavioral and synaptic function.
Unfortunately BDNF itself, due to its chemical composition, has posed significant pharmacokinetic problems. The plasma half life of intravenous or subcutaneous BDNF administration (only 1 minute in rats [16]) has been cited as the main reason for its failure during neurological clinical trials [17]. In addition it has been noted that peripherally administered BDNF does not efficiently cross the blood-brain barrier [18]. To counteract these problems with bioavailability several approaches have been taken, for example several small peptide mimetics have been derived to pharmacologically activate or inhibit the TrkB receptor [19, 20]. Many of these structures developed possess small cyclic pentapeptide compositions that still facilitate activation of the TrkB receptor and engender neuroprotective activity. These cyclic pentapeptides, compared to native BDNF, possess a tremendous increase in plasma stability, similar potency and an ability to cross model biological membranes (Fig. 1). Although peripherally injected BDNF does not cross the blood-brain barrier, synthetic full length native BDNF protein can be enabled to cross the blood-brain barrier when it is fused with a human insulin receptor monoclonal antibody [18]. The activity and clearance rate of the BDNF fusion protein appears to be similar to that of the endogenous protein. In this manner, it may be possible to recapitulate the effects of exercise on the brain using BDNF fusion proteins. However, no studies to date have applied blood-brain barrier permeable, BDNF fusion proteins to investigate whether they function similarly to the endogenous protein in promoting neurogenesis and synaptic plasticity. The use of blood-brain barrier permeable neurotrophin fusion proteins has potential for the treatment of neurodegeneration and mood disorders.
Figure 1. BDNF and cyclic pentapeptide analogs.

Panel A depicts the amino acid primary sequence of brain-derived neurotrophic factor (BDNF). Panel B depicts multiple cyclic pentapeptide agonistic analogs of BDNF that can selectively activate the TrkB tyrosine kinase receptor responsible for the multiple physiological effects of BDNF. Multiple diverse amino acid substitutions (Xxx) can be introduced to the cyclic pentapeptide core to modulate both maximal agonistic potency and neuroprotective capacity [20].(Nle, Norleucine).
Rather than employing direct ligand activation of the TrkB receptors to mimic the beneficial effects of physical activity, indirect pharmacological mechanisms have been shown to also be practical and efficacious. For example, antidepressants promote increased BDNF levels. This effect is not specific to BDNF, as multiple neurotrophic factors are enhanced following antidepressant treatment [21, 22]. The widespread effects of exercise on expression of a variety of neurotrophic factors parallels the widespread effects of antidepressant treatment on neurotrophins, leading to speculation that exercise may have antidepressant effects. Pharmacologically, antidepressant effects are typically assessed in rodents using a number of behavioral tasks, including the open field and elevated plus maze. In the open field, exploration of a novel environment is quantified using photobeam breaks, with increased or decreased activity indicative of a change in anxiety-like behavior. In the elevated plus maze, the amount of exploration in the open arms, relative to the closed arms of the apparatus, measures anxiety-like behavior.
Interestingly the effects of running parallel those of pharmacological antidepressant treatment on these tasks. Voluntary running alters open field activity in some studies [23, 24], but not all [6, 25]. These differences may be attributable to differences in the amount of running, species, and strain used in the studies. The effects of running on open field behavior are similar but not identical to those of antidepressant treatment; while treatment with the antidepressant drug fluoxetine (Prozac) reliably reduces open field exploration [26], voluntary running has been shown to reduce [23, 24] or have no effect on open field behavior [6, 25]. Based on the reliable results of antidepressants on open field behavior, and the somewhat equivocal results of running on open field activity, voluntary running may be considered a weaker anxiolytic stimulus.
Running increases the amount of time spent in the open arms of the elevated plus maze, which is suggestive of reduced anxiety [23]. This effect is consistent with the effects of chronic treatment with the antidepressant fluoxetine [27]. Because the effects of running and antidepressant treatment are similar on two different tests of anxiety-like behavior in rodents, the neural substrates for running and pharmacological antidepressant treatment may also be similar. Voluntary running also appears to mimic antidepressant treatment in another respect – both running and antidepressant treatment enhance adult neurogenesis. The hippocampal dentate gyrus continues to generate new neurons throughout the lifespan, and the rate at which this occurs can be influenced by a number of factors. Specific inhibition of adult neurogenesis disrupts the behavioral response to antidepressants [28]; similarly, disrupting exercise-induced neurogenesis prevents changes in learning with physical activity [29]. The study by Santarelli and colleagues [28] evaluated the role of adult neurogenesis in changes in anxiety-like behavior in mice, while the study by Clark et al [29] characterized the role of adult neurogenesis in changes in learning behavior following running. Although these two studies have different behavioral endpoints, the outcome is similar in that adult-generated neurons contribute to the behavioral phenotype following running and antidepressant treatment. A recent report suggests that the anxiolytic effects of exercise and antidepressants are both dependent on adult neurogenesis [30]. In detail, this study employed a specific knockdown of the TrkB BDNF receptor in neural progenitor cells of the adult hippocampus. This manipulation abrogated the effects of exercise and antidepressants on adult neurogenesis, and also prevented the behavioral improvement on tests of anxiety and depression. This study strongly supports the concept of a common neurogenic mechanism for the behavioral consequences of exercise and antidepressants.
Exercise and Central Monoamines
Voluntary running activity also influences serotonin levels. Specifically, voluntary running increases expression of tryptophan hydroxylase, the rate-limiting enzyme for serotonin synthesis, in the raphe nuclei [31]. Involuntary treadmill training increases extracellular serotonin in the hippocampus [32]. Voluntary running also enhanced levels of the serotonin metabolite, 5-hydroxyindole acetic acid, in the hippocampus of animals subjected to footshock [33]. Taken together, these data suggest that the effects of exercise on resistance to psychological stress may be mediated, in part, by changes in serotonergic neurotransmission. In addition to modulation of serotonin, coordinated physical exercise also exerts potent effects upon levels of noradrenaline. Rats selected across generations for high levels of wheel-running exhibit elevated levels of extracellular noradrenaline in the hippocampus [34]. While serotonergic lesions [35] or transient blockade of serotonin receptors [36] had no effect on the exercise-induced upregulation of hippocampal BDNF, noradrenergic lesions or blockade of noradrenergic receptors prevented the effects of exercise on BDNF [35, 36]. If we assume that the enhancement of synaptic plasticity following exercise is dependent on the actions of BDNF, then it becomes tempting to speculate that changes in noradrenergic signaling may act upstream of BDNF to modulate synaptic plasticity and memory.
Exercise Pharmacomimetic Effects on Monoamines
The pharmacological agents that most effectively mimic the effects of exercise on central monoamines so far are the selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs). SSRIs include citalopram (Celexa), escitalopram (Lexapro), fluoxetine (Prozac), fluvoxamine (Luvox), paroxetine (Paxil) and sertraline (Zoloft). SNRIs include venlafaxine (Effexor) and duloxetine (Cymbalta), among others (Fig. 2). The mechanism of action for SSRIs and SNRIs involves inhibition of neurotransmitter reuptake, thereby prolonging the actions of the neurotransmitter at the synapse.
Figure 2. Neurotransmitter re-uptake inhibitor anti-depressants.
