

Abstract
(4-Phenoxyphenylsulfonyl)methylthiirane (SB-3CT) is the selective inhibitor of matrix metalloproteinase 2 (MMP2). The inhibition mechanism of MMP2 by SB-3CT involves C–H deprotonation with concomitant opening of the three-membered heterocycle. In this study, the energetics of the deprotonation-induced ring opening of (4-phenoxyphenylsulfinyl)methylthiirane, the sulfoxide analogue of SB-3CT, are examined computationally using DFT and QM/MM calculations. A model system, 2-(methylsulfinylmethyl)thiirane, is used to study the stereoelectronic and conformational effects of reaction barriers in methanol. For the model system in methanol solution (using polarizable continuum model), the reaction barriers range from 17 to 23 kcal/mol with significant stereoelectronic effects. However, the lowest barriers of the (R, R) and (S, R) diastereomers are similar. Two diastereomers of the sulfoxide analogue of SB-3CT are studied in the active site of MMP2 by QM/MM methods with an accurate partial charge fitting procedure. The ring opening reactions of these two diastereomers have similar reaction energetics. Both are exothermic from the reactant to the ring opening product (thiolate). The protonation of the thiolate by a water molecule is endothermic in both cases. However, the deprotonation / ring opening barriers in the MMP2 active site using QM/MM methods for the (R, R) and (S, R) inhibitions are quite different (23.3 and 28.5 kcal/mol, respectively). The TSs identified in QM/MM calculations were confirmed by vibrational frequency analysis and reaction path following. The (R, R) diastereomer has hydrogen bond between the sulfoxide oxygen and the backbone NH of Leu191, while the (S, R) has hydrogen bond between the sulfoxide oxygen and a water molecule. The dissimilar strengths of these hydrogen bonds as well as minor differences in the TS structures contribute to the difference between the barriers. Compared to SB-3CT, both diastereomers of the sulfoxide analogue have higher reaction barriers, and have less exothermic reaction energies. This agrees well with the experiments that SB-3CT is a more effective inhibitor of MMP2 than its sulfoxide analogue.
Keywords: MMP2, SB-3CT, Sulfoxide, Analogue, QM/MM, ONIOM, Inhibition, RESP, Charge fitting
Introduction
Matrix metalloproteinases (MMPs) regulate functions of the extracellular matrix (ECM). These zinc-dependent endopeptidases are involved in many biological processes, such as embryonic development,1 tissue remodeling and repair,2 neurophathic pain processes,3 cancers,4,5 and other diseases.6-9 Therefore, this group of proteins is a primary target for drug design.
Gelatinase A (MMP2) is an MMP which digests type IV collagens.10 Excessive activities of this enzyme are implicated in tumor metastasis and angiogenesis. Based on the studies of structures and catalytic mechanisms of MMP2,11-17 many inhibitors have been developed against MMP2 activity.14,18-23 SB-3CT is one such inhibitor that selectively inhibits MMP2 with high activity.24
Recent experiments from the Mobashery lab demonstrate that the mechanism for SB-3CT inhibition of MMP2 involves proton transfer coupled to ring opening (Scheme 1).25 In this mechanism, a hydrogen from the methylene group between the sulfone and the thiirane is abstracted by the carboxylate of glutamate-404. This deprotonation initiates ring opening of the thiirane ring, and produces a thiolate that can coordinate to the zinc ion at the active site. This mechanism was supported by the observation of a primary kinetic isotope effect for the methylene hydrogen. The experiments indicate that the sulfoxide analogue of SB-3CT is a linear competitive inhibitor of MMP2 that does not undergo ring opening, despite the structural similarity between these two species. The acidity of alkylarylsulfoxides (approximate pKa = 33)26 is significantly weaker than that of the corresponding sulfone (approximate pKa = 29).27 This computational study tests whether the reduced carbon acidity of the sulfoxide analogue of SB-3CT could account for the fact that it is a poorer inhibitor of MMP2.
Scheme 1.
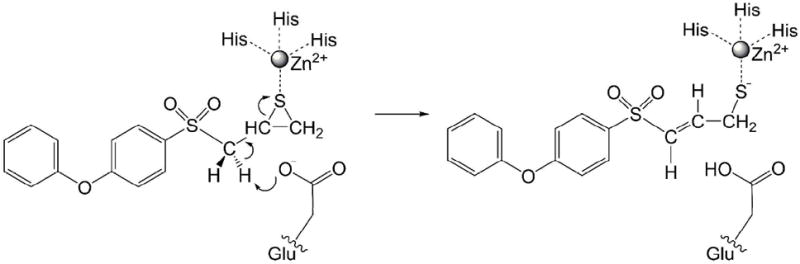
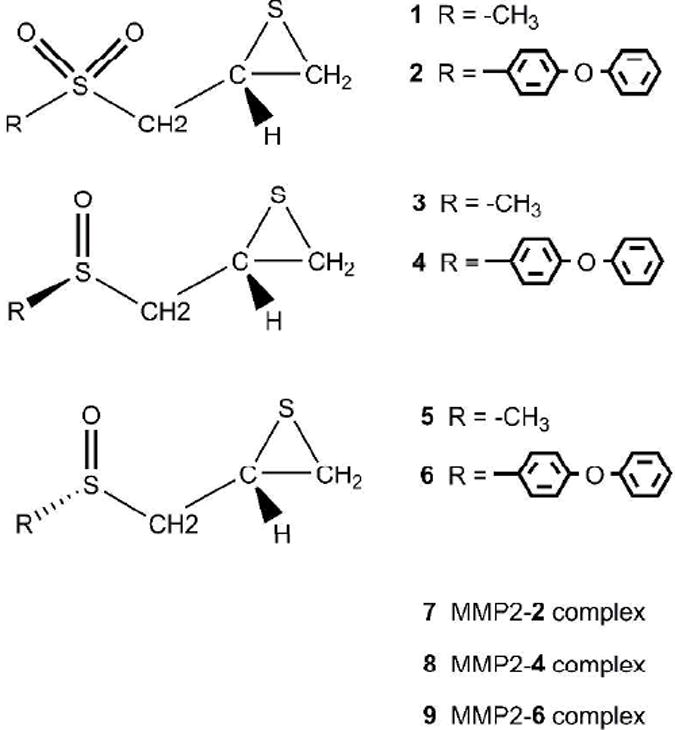
Previously, we used density functional theory (DFT) calculations to investigate the coupled deprotonation and ring opening of (R)-2-(methylsulfonylmethyl)-thiirane in solution (1 in Scheme 2) as model for (R)-SB-3CT (2).28 This prepared the way for a more extensive study of the deprotonation / ring opening mechanism for the inhibition of MMP2 by SB-3CT and its oxirane analogue, using combined quantum mechanics / molecular mechanics (QM/MM) calculations.29 We apply a similar strategy to investigate the energetics of sulfoxide analogue of SB-3CT (4 and 6) and to compare its behavior to SB-3CT as an inhibitor of MMP2. The investigation of the sulfoxide analogue of SB-3CT has been carried out in two stages. First, 3 and 5 were used as models of 4 and 6, respectively, and studied in methanol solution using a polarizable continuum model (PCM). Second, the energetics of the ring opening reaction of 4 and 6 in the active site of MMP2 were examined by QM/MM methods.
Scheme 2.
Computational Methods
Calculations were carried out with the development version of the Gaussian series of programs.30 B3LYP density functional theory31-33 was chosen since it performs better than other level of theories when compared to CBS-QB334,35 calculations on these systems.28 The 6-31+G(d) basis set was used for geometry optimization followed by single point calculations with a 6-311+G(d,p) basis set. All calculations were carried out in solution using the integral-equation-formalism of the polarizable continuum model (IEF-PCM),36 and methanol (dielectric constant ε = 32.63) as solvent. Transition states (TS) were confirmed by vibrational frequency analysis and had only one imaginary frequency. The solution phase enthalpies reported in this study are based on IEF-PCM/B3LYP/6-311+G(d,p) energies with optimized geometries and zero point energies obtained with the 6-31+G(d) basis set.