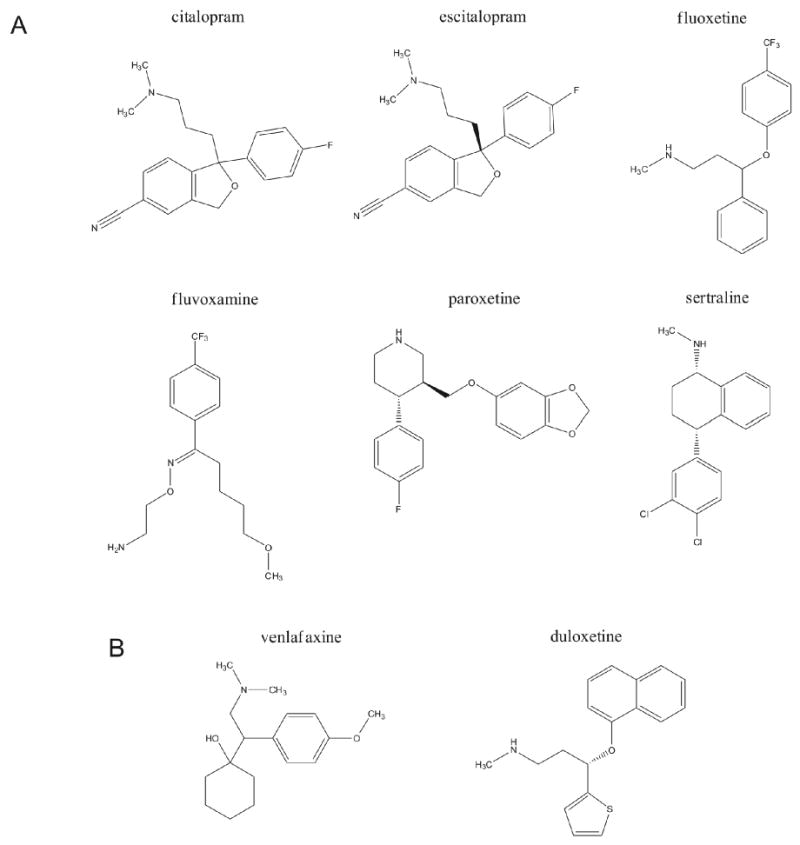
Panel A indicates the chemical structures of multiple forms of selective serotonin re-uptake inhibitors (SSRI: citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, sertraline). Panel B depicts two selective norepinephrine re-uptake inhibitor chemical structures (SNRI: venlafaxine, duloxetine). Both examples of these classes of compound can directly or indirectly mimic the beneficial effects of coordinated physical activity.
Of specific interest and importance for the design of future exercise pharmacomimetics is the fact that the effects of exercise and SSRIs are quite distinct at the synaptic level. Exercise increases hippocampal serotonin concentrations [32], while SSRIs prolong the actions of serotonin by blocking its reuptake. However, both exercise and SSRI treatment are associated with a net increase in serotonergic signaling on postsynaptic cells. Questions remain as to whether exercise-induced regulation of serotonin levels and SSRI-induced alterations in serotonergic neurotransmission will have similar effects on pre-synaptic autoreceptor sensitivity and postsynaptic serotonin receptor expression. Most postsynaptic serotonin receptors are coupled to G-proteins, and it also remains to be determined whether G-protein receptor coupling to intracellular second messenger cascades are similar following exercise and SSRI treatment. There is now considerable evidence to suggest that distinctions not only in G protein coupling of the receptor but alterations in the intracellular protein milieu, with specific regards to proteins that interact with the G protein-coupled receptors, can have dramatic effects upon ligand selectivity and signaling output [37-39]. An in-depth appreciation of the subtle proteinacious changes that occur pre- or post-synaptically during coordinated physical exercise may therefore facilitate the creation of more specific exercise pharmacomimetics. As the effects of exercise and SNRI treatment converge on hippocampal BDNF production (as noradrenergic neurotransmission is necessary for exercise-induced upregulation of hippocampal BDNF concentrations [35, 36] and SNRI treatment has also been shown to increase hippocampal BDNF [40]), it appears likely that BDNF serves as a signaling nexus for the effects of SSRIs, SNRIs, and exercise.
Exercise Pharmacomimetics and Insulin-Like Growth Factors
Insulin-like growth factor-1 (IGF-1) is a potent peripheral and centrally acting growth factor and neurotrophic agent. Coordinated physical activity increases IGF-1 concentrations in humans [41] and rodents [42, 43]. IGF-1 is synthesized and released both locally in the central nervous system and in multiple peripheral organs including the liver. IGF-1 agonistically stimulates its cognate tyrosine kinase IGF-1 receptor (IGF-1R) as well as the structurally-related insulin receptor. IGF-1 mediated activation of IGF-1 receptors activates multiple molecular cascades including the phosphoinositide-3 kinase/protein kinase B pathway and the c-Src non-receptor tyrosine kinase [44], thereby modulating cell proliferation and cellular metabolism. IGF-1 receptor activity also appears to be closely linked to the actions of BDNF in translating physical activity to neurological benefits. For example, IGF-1 signaling via the IGF-1 receptor is necessary for the exercise-induced upregulation of BDNF [45]. Increased IGF-1 production following exercise training may interact with BDNF to modulate synaptic plasticity, but the area of overlap between the exercise-induced regulation of BDNF and IGF-1 is still being characterized [11]. Administration of IGF-1 blocking antibodies prevented the exercise-induced upregulation of BDNF [45, 46]. On the other hand, blocking BDNF signaling prevented the exercise-induced upregulation of IGF-1 in the hippocampus [47]. Based on these results, it is unclear whether exercise increases BDNF through IGF-1, or vice versa. However, in spite of this unresolved interaction, it is likely that IGF-1 and BDNF interact to regulate changes in hippocampus-dependent learning following exercise [47].
Pharmacological Mimetics of the Effects of Exercise on IGF-1
Full-length IGF-1 protein can be administered peripherally, and crosses the blood-brain barrier. Therefore, administration of exogenous IGF-1 could be considered an exercise pharmacomimetic. IGF-1 has indeed been employed as an injectable drug for multiple human disorders [48]. Despite possessing clinical efficacy, the size and complexity of the hormone presents multiple issues concerning cost of production, bioavailability and selectivity. As with most endogenous growth factor-like molecules the creation of small molecule IGF-1 mimetics has proved to be complicated [49]. This is partly due to the large size of the native IGF-1 molecule and also the fact that it also must undergo a three-dimensional alteration in structure to productively engage and fully activate the IGF-1R [50, 51]. In addition to these structural nuances, the circulatory bioavailability of IGF-1 molecules is also controlled by a family of plasma-borne IGF binding proteins (IGFBPs) [52, 53]. Six distinct types of IGFBPs have been identified in mammals and binding of IGF-1 to some of these proteins effectively blocks their interaction with the IGF-1R [54]. Therefore one mechanism of modulating IGF-1 for therapeutic actions can involve the creation of molecular mimetics of IGF-1 that specifically bind the inhibitory IGFBPs and therefore potentiate the activity of circulatory IGF-1 [55].
One of the other primary challenges to the widespread use of IGF-1 is the fact that administration of exogenous IGF-1 induces hypoglycemia, which has important and detrimental consequences for hippocampal function [11]. Moreover, IGF-1 exerts trophic effects not only on neurons, but also in multiple peripheral cell lineages including tumor cells. Despite this setback, mechanistic windows that may facilitate the creation of cell-type-specific receptor activation have been recently elucidated for transmembrane receptors that control complex intracellular signaling cascades [56]. The ability of cell-type specific receptor-interacting proteins to alter both ligand sensitivity and functionality has been demonstrated for multiple forms of transmembrane receptor [37] and therefore are highly likely to be applicable to the IGF-1R.