Since the previous study focused on the R isomer of SB-3CT (2), all the sulfoxide analogues in this study have the R configuration of the thiirane group (3, 4, 5, and 6). The initial structures of complexes of MMP2 with 4 and 6 (designated as 8 and 9, respectively) were based on the structure of the MMP2 complex with SB-3CT (7), which was constructed using docking and molecular dynamics methods.29
A two-layer ONIOM method37-44 was used for the QM/MM study of the inhibition mechanism of 4 and 6. Similar to the previous study, the zinc ion, the three imidazole rings from His403, His407 and His413, the CH2CO2− part of the Glu404 side chain, the thiirane with the SOCH2 group, and one water molecule coordinating with zinc in MD simulation were included in QM region (45 atoms). The B3LYP/6-31G(d) level of DFT was employed to describe the QM part of the system, and the parm96 parameter set of the Amber force field45 used to describe the MM part of the system. QM/MM geometry optimization was carried by a mechanical embedding scheme (electrostatic interactions between the QM and MM regions are handled by MM). Single point calculations with electronic embedding44 in this scheme were used for the final QM/MM energies calculated at the ONIOM(B3LYP/6-311+G(d,p):AMBER) level of theory. The QM/MM energetic profiles reported here were generated using these single point energies of reactant, TS and product for each analogue. All ONIOM calculations were carried out with the development version of Gaussian.30
The TSs identified in QM/MM study were confirmed by following methods. Since a full vibrational frequency analysis is not practical for the entire MMP2-inhibitor system at this time, only the two lowest vibrational frequencies of TSs were calculated. The presence of only one imaginary frequency confirmed that these structures are transition states. Secondly, a partial protein model was constructed containing all of the QM region atoms, the moving parts of the MM region, and enough frozen parts of the MM region that surround the first two parts. Full frequency analysis was carried using the partial models of both TSs. There are numerous dangling bonds in the frozen MM region of the partial model, but these missing bonds have little effect on the frequency calculations since these atoms are frozen during optimization. The vibrational normal modes of TSs were inspected visually.
To obtain accurate energies, we developed a method for obtaining partial charges for the reactive system that alternates between QM/MM geometry optimization and Resp46,47 charge fitting.29 This procedure is described as following. (1) A preliminary set of partial charges was obtained for the substrates in the gas phase using the Resp procedure. (2) The reactant, TS and product were optimized by ONIOM calculations using mechanical embedding with the preliminary partial charges. (3) The QM atoms of the reactant, TS and product from the ONIOM geometry optimizations in the second step were used to obtain an improved set of partial charges using the Resp procedure, and the hydrogen atoms added to cap the dangling bonds were constrained to have zero charges. (4) The reactant, TS and product were optimized in the active site by ONIOM calculations using mechanical embedding with the improved charges. (5) Step 3 and 4 were repeated until the difference in the ONIOM total energies between the last two rounds of optimizations was less than 0.1 kcal/mol. (6) The converged geometries and charges from the last step were used for single point calculations with electronic embedding.
Results and Discussion
Model systems in solution
The sulfoxide analogues of (R)-2-(methylsulfonylmethyl)thiirane 1 can adopt two configurations leading to two diastereomers, (R,R)-(3) and (S,R)-(5) in Scheme 2. The conformers of model molecules 3 and 5 have been studied in methanol. For each diastereomer, the rotations about C–S and C–C bonds produce multiple conformers. Nine conformational minima were identified for 3 (Scheme 3). The energy differences of these conformers are between 0.2 and 4.8 kcal/mol. In the lowest energy conformer, 3d, the methyl group attached to sulfoxide is anti to the thiirane group with regard to the rotation of the C–S bond, and CH2 group of thiirane is anti to the sulfoxide sulfur with regard to the rotation of the C–C bond. Four other conformers are within 1.0 kcal/mol of the lowest minimum. The highest energy conformers, 3h and 3i, have obvious steric interactions between methyl and thiirane groups. Similar results were found for 5 (see Scheme S1 in the Supporting Information). The lowest energy conformer also has the methyl group anti to the thiirane group, and the highest energy conformers have significant steric interactions.
Scheme 3.
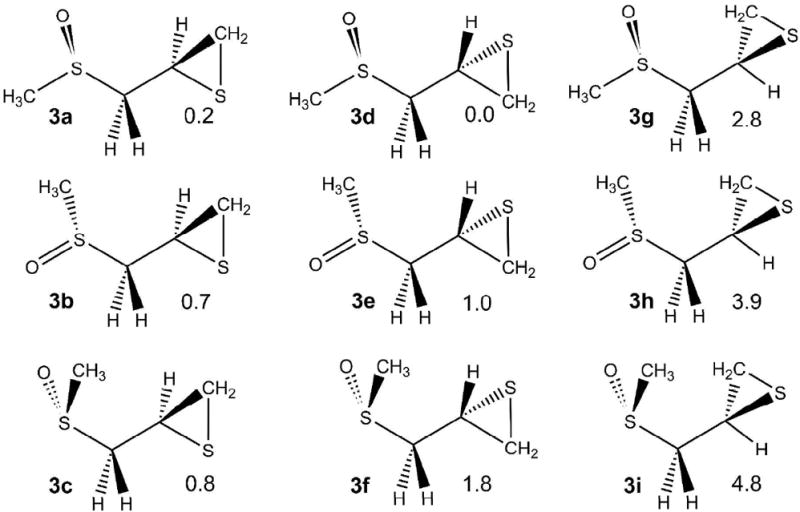
(R,R)-2-methylsulfinylmethyl)thiirane (relative energies in kcal/mol)
For the reaction of the model systems calculated in methanol solution, acetate is the proton acceptor, and abstracts the pro-S hydrogen of methylene group, resulting in opening of the thiirane ring (Scheme 4). This is the same hydrogen that is abstracted at the active site of MMP2. Starting from all of the reactant conformers, five TSs were identified for the reaction of the (R,R) diastereomer (3-TSa-e, Scheme 4). The reaction barriers were calculated with respect to the lowest minimum in Scheme 3. The barriers range from 16.9 to 23.3 kcal/mol. Selected geometrical parameters for the lowest energy reactant complex and TS are shown in Figure S1 of the Supporting Information. Similar to the sulfone, the TS with lowest barrier for deprotonation initiated ring opening has an anti configuration between the two breaking bonds, but has a much higher barrier than the sulfone (16.9 for 3-TSd vs 11.3 kcal/mol for 1).28 For a given orientation of the sulfoxide group in the (R,R) diastereomer, the TSs with the leaving hydrogen anti to the breaking C–S bond in thiirane ring (3-TSd and e) have lower energies than the TSs with syn orientation (3-TSa-c). A carbanion next to a sulfoxide group prefers to be anti to the sulfoxide oxygen.48,49 Thus, the TSs with breaking C–H bonds anti to the sulfoxide oxygen (3-TSa and d) have lower energies than other orientations. With the methyl group anti to the leaving proton (3-TSb and e), the barriers of ring opening are about 4 kcal/mol higher. Using the infinitely separated ring opening product and acetic acid, the reaction energies are 2.6 and 4.5 kcal/mol for the E and Z products, respectively (Figure S2, Supporting Information). These energies are nearly the same as the heats of reaction for the sulfone system (2.8 and 5.3 kcal/mol for the E and Z products respectively.28
Scheme 4.
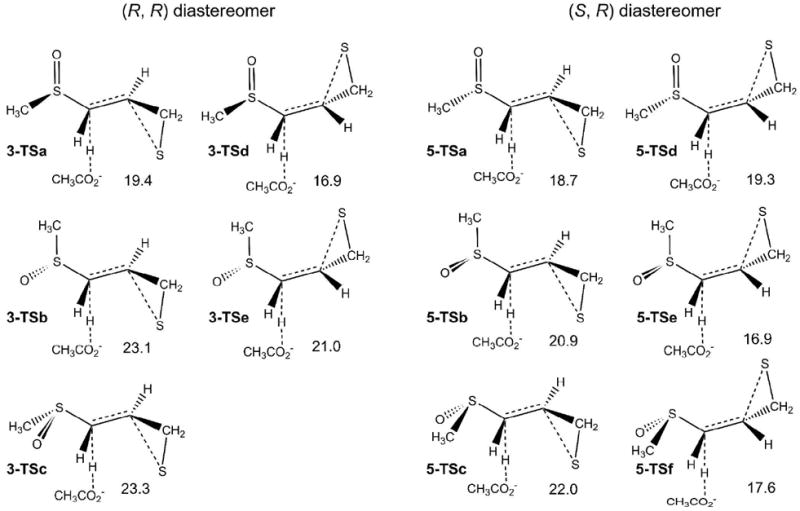
2-(Methylsulfinylmethyl)thiirane transition states (barrier heights relative to the most stable reactant conformation in kcal/mol)
The barriers for the (S, R) diastereomer (5) range from 16.9 to 22.0 kcal/mol, which is close to the values obtained for 3. However, the TS with the breaking C–H bond anti to both the sulfoxide oxygen and the breaking C–S bond (5-TSd) does not have the lowest barrier due to the steric interaction between the methyl group attached to sulfoxide and the methylene group in thiirane ring. In the conformation with the breaking C–H bond syn to the breaking C–S bond (5-TSa-c), this steric interaction is absent and the breaking C–H bond prefers to be anti to the sulfoxide oxygen. The TS with lowest barrier (16.9 kcal/mol, 5-TSe) has the breaking C–H bond anti to the breaking C–S bond and anti to the methyl group attached to sufoxide group. The reaction energies are similar to the (R,R) diastereomer, 1.7 and 3.4 kcal/mol for the E and Z products, respectively (Figure S3, Supporting Information).