Although the direct treatment of IGF-1 may be fraught with complications, there are a number of other pharmacological treatments that can increase endogenous production of IGF-1. It has been demonstrated that SSRIs increase IGF-1 receptor expression in the frontal cortex of rats [57]. Therefore it seems that many of the physiological pathways that connect physical activity to the promotion of neurological benefit are closely intertwined.
Glucocorticoid Receptor Signaling and Physical Activity
Glucocorticoid hormones are produced in the adrenal glands and act on the liver to stimulate gluconeogenesis. Steroid hormones such as cortisol in humans and corticosterone in rodents form an essential part of the homeostatic stress response. Perceived stress induces the synthesis and release of corticotrophin-releasing factor (CRF) from the hypothalamic paraventricular nucleus. Elevated levels of CRF then signal to the anterior pituitary to produce adrenocorticotrophic hormone (ACTH). Release of ACTH into the bloodstream then acts on the adrenal gland to synthesize and release glucocorticoids. At each level of the hypothalamic-pituitary-adrenal axis (HPA axis), glucocorticoid receptors mediate negative feedback, allowing for a rapid ‘fight or flight’ response that terminates upon cessation of stress. The glucocorticoid receptor (GR) activation process is initiated after the steroid enters a cell binding to and forming a stable complex with the GR. The ligand-bound GR then translocates to the cellular nucleus where it can either indirectly modulate transcription factor activity or directly bind specific DNA sequences to modulate mRNA synthesis. The primary transcription factor targets for the liganded GR tend to be involved in inflammatory processes, e.g. NF-κB and AP-1 that have been implicated in neuropathological activities [56]. With respect to glucocorticoid actions, the liganded GR after binding to these factors effectively inhibits their activity, thus depressing the production of inflammatory mediators such as TNF-α and IL-6. This inhibitory activity of the ligand-GR complex is termed ‘transrepression’. The direct DNA-binding activity of the liganded GR complex (often termed ‘transactivation’) is often considered to mediate the deleterious side effects of glucocorticoids [58].
Protracted exposure to elevated glucocorticoid hormones has negative consequences for hippocampal plasticity. Specifically, elevated corticosterone levels impair synaptic plasticity [59], induce neuronal atrophy [60], and reduce adult neurogenesis [61]. Interestingly, not all manipulations that elevate glucocorticoids have a negative impact on hippocampal structure and function. Voluntary exercise increases circulating glucocorticoids in humans [62] and in rodent models [5, 63, 64]. Despite increases in circulating glucocorticoids, the effects of exercise on hippocampal structure and function are generally positive. Voluntary running has also been demonstrated to improve blood flow to the hippocampus in humans [65], and increases adult neurogenesis and dendritic spine density in the hippocampus of rodents [5, 66-69]. It is possible therefore that the temporal nature of hormone release (pulsatility or duration of release) may allow glucocorticoids to exert these distinct (beneficial or detrimental) pharmacological effects [56].
If the stress of running is biologically different from other kinds of stress, how is it different? Voluntary running elicits elevations in corticosterone-binding globulin (CBG), which inhibits the biological actions of glucocorticoid hormones [63]. Therefore, CBG is one potential target for the development of exercise mimetics. Voluntary running has been reported to have anxiolytic effects [24], although this has not been the case in all experiments [6]. If voluntary running has antidepressant and anxiolytic effects, then one potential mechanism for these effects involves increased CBG. It is conceivable that exercise-induced elevations in CBG could protect against different types of stressors. On the other hand, it is also conceivable that exercise-induced elevations in corticosterone could sensitize individuals to other forms of stress. Clearly, the modulation of HPA axis functioning by exercise is still being elucidated.
Glucocorticoid-Based Exercise Pharmacomimetic Strategies
As the effects of exercise on glucocorticoid signaling are still being characterized, it is challenging to conceive of pharmacomimetics for the effects of exercise on glucocorticoid levels. However, by examining the effects of exercise at the level of glucocorticoid receptor function, it may be possible to speculate on drug interventions that mimic the effects of exercise. Corticosterone in rodents, and cortisol in humans, binds to two classes of receptors that differ in their expression and affinity. The type I glucocorticoid receptor or mineralocorticoid receptor (MR), binds corticosteroids with high affinity and is tonically occupied. The type II glucocorticoid receptor (GR) has a lower affinity for corticosteroids and is occupied during stress and at the high points of the circadian cycle. As chronic effects of glucocorticoids can often be detrimental, the ability to control the plasma half-life is an important consideration for GR-based pharmacotherapeutics. In addition, due to the wide range of activities that steroidal molecules possess due to binding to other steroid receptors, it would desirable to create non-steroidal GR ligands. With the careful design of these agents it may be feasible that the avoidance of the unwanted effects of GR activation could be avoided. Several routes for the creation of non-steroidal mimetics have been pursued, e.g. quinol-4-ones [70] and aryl pyrazoles [71] (Fig. 3). These non-steroidal derivatives are thought to potentially possess less side effects compared to actual steroidal agents. An additional mechanism to reduce the ‘transactivation’ phenomena for better pharmacological profiles is the creation of so-called selective glucocorticoid receptor agonists (SEGRA) agents (Fig. 3). These SEGRAs are non-steroidal glucocorticoid mimetics that possess a selective activity at the GR and a signalling bias towards ‘transrepression’ and away from ‘transactivation’ [72, 73].
Figure 3. Chemical modulators of steroid hormone receptor activity.
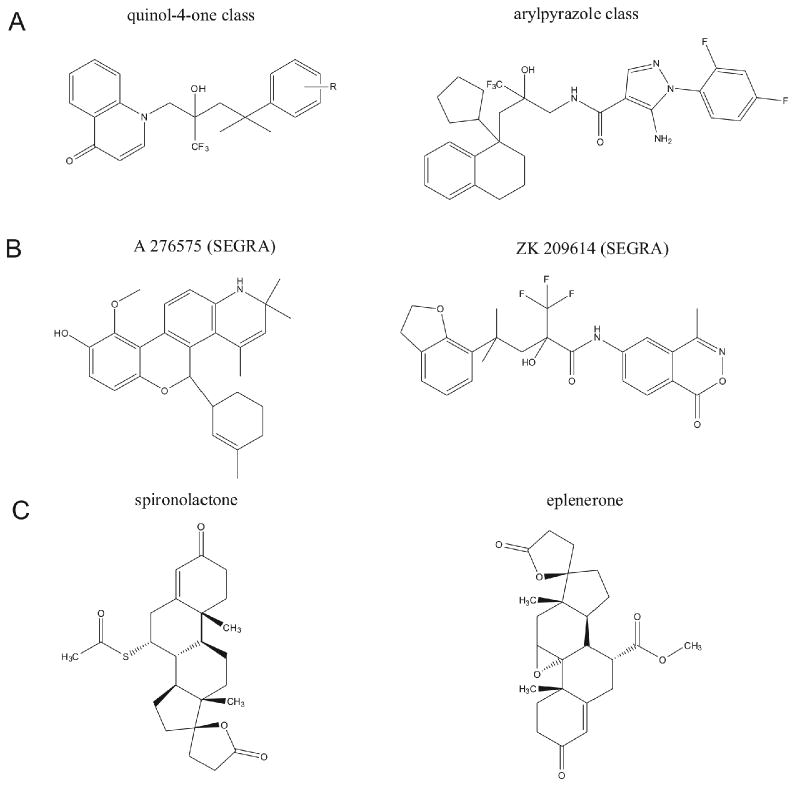
Panel A depicts two base-structure non-steroidal A-ring mimetic glucocorticoid receptor agonists. Multiple modifications can be made to these base structures to modulate their pharmacodynamics and receptor efficacy (A-ring mimetic - quinol-4-one [70]: aryl pyrazole [71]). Panel B depicts the chemical structures of two selective non-steroidal selective glucocorticoid receptor agonists (SEGRAs: A 276575 [72]; ZK 209614 [73]). Panel C depicts the chemical structures of two selective mineralocorticoid receptor antagonistic agents.
Voluntary running reduces MR binding capacity and expression in the hippocampus of mice [63, 74]. It is uncertain whether this is a positive adaptation that mediates the enhancement of cognitive function by exercise, or a negative adaptation that would exert deleterious effects if compensatory mechanisms were not present. As running reduces binding and expression of the MR, it may be possible to mimic the effects of running using MR antagonists, e.g. spironolactone or eplenerone (Fig. 3). It has been experimentally demonstrated that spironolactone potentiates the running-induced enhancement of adult neurogenesis [74]. One possible mechanism for this effect involves reduced glucocorticoid exposure, as blockade of MR would lead to greater occupancy of GR, thereby facilitating negative feedback on the hypothalamic-pituitary-adrenal axis. Another hypothesis would suggest that MR blockade directly facilitates the differentiation, maturation, and/or survival of newly generated neurons. These two hypotheses are not mutually exclusive, but require further exploration to determine the feasibility of targeting glucocorticoid receptors as physical activity mimetics.
Physical Activity Modulates Mitochondrial Function
Mitochondria play a critical role in health and disease. As the source of energy for the cell, they stand at the intersection of plasticity and homeostasis. Based on their function, it is clear that they would be responsive to exercise – a manipulation that places a greater demand on energy metabolism. In rodents, voluntary running increases the expression of ubiquitous mitochondrial creatine kinase (uMtCK) and mitochondrial uncoupling protein 2 (UCP2) [47]. Indeed, the effects of running on synaptic structure are dependent on the presence of UCP2 [75]. In a microarray study, several mitochondrial genes were altered by lifelong running in the hippocampus of aged mice, including mitochondrial ribosomal protein S6 (Mrps6, Entrez Gene ID: 121022) mitochondrial ribosomal protein L22 (Mrpl22, Entrez Gene ID: 216767), and electron transferring flavoprotein (Etfa, Entrez Gene ID: 110842) [76]. These studies point to the mitochondria as targets for the effects of running in the adult and aging brain.
Pharmacological Activators of Mitochondrial Stress Proteins
Potassium influx through adenosine-triphosphate sensitive potassium channels (KATP channels) activates the mitochondrial stress response. Diazoxide is a potent activator of mitochondrial KATP channels. Similar to running activity, treatment with diazoxide attenuates damage following stroke [77, 78] (Fig. 4). It has not yet been determined whether the protective effects of running and diazoxide converge on mitochondrial stress proteins.
Figure 4. Energy-regulatory and developmentally-targeted pharmacomimetics of coordinated physical activity.
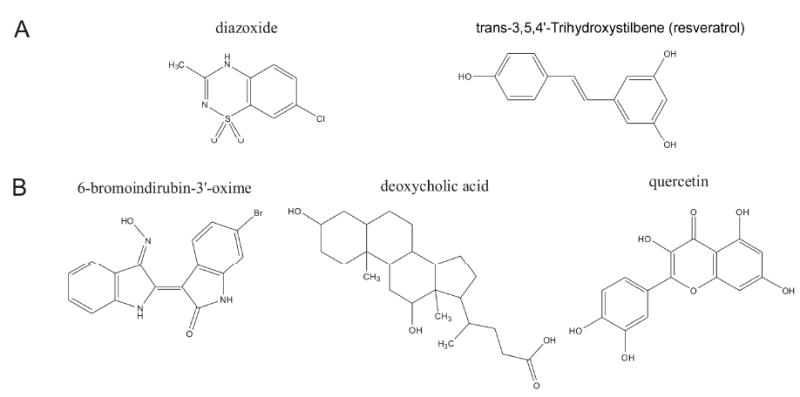
Panel A depicts the chemical structures of two agents that possess exercise pharmacomimetic activity by controlling stress-response pathways. Panel B depicts the diverse chemical structures of pharmacologically active agents that can effectively modulate the Wnt pathway in hippocampal tissue.
The presence of mitochondrial UCP2 is necessary for exercise-induced synaptogenesis [75]. Just as running activity protects against damage following stroke [77], treatment with resveratrol induces a preconditioning mechanism that is neuroprotective following ischemia [79]. The protective effects of resveratrol and the synaptogenic effects of exercise both converge on mitochondrial UCP2 [75, 79] (Fig. 4). However, it remains to be seen whether changes in UCP2 are necessary for the protective effects of exercise and resveratrol against ischemic damage. Moreover, the possibility of an additive neuroprotective effect of exercise and resveratrol treatment remains to be addressed.
Physical Activity Stimulates the Wnt Pathway
The Wnt pathway is involved in brain development and synaptic plasticity [80]. New data suggest that this pathway is also responsive to physical activity. Specifically, genes in the Wnt pathway were differentially modulated in the brains of mice exposed to lifelong running [76]. These genes include Cacybp (calcyclin binding protein, Entrez Gene ID: 12301), Nlk (nemo-like kinase, Entrez Gene ID: 18099), Camk2a (calcium-calmodulin dependent protein kinase alpha, Entrez Gene ID: 12322), Prkcc (protein kinase C gamma, Entrez Gene ID: 18752), and Ppp3ca (protein phosphatase 3, catalytic subunit, alpha isoform, Entrez Gene ID: 19055). Calcyclin binding protein ubiquitinates β-catenin, a critical component of the Wnt pathway [81]. This leads to proteasomal degradation of β-catenin, thereby modulating Wnt signaling. Nemo-like kinase inhibits the actions of lymphoid enhancer binding factor 1 (TCF/LEF), downstream of β-catenin in the Wnt signaling pathway [82]. Wnt5 signals via frizzled to activate calcium-calmodulin dependent protein kinase alpha in the presence of elevated calcium [83]. Alterations in the expression of calcium-calmodulin dependent protein kinase alpha has implications for synaptic plasticity, as Camk2a signaling is essential for long-term potentiation [84]. Protein kinase C-γ is also involved in synaptic plasticity and long-term potentiation. Mutant mice lacking protein kinase C-γ display impaired long-term potentiation with intact long-term depression [85], in support of a possible role in learning and memory [86]. Protein kinase C is also implicated in the Wnt signaling pathway [87]. Expression of protein phosphatase 3 is also enhanced in aged runners, and this gene is involved in Wnt signaling [88]. Our study [76] is the first study that reports an interaction between voluntary running and Wnt signaling. Based on the many interactions between Wnt signaling, synaptic plasticity, and cognition, it is possible that running-induced enhancement of cognitive function may involve changes in Wnt signaling.