To summarize model system calculations in solution, the lowest barriers for the sulfoxides 3 and 5 are approximately 17 kcal/mol, about 6 kcal/mol higher than for the sulfone 1.28 This observation is in agreements that the acidity of alkylarylsulfoxides (pKa ~ 33)26 is much weaker than that of the corresponding sulfone (pKa ~ 29).27 Like the sulfone, the sulfoxide group exerts a significant stereoelectronic effect on the barrier height.
QM/MM study
The structures and energetics for deprotonation initiated ring opening reactions of sulfoxide analogues of SB-3CT at MMP2 active site were investigated by QM/MM methods. To facilitate comparison with the previous QM/MM results for SB-3CT (2),29 the initial structures for the sulfoxide reactant complexes with MMP2 were generated by deleting an oxygen from the sulfone reactant complex with MMP2. In the complex of MMP2 and 2 (7), there is a hydrogen bond between one oxygen of the sulfone and the backbone amide hydrogen of Leu191. A similar hydrogen bond is found in various other MMP2 inhibitors.50,51 The complex of MMP2 with sulfoxide 4 (designated as 8) retains this hydrogen bond. The other oxygen in the sulfone-MMP2 complex is hydrogen bonded to a water. The complex of MMP2 with sulfoxide 6 (designated as 9) also has this hydrogen bond to a water molecule but lacks the hydrogen bond to Leu191. The QM/MM energies of 7, 8 and 9 are given in Table 1, and illustrated in Figure 1.
Table 1.
QM/MM calculations of the energetics for the ring-opening reactions of SB-3CT (2) and the sulfoxide analogues of SB-3CT (4 and 6) of in the active site of MMP2a
| inhibitor | barrier height | reaction enthalpy |
|
|---|---|---|---|
| P1 (unprotonated product) | P2 (protonated product) | ||
| 7 | 19.9 | −21.1 | −0.7 |
| 8 | 23.3 | −17.2 | −2.5 |
| 9 | 28.5 | −17.3 | −1.6 |
ONIOM(B3LYP/6-311+G(d,p):AMBER)//ONIOM(B3LYP/6-31G(d):AMBER) with electronic embedding; energies in kcal/mol. (See Figures 1-3, and Figures S4 and S5 in Supporting Information)
Figure 1.
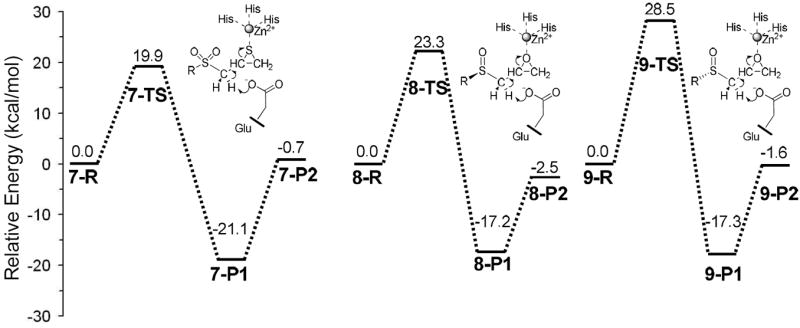
Energy profiles for SB-3CT (2) and its sulfoxide analogues (4 and 6) in the MMP2 active site. Relative energies (in kcal/mol) were calculated at ONIOM(B3LYP/6-311+G(d,p):AMBER) using electronic embedding with the reactant complexes used as reference states.