Pharmacological Manipulations that Activate the Wnt Pathway
Novel small molecules that regulate cellular function have been identified using cell-based screens of natural products and existing drugs. Using this technique, several small molecules that regulate Wnt/β-catenin signaling have been identified. These molecules include the agonists 6-bromoindirubin-3′-oxime (BIO) [89], lithium chloride [90], deoxycholic acid [91], and the antagonist quercetin [92] (Fig. 4). BIO is a glycogen synthase kinase-3 [GSK-3] inhibitor that activates Wnt/β-catenin signaling. Lithium chloride is a neuropsychiatric drug used to treat bipolar disorder; this drug also activates the Wnt/β-catenin pathway. Deoxycholic acid activates Wnt/β-catenin, and exacerbates cancer cell growth [91]. In contrast, quercetin reduces activation of Wnt/β-catenin and arrests the growth of cancer cells [92]. It remains to be determined whether the actions of running on hippocampal neuroplasticity could be recapitulated following local administration of Wnt/β-catenin activators in the hippocampus. Moreover, the possibility that the effects of running on hippocampal structure and function could be blocked following local application of inhibitors of Wnt signaling remains to be addressed.
Physical Activity and Nitric Oxide Signaling
Nitric oxide (NO) is a gaseous second messenger that mediates retrograde signaling at synapses. It is involved in synaptic plasticity [93], learning [94] and adult neurogenesis [95]. Nitric oxide signaling also regulates BDNF [96] and the effects of exercise on hippocampal BDNF can be prevented by inhibition of nitric oxide [97]. Similarly, the enhancement of adult neurogenesis by exercise can be abrogated following pretreatment with a nitric oxide inhibitor [98]. In a microarray study [76], exercise was reported to regulate levels of dimethylarginine dimethylaminohydrolase 1 [Ddah1; Entrez Gene ID: 64157], the enzyme that metabolizes dimethylarginine, a component of the nitric oxide synthesis pathway. Based on these data, it is apparent that nitric oxide signaling contributes to the regulation of BDNF levels and adult neurogenesis by exercise.
Pharmacological Manipulation of the Nitric Oxide System
As little is known regarding the effects of exercise on specific components of the nitric oxide (NO) synthesis pathway, it is challenging to identify potential pharmacological mimetics for the effects of exercise on nitric oxide signaling. Indeed, because the studies mentioned above employed global inhibitors of NO signaling [97, 98], it remains to be determined whether these effects are centrally mediated. The classical series of compounds that have been clinically employed to control the NO system include: NO precursors such as L-arginine [99]; NO donors such as nitroglycerin [100] and phosphodiesterase 5 inhibitors [101]. However, based on our observation of changes in enzymes that metabolize demethylarginine following running in aged mice [76], it is apparent that at least some components of the NO signaling pathway are altered in the hippocampus. Before exercise mimetics targeted to NO signaling can be identified, it will be necessary to determine whether exercise-induced regulation of NO signaling is centrally mediated.
Advantages and Limitations to Exercise Pharmacomimetics Targeted to the Hippocampus
There are several advantages and limitations to the use of pharmacological mimetics for the effects of exercise on the brain. The obvious advantage involves the low adherence rate for voluntary exercise programs as compliance would likely be far higher for an exercise pharmacomimetic. Other benefits of pharmacomimetics include the potential for widespread use among individuals who are physically unable to engage in vigorous physical activity. The drawback of pharmacological interventions that mimic the effects of exercise involves the generation of potentially multiple side-effects. This is likely due to the multifactorial nature of the processes chemically translating coordinated physical movement to improved neurophysiological activity. SSRIs and SNRIs, although they effectively reproduce some of the behavioral and neurochemical effects of exercise in rodents, also induce a variety of side effects, including unintended weight gain. Changes in body weight with antidepressant treatment may abrogate any potential benefit of antidepressants as an exercise mimetic. Similarly, interfering with glucocorticoid signaling has consequences for a variety of physiological functions, including the immune response [102]. This could obscure any benefit following anti-glucocorticoid treatment. Administration of IGF-1, while initially having a positive impact on central function and peripheral metabolism, may over the long term contribute to tumorigenesis [103]. And while less is known about the potential side effects of mitochondrial stress protein activators and Wnt signaling upregulators, it is likely that any drug-based mimetic for the effects of exercise will have unintended consequences.
Summary and Conclusion
Here we present evidence suggesting that central IGF-1, monoamines, Wnt signaling, glucocorticoids, mitochondrial stress proteins, and the nitric oxide pathway may be amenable as targets for the development of physical activity mimetics. In addition to BDNF signaling, IGF-1 signaling in particular is likely to act throughout the organism to enhance overall somatic function. The effects of exercise on serotonin and norepinephrine can also be recapitulated using SSRIs and SNRIs, respectively. Targeting central glucocorticoid signaling is a more difficult proposition, as glucocorticoids have multiple functions throughout the organism. In order to specifically regulate central glucocorticoid signaling, intranasal drug delivery could be employed. A similar mode of delivery to locally modulate nitric oxide in the brain and regulate Wnt signaling would be ideal, as these pathways also contribute to multiple physiological functions throughout the body. Understanding the molecular targets for exercise-induced regulation of neuroplasticity may lead to the development of novel therapeutic treatments for psychiatric and neurodegenerative disorders.
Figure 5. Multiple mechanistic routes for current and future hippocampally-targeted pharmacomimetics of exercise.
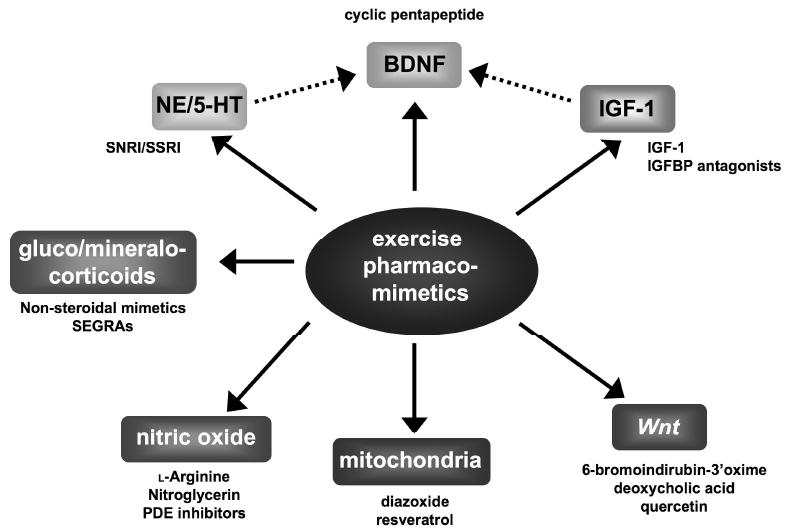
It is clear that amongst the currently studied neurochemical pathways that translate the beneficial actions of coordinated physical activity to neuronal health there are significant points of convergence, e.g. the pharmacological effects of IGF-1 or norepinephrine/serotonin modulation upon BDNF. With furthere research additional functional connections may also arise between the other mechanistic pathways delineated in the figure, thus providing us with additional key loci of exercise pharmacomimesis. It is likely in the future that many more efficacious and tractable pathways will also emerge to allow additional therapeutic compounds to be developed that will potentienally better mimic the beneficial effects if coordinated physical activity.
Acknowledgments
This work was supported by a training grant from the National Institute on Aging administered through Johns Hopkins University to A.M.S., and by the Intramural Program at the National Institute on Aging.
References
- 1.Hillman CH, Erickson KI, Kramer AF. Nat Rev Neurosci. 2008;9:58. doi: 10.1038/nrn2298. [DOI] [PubMed] [Google Scholar]
- 2.Colcombe SJ, Erickson KI, Scalf PE, Kim JS, Prakash R, McAuley E, Elavsky S, Marquez DX, Hu L, Kramer AF. J Gerontol A Biol Sci Med Sci. 2006;61:1166. doi: 10.1093/gerona/61.11.1166. [DOI] [PubMed] [Google Scholar]
- 3.Erickson KI, Prakash RS, Voss MW, Chaddock L, Hu L, Morris KS, White SM, Wójcicki TR, McAuley E, Kramer AF. Hippocampus. 2009 doi: 10.1002/hipo.20547. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Winter B, Breitenstein C, Mooren FC, Voelker K, Fobker M, Lechtermann A, Krueger K, Fromme A, Korsukewitz C, Floel A, Knecht S. Neurobiol Learn Mem. 2007;87:597. doi: 10.1016/j.nlm.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Stranahan AM, Khalil D, Gould E. Nature Neuroscience. 2006;9:526. doi: 10.1038/nn1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stranahan AM, Lee K, Martin B, Maudsley S, Golden E, Cutler RG, Mattson MP. Hippocampus. 2009 doi: 10.1002/hipo.20577. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin B, Mattson MP, Maudsley S. Ageing Res Rev. 2006;5:332–353. doi: 10.1016/j.arr.2006.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin B, Pearson M, Kebejian L, Golden E, Keselman A, Bender M, Carlson O, Egan J, Ladenheim B, Cadet JL, Becker KG, Wood W, Duffy K, Vinayakumar P, Maudsley S, Mattson MP. Endocrinology. 2007;148:4318–4333. doi: 10.1210/en.2007-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. Neuron. 1999;24:401. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- 10.Mattson MP, Maudsley S, Martin B. Trends Neurosci. 2004;27:589–594. doi: 10.1016/j.tins.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 11.Mattson MP, Maudsley S, Martin B. Ageing Res Rev. 2004;3:445–464. doi: 10.1016/j.arr.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 12.Tang SW, Chu E, Hui T, Helmeste D, Law C. Neurosci Lett. 2008;431:62. doi: 10.1016/j.neulet.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 13.Neeper SA, Gómez-Pinilla F, Choi J, Cotman C. Nature. 1995;373:109. doi: 10.1038/373109a0. [DOI] [PubMed] [Google Scholar]
- 14.Vaynman S, Ying Z, Gomez-Pinilla F. Eur J Neurosci. 2004;20:2580. doi: 10.1111/j.1460-9568.2004.03720.x. [DOI] [PubMed] [Google Scholar]
- 15.Messaoudi E, Ying SW, Kanhema T, Croll SD, Bramham CR. J Neurosci. 2002;22:7453. doi: 10.1523/JNEUROSCI.22-17-07453.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poduslo JF, Curran GL. Brain Res Mol Brain Res. 1996;36:280–286. doi: 10.1016/0169-328x(95)00250-v. [DOI] [PubMed] [Google Scholar]
- 17.The BDNF study Group (Phase III) Neurology. 1999;52:1427–1433. doi: 10.1212/wnl.52.7.1427. [DOI] [PubMed] [Google Scholar]
- 18.Boado RJ, Zhang Y, Zhang Y, Pardridge WM. Biotechnol Bioeng. 2007;97:1376. doi: 10.1002/bit.21369. [DOI] [PubMed] [Google Scholar]
- 19.Fletcher JM, Hughes RA. J Pept Sci. 2006;12:515–524. doi: 10.1002/psc.760. [DOI] [PubMed] [Google Scholar]
- 20.Fletcher JM, Hughes RA. Bioorg Med Chem. 2009;17:2695–2702. doi: 10.1016/j.bmc.2009.02.053. [DOI] [PubMed] [Google Scholar]
- 21.Nibuya M, Morinobu S, Duman RS. J Neurosci. 1995;15:7539. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nelson RL, Guo Z, Halagappa VM, Pearson M, Gray AJ, Matsuoka Y, Brown M, Martin B, Iyun T, Maudsley S, Clark RF, Mattson MP. Exp Neurol. 2007;205:166–176. doi: 10.1016/j.expneurol.2007.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Binder E, Droste SK, Ohl F, Reul JM. Behav Brain Res. 2004;155:197. doi: 10.1016/j.bbr.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 24.Salam JN, Fox JH, Detroy EM, Guignon MH, Wohl DF, Falls WA. Behav Brain Res. 2009;197:31. doi: 10.1016/j.bbr.2008.07.036. [DOI] [PubMed] [Google Scholar]
- 25.Leasure JL, Jones M. Neuroscience. 2008;156:456. doi: 10.1016/j.neuroscience.2008.07.041. [DOI] [PubMed] [Google Scholar]
- 26.Dulawa SC, Holick KA, Gundersen B, Hen R. Neuropsychopharmacology. 2004;29:1321. doi: 10.1038/sj.npp.1300433. [DOI] [PubMed] [Google Scholar]
- 27.Kurt M, Arik AC, Celik S. J Basic Clin Physiol Pharmacol. 2000;11:173. doi: 10.1515/jbcpp.2000.11.2.173. [DOI] [PubMed] [Google Scholar]
- 28.Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C, Hen R. Science. 2003;301:805. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- 29.Clark PJ, Brzezinska WJ, Thomas MW, Ryzhenko NA, Toshkov SA, Rhodes JS. Neuroscience. 2008;155:1048. doi: 10.1016/j.neuroscience.2008.06.051. [DOI] [PubMed] [Google Scholar]
- 30.Li Y, Luikart BW, Birnbaum S, Chen J, Kwon CH, Kernie SG, Bassel-Duby R, Parada LF. Neuron. 2008;59:399. doi: 10.1016/j.neuron.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malek ZS, Sage D, Pévet P, Raison S. Endocrinology. 2007;148:5165. doi: 10.1210/en.2007-0526. [DOI] [PubMed] [Google Scholar]
- 32.Gomez-Merino D, Béquet F, Berthelot M, Chennaoui M, Guezennec CY. Neurosci Lett. 2001;301:143. doi: 10.1016/s0304-3940(01)01626-3. [DOI] [PubMed] [Google Scholar]
- 33.Dishman RK, Renner KJ, Youngstedt SD, Reigle TG, Bunnell BN, Burke KA, Yoo HS, Mougey EH, Meyerhoff JL. Brain Res Bull. 1997;42:399. doi: 10.1016/s0361-9230(96)00329-2. [DOI] [PubMed] [Google Scholar]
- 34.Morishima M, Harada N, Hara S, Sano A, Seno H, Takahashi A, Morita Y, Nakaya Y. Neuropsychopharmacology. 2006;31:2627. doi: 10.1038/sj.npp.1301028. [DOI] [PubMed] [Google Scholar]