MMP2 complex with 4
The reactant, TS, and product structures for the ring open reaction of 4 at active site of MMP2 are shown in Figure 2 and Figure S4. The orientation and key intermolecular contacts between 4 and the MMP2 active site in reactant complex, 8-R, closely resemble the complex structure of MMP2 and SB-3CT29 and crystallographic structures of other MMP inhibitors.16 The phenoxyphenyl side chain of 4 is located as expected in the S1’ pocket, and a strong (1.89 Å) hydrogen bond from the backbone NH of Leu191 to the sulfoxide oxygen is preserved in the reactant complex as mentioned above (Figure 2 and S4).
Figure 2.
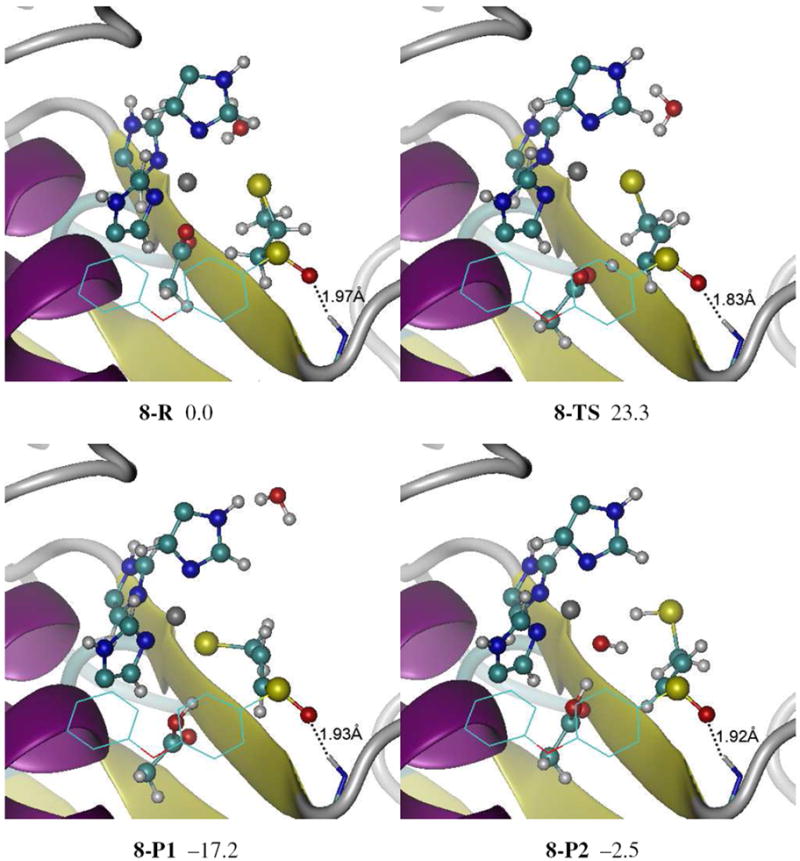
Reactant, transition state and products for the (R, R) sulfoxide analogue of SB-3CT (4) in the MMP2 active site (8) optimized at the ONIOM(B3LYP/6-31G(d):AMBER) level of theory. Energies (in kcal/mol) were calculated by ONIOM(B3LYP/6-311+G(d,p):AMBER) using electronic embedding with the reactant complex used as reference state. The sulfoxide group of 4 forms a hydrogen bond with the backbone NH of Leu191. Complex 8-P1 is the unprotonated ring opening product. In complex 8-P2, the ring opening product thiolate is protonated by a water molecule, and the resulting hydroxide anion coordinates with the zinc. See Figure S4 in the Supporting Information for details. Atoms are colored according to atom types (H, C, N, O, S, Zn, shown in white, cyan, blue, red, yellow, and grey, respectively).
In reactant complex, the zinc shows tetrahedral coordination with the three histidines (2.07 Å, 2.14 Å, and 2.25 Å) and an oxygen of the Glu404 carboxylate (1.99 Å). The water molecule included in QM region is not coordinated to zinc (3.59 Å). The substrate 4 has a conformation in which the lone pair of sulfoxide group is anti to thiirane, and CH2 group of thiirane is anti to sulfoxide group. This conformer approximately corresponds to the conformer 3f in Scheme 3 that is 1.8 kcal/mol above the lowest minimum.