- 35.Garcia C, Chen MJ, Garza AA, Cotman CW, Russo-Neustadt A. Neuroscience. 2003;119:721. doi: 10.1016/s0306-4522(03)00192-1. [DOI] [PubMed] [Google Scholar]
- 36.Ivy AS, Rodriguez FG, Garcia C, Chen MJ, Russo-Neustadt AA. Pharmacol Biochem Behav. 2003;75:81. doi: 10.1016/s0091-3057(03)00044-3. [DOI] [PubMed] [Google Scholar]
- 37.Maudsley S, Martin B, Luttrell LM. J Pharmacol Exp Ther. 2005;314:485–494. doi: 10.1124/jpet.105.083121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maudsley S, Davidson L, Pawson AJ, Chan R, López de Maturana R, Millar RP. Cancer Res. 2004;64:7533–7544. doi: 10.1158/0008-5472.CAN-04-1360. [DOI] [PubMed] [Google Scholar]
- 39.Maudsley S, Martin B, Luttrell LM. Curr Alzheimer Res. 2007;4:3–19. doi: 10.2174/156720507779939850. [DOI] [PubMed] [Google Scholar]
- 40.Calabrese F, Molteni R, Maj PF, Cattaneo A, Gennarelli M, Racagni G, Riva MA. Neuropsychopharmacology. 2007;32:2351. doi: 10.1038/sj.npp.1301360. [DOI] [PubMed] [Google Scholar]
- 41.Copeland JL, Heggie L. Int J Sports Med. 2008;29:182. doi: 10.1055/s-2007-965114. [DOI] [PubMed] [Google Scholar]
- 42.Trejo JL, Carro E, Torres-Aleman I. J Neurosci. 2001;21:1628–34. doi: 10.1523/JNEUROSCI.21-05-01628.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Llorens-Martín M, Torres-Alemán I, Trejo JL. Neuromolecular Med. 2008;10:99. doi: 10.1007/s12017-008-8026-1. [DOI] [PubMed] [Google Scholar]
- 44.Martin B, Brenneman R, Golden E, Walent T, Becker KG, Prabhu VV, Wood W, 3rd, Ladenheim B, Cadet JL, Maudsley S. J Biol Chem. 2009;284:2493–2511. doi: 10.1074/jbc.M804545200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ding Q, Vaynman S, Akhavan M, Ying Z, Gomez-Pinilla F. Neuroscience. 2006;140:823. doi: 10.1016/j.neuroscience.2006.02.084. [DOI] [PubMed] [Google Scholar]
- 46.Chen MJ, Russo-Neustadt AA. Growth Factors. 2007;25:118. doi: 10.1080/08977190701602329. [DOI] [PubMed] [Google Scholar]
- 47.Gomez-Pinilla F, Vaynman S, Ying Z. Eur J Neurosci. 2008;28:2278. doi: 10.1111/j.1460-9568.2008.06524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clark RG, Robinson ICAF. Cytokine Growth Factor Rev. 1996;7:65–80. doi: 10.1016/1359-6101(96)00006-8. [DOI] [PubMed] [Google Scholar]
- 49.De Meyts P. Diabetologia. 1994;37:135–148. doi: 10.1007/BF00400837. [DOI] [PubMed] [Google Scholar]
- 50.Gill R, Wallach B, Verma C, Urso B, DeWolf E, Grotzinger J, Murray-Rust J, Pitts J, Wollmer A, De Meyts P, Woods S. Protein Eng. 1996;9:1011–1019. doi: 10.1093/protein/9.11.1011. [DOI] [PubMed] [Google Scholar]
- 51.Jansson M, Uhlen M, Nilsson B. Biochemistry. 1997;36:4108–4117. doi: 10.1021/bi961553i. [DOI] [PubMed] [Google Scholar]
- 52.Jones JI, Clemmons DR. Endocr Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 53.Bach LA, Rechler MM. Diabetes Rev. 1995;3:38–61. [Google Scholar]
- 54.Baxter RC, Martin JL. Proc Natl Acad Sci U S A. 1989;86:6898–6902. doi: 10.1073/pnas.86.18.6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lowman HB, Chen YM, Skelton NJ, Mortensen DL, Tomlinson DL, Sadick MD, Robinson ICAF, Clark RG. Biochemistry. 1998;37:8870–8878. doi: 10.1021/bi980426e. [DOI] [PubMed] [Google Scholar]
- 56.Chadwick W, Magnus T, Martin B, Keselman A, Mattson MP, Maudsley S. Trends Neurosci. 2008;31:504–511. doi: 10.1016/j.tins.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grunbaum-Novak N, Taler M, Gil-Ad I, Weizman A, Cohen H, Weizman R. Eur Neuropsychopharmacol. 2008;18:431. doi: 10.1016/j.euroneuro.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 58.Burnstein KL, Cidlowski JA. Mol Cell Endocrinol. 1992;83:1–8. doi: 10.1016/0303-7207(92)90187-b. [DOI] [PubMed] [Google Scholar]
- 59.Pavlides C, Watanabe Y, McEwen BS. Hippocampus. 1993;3:183. doi: 10.1002/hipo.450030210. [DOI] [PubMed] [Google Scholar]
- 60.Woolley CS, Gould E, McEwen BS. Brain Res. 1990;531:225. doi: 10.1016/0006-8993(90)90778-a. [DOI] [PubMed] [Google Scholar]
- 61.Gould E, Cameron HA, Daniels DC, Woolley CS, McEwen BS. J Neurosci. 1992;12:3642. doi: 10.1523/JNEUROSCI.12-09-03642.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hill EE, Zack E, Battaglini C, Viru M, Viru A, Hackney AC. J Endocrinol Invest. 2008;31:587. doi: 10.1007/BF03345606. [DOI] [PubMed] [Google Scholar]
- 63.Droste SK, Gesing A, Ulbricht S, Müller MB, Linthorst AC, Reul JM. Endocrinology. 2003;144:3012. doi: 10.1210/en.2003-0097. [DOI] [PubMed] [Google Scholar]
- 64.Droste SK, Chandramohan Y, Hill LE, Linthorst AC, Reul JM. Neuroendocrinology. 2007;86:26. doi: 10.1159/000104770. [DOI] [PubMed] [Google Scholar]
- 65.Pereira AC, Huddleston DE, Brickman AM, Sosunov AA, Hen R, McKhann GM, Sloan R, Gage FH, Brown TR, Small SA. Proc Natl Acad Sci U S A. 2007;104:5638. doi: 10.1073/pnas.0611721104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Farmer J, Zhao X, van Praag H, Wodtke K, Gage FH, Christie BR. Neuroscience. 2004;124:71. doi: 10.1016/j.neuroscience.2003.09.029. [DOI] [PubMed] [Google Scholar]
- 67.Eadie BD, Redila VA, Christie BR. J Comp Neurol. 2005;486:39. doi: 10.1002/cne.20493. [DOI] [PubMed] [Google Scholar]
- 68.Redila VA, Christie BR. Neuroscience. 2006;137:1299. doi: 10.1016/j.neuroscience.2005.10.050. [DOI] [PubMed] [Google Scholar]
- 69.Stranahan AM, Khalil D, Gould E. Hippocampus. 2007;17:1017. doi: 10.1002/hipo.20348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Regan J, Lee TW, Zindell RM, Bekkali Y, Bentzien J, Gilmore T, Hammach A, Kirrane TM, Kukulka AJ, Kuzmich D, Nelson RM, Proudfoot JR, Ralph M, Pelletier J, Souza D, Zuvela-Jelaska L, Nabozny G, Thomson DS. J Med Chem. 2006;49:7887–7896. doi: 10.1021/jm061273t. [DOI] [PubMed] [Google Scholar]