In the QM/MM calculations of the TS, 8-TS in Figure 2 and Figure S4, the transferring proton is half way between the acceptor oxygen and donor carbon (1.33 Å from each). The breaking C–S bond is elongated to 2.32 Å in 8-TS. The Glu404 side chain has moved away from zinc in order to abstract the proton. The hydrogen bond between the backbone NH of Leu191 to the sulfoxide oxygen is preserved (1.83 Å) and is slightly shorter than in the reactant complex. In the TS, the Glu404 side chain moves away from the zinc (dZn-O 4.01 Å and 4.67 Å), the thiirane ring opens and the sulfur becomes strongly coordinated to the zinc (2.31 Å). The Glu404 sidechain abstracts one of the methylene protons. Because of the constraints imposed by binding in the active site, the breaking C–H bond in the TS is neither syn nor anti to the breaking C–S bond as observed in model systems in Scheme 4, but in between these two orientations. The breaking C–H bond is approximately eclipsed to the C–S bond between sulfoxide and phenyl group. It is noteworthy that the water molecule in the QM region forms a loose hydrogen bond with thiirane sulfur (2.35 Å).
The two lowest vibrational frequencies were calculated for 8-TS (1287.8i cm−1 and 20.9 cm−1) and confirmed that there is only one imaginary frequency for this TS. A full vibrational frequency analysis was carried out for the partial model which contains all of the QM region, the moving parts of the MM region, and enough frozen parts of the MM region that surround the first two parts (Figure S6). Only one imaginary frequency was found (1288.6i cm−1), which also confirmed the identity of this TS. The intrinsic reaction coordinate (IRC) was calculated for 8-TS using the partial model to generate the reaction path from reactant to product through 8-TS. Animations of the normal mode corresponding to imaginary frequency and the IRC path are provided in the supporting information.
In the product, 8-P1, the thiolate sulfur remains tightly coordinated to the zinc. The protonated Glu404 stays away from zinc. As in the reactant complex and TS, the zinc has tetrahedral coordination. Beside three histidines, the fourth ligand of zinc in 8-P1 is the thiolate. The water molecule in the QM region does not interact with the zinc in this structure. Because a substantial portion of the active site of MMP2 is exposed to solvent, the previous QM/MM study also examined the protonation of the ring opening products of SB-3CT at active site of MMP2 by the adjacent QM water molecule.29 For the sulfoxide product complex, this proton transfer yields 8-P2 in Figure 2 and Figure S4. The hydroxide anion produced by deprotonation of water coordinates with zinc in 8-P2 (1.86 Å), displacing the thiol (dZn-S = 4.44 Å).
The barrier for the ring opening of 4 at the active site of MMP2 is 23.3 kcal/mol, which is 3.4 kcal/mol higher than the barrier of SB-3CT (19.9 kcal/mol).29 The reaction from 8-R to 8-P1 is exothermic with an energy of −17.2 kcal/mol. However, the protonation of thiolate by water molecule (from 8-P1 to 8-P2) is very endothermic. Therefore, the ring opening product of 8 will favor the thiolate state (8-P1) over thiol state (8-P2).
MMP2 complex with 6
The reactant, TS, and product structures for the ring open reaction of 6 at active site of MMP2 are shown in Figure 3 and Figure S5. The MMP2-6 reactant complex is similar to the MMP2-4 reactant complex except for the absence of hydrogen bond between the backbone NH of Leu191 and the sulfoxide oxygen due to the different configuration of sulfoxide groups in 4 and 6. Instead, the sulfoxide oxygen forms a hydrogen bond with a water molecule (shown in tube structural representation in Figure 3). This water molecule also forms a hydrogen bond with carbonyl oxygen of Pro423 backbone (1.68 Å).
Figure 3.