- 71.Clackers M, Coe DM, Demaine DA, Hardy GW, Humphreys D, Inglis GG, Johnston MJ, Jones HT, House D, Loiseau R, Minick DJ, Skone PA, Uings I, McLay IM, Macdonald SJ. Bioorg Med Chem Lett. 2007;17:4737–4745. doi: 10.1016/j.bmcl.2007.06.066. [DOI] [PubMed] [Google Scholar]
- 72.Lin CW, Nakane M, Stashko M, Falls D, Kuk J, Miller L, Huang R, Tyree C, Miner JN, Rosen J, Kym PR, Coghlan MJ, Carter G, Lane BC. Mol Pharmacol. 2002;62:297–303. doi: 10.1124/mol.62.2.297. [DOI] [PubMed] [Google Scholar]
- 73.Schäcke H, Schottelius A, Döcke Wd, Strehlke P, Jaroch S, Schmees N, Rehwinkel H, Hennekes H, Asadullah K. Proc Natl Acad Sci U S A. 2004;101:227–232. doi: 10.1073/pnas.0300372101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chang YT, Chen YC, Wu CW, Yu L, Chen HI, Jen CJ, Kuo YM. Psychoneuroendocrinology. 2008;33:1173. doi: 10.1016/j.psyneuen.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 75.Dietrich MO, Andrews ZB, Horvath TL. J Neurosci. 2008;28:10766. doi: 10.1523/JNEUROSCI.2744-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stranahan AM, Lee K, Becker KG, Zhang Y, Maudsley S, Martin B, Cutler RG, Mattson MP. Neurobiology of Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.10.016. Epub ahead of print; doi: 10.1016-j.neurobiolaging.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stummer W, Weber K, Tranmer B, Baethmann A, Kempski O. Stroke. 1994;25:1862. doi: 10.1161/01.str.25.9.1862. [DOI] [PubMed] [Google Scholar]
- 78.Shake JG, Peck EA, Marban E, Gott VL, Johnston MV, Troncoso JC, Redmond JM, Baumgartner WA. Ann Thorac Surg. 2001;72:1849. doi: 10.1016/s0003-4975(01)03192-7. [DOI] [PubMed] [Google Scholar]
- 79.Della-Morte D, Dave KR, Defazio RA, Bao YC, Raval AP, Perez-Pinzon MA. Neuroscience. 2009;159:993. doi: 10.1016/j.neuroscience.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tang SJ. Synapse. 2007;61:866. doi: 10.1002/syn.20434. [DOI] [PubMed] [Google Scholar]
- 81.Fukushima T, Zapata JM, Singha NC, Thomas M, Kress CL, Krajewska M, Krajewski S, Ronai Z, Reed JC, Matsuzawa S. Immunity. 2006;24:29. doi: 10.1016/j.immuni.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 82.Yamada M, Ohnishi J, Ohkawara B, Iemura S, Satoh K, Hyodo-Miura J, Kawachi K, Natsume T, Shibuya H. J Biol Chem. 2006;281:20749. doi: 10.1074/jbc.M602089200. [DOI] [PubMed] [Google Scholar]
- 83.Kohn AD, Moon RT. Cell Calcium. 2005;38:439. doi: 10.1016/j.ceca.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 84.Miller S, Yasuda M, Coats JK, Jones Y, Martone ME, Mayford M. Neuron. 2002;36:507. doi: 10.1016/s0896-6273(02)00978-9. [DOI] [PubMed] [Google Scholar]
- 85.Abeliovich A, Chen C, Goda Y, Silva AJ, Stevens CF, Tonegawa S. Cell. 1993;75:1253. doi: 10.1016/0092-8674(93)90613-u. [DOI] [PubMed] [Google Scholar]
- 86.Colombo PJ, Gallagher M. Hippocampus. 2002;12:285. doi: 10.1002/hipo.10016. [DOI] [PubMed] [Google Scholar]
- 87.Spinsanti P, De Vita T, Caruso A, Melchiorri D, Misasi R, Caricasole A, Nicoletti F. J Neurochem. 2008;104:1588. doi: 10.1111/j.1471-4159.2007.05111.x. [DOI] [PubMed] [Google Scholar]
- 88.Saneyoshi T, Kume S, Amasaki Y, Mikoshiba K. Nature. 2002;417:295. doi: 10.1038/417295a. [DOI] [PubMed] [Google Scholar]
- 89.Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH. Nat Med. 2004;10:55. doi: 10.1038/nm979. [DOI] [PubMed] [Google Scholar]
- 90.Klein PS, Melton DA. Proc Natl Acad Sci U S A. 1996;93:8455. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pai R, Tarnawski AS, Tran T. Mol Biol Cell. 2004;15:2156. doi: 10.1091/mbc.E03-12-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Park CH, Chang JY, Hahm ER, Park S, Kim HK, Yang CH. Biochem Biophys Res Commun. 2005;328:227. doi: 10.1016/j.bbrc.2004.12.151. [DOI] [PubMed] [Google Scholar]
- 93.O'Dell TJ, Huang PL, Dawson TM, Dinerman JL, Snyder SH, Kandel ER, Fishman MC. Science. 1994;265:542. doi: 10.1126/science.7518615. [DOI] [PubMed] [Google Scholar]
- 94.Böhme GA, Bon C, Lemaire M, Reibaud M, Piot O, Stutzmann JM, Doble A, Blanchard JC. Proc Natl Acad Sci U S A. 1993;90:9191. doi: 10.1073/pnas.90.19.9191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Packer MA, Stasiv Y, Benraiss A, Chmielnicki E, Grinberg A, Westphal H, Goldman SA, Enikolopov G. Proc Natl Acad Sci U S A. 2003;100:9566. doi: 10.1073/pnas.1633579100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cheng A, Wang S, Cai J, Rao MS, Mattson MP. Dev Biol. 2003;258:319. doi: 10.1016/s0012-1606(03)00120-9. [DOI] [PubMed] [Google Scholar]
- 97.Chen MJ, Ivy AS, Russo-Neustadt AA. Brain Res Bull. 2006;68:257. doi: 10.1016/j.brainresbull.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 98.Moon M, Huh Y, Park C. Neuroreport. 2006;17:1121. doi: 10.1097/01.wnr.0000224767.84830.1b. [DOI] [PubMed] [Google Scholar]
- 99.Kubota M, Sakakihara Y, Mori M, Yamagata T, Momoi-Yoshida M. Brain Dev. 2004;26:481–483. doi: 10.1016/j.braindev.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 100.Willmot M, Ghadami A, Whysall B, Clarke W, Wardlaw J, Bath PM. Hypertension. 2006;47:1209–1215. doi: 10.1161/01.HYP.0000223024.02939.1e. [DOI] [PubMed] [Google Scholar]
- 101.Toda N, Ayajiki K, Okamura T. J Physiol. 1997;498:453–461. doi: 10.1113/jphysiol.1997.sp021871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Perretti M, D'Acquisto F. Nat Rev Immunol. 2009;9:62. doi: 10.1038/nri2470. [DOI] [PubMed] [Google Scholar]
- 103.Fürstenberger G, Senn HJ. Lancet Oncol. 2002;3:298. doi: 10.1016/s1470-2045(02)00731-3. [DOI] [PubMed] [Google Scholar]