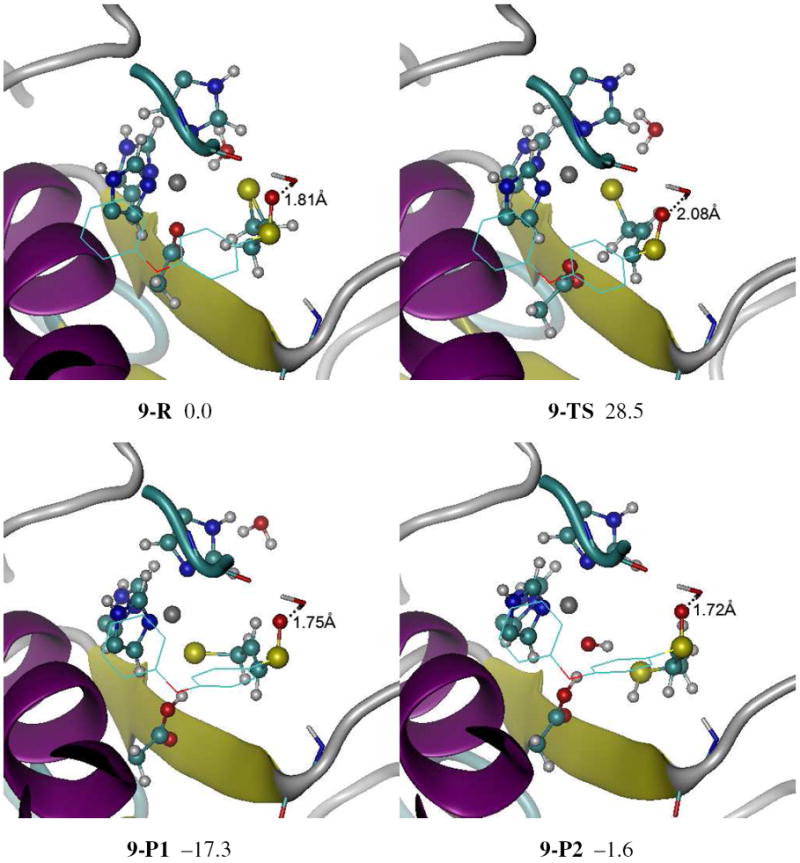
Reactant, transition state and products for the (S, R) sulfoxide analogue of SB-3CT (6) in the MMP2 active site (9) optimized at the ONIOM(B3LYP/6-31G(d):AMBER) level of theory. Energies (in kcal/mol) were calculated by ONIOM(B3LYP/6-311+G(d,p):AMBER) using electronic embedding with the reactant complex used as reference state. Sulfoxide group of 6 forms hydrogen bond not with the backbone NH of Leu191, but with a water molecule (shown in tube). Complex 9-P1 is the unprotonated ring opening product. In complex 9-P2, the ring opening product thiolate is protonated by water molecule, and the resulting hydroxide anion coordinates with the zinc. See Figure S5 in the Supporting Information for details. The color scheme is the same as Figure 2.
In reactant complex, the zinc shows tetrahedral coordination with the three histidines and an oxygen the Glu404 carboxylate (1.97 Å). The substrate 6 has a conformation in which phenyl group of sulfoxide group is anti to thiirane, and CH2 group of thiirane is anti to sulfoxide group. This corresponds approximately to the conformer which is 0.9 kcal/mol above the lowest minimum in Scheme S1 (5a).
In the QM/MM calculations of the TS, 9-TS in Figure 3 and Figure S5, the transferring proton is 1.50 Å from the acceptor oxygen, and 1.24 Å from the donor carbon. The breaking C–S bond is elongated to 2.46 Å in 9-TS. Compared to 8-TS, this TS is earlier in terms of proton transferring and later in ring opening. The Glu404 side chain moves away from zinc, and the thiirane sulfur is strongly coordinated to the zinc (2.31 Å) in the TS. Similar to the 8-TS, the water molecule in the QM region also forms a hydrogen bond with thiirane sulfur (2.34 Å) in the TS. The thiolate sulfur remains tightly coordinated to the zinc in 9-P1 (2.30 Å). The zinc has tetrahedral coordination with the three histidines and the thiolate. The proton transfer to the product leads to 9-P2 in Figure 3 and Figure S5.
The reaction energy from 9-R to 9-P1 (−17.3 kcal/mol) and 9-P2 (−1.6 kcal/mol) are very close to the results of complex 8. The similarity of reaction energies between 8 and 9 indicates that the hydrogen bonds between the backbone NH of Leu191 or water molecule and the substrate do not affect the thermodynamics of the ring opening reaction at the active site of MMP2. However, the barrier for the ring opening of 6 at the active site of MMP2 (complex 9-TS) is 28.5 kcal/mol, which is 5.2 kcal/mol higher than the barrier of complex 8. The major differences between 9-TS and 8-TS are the distances of hydrogen bond involved with sulfoxide oxygen. In complex 8, the sulfoxide oxygen forms a hydrogen bond with the backbone NH of Leu191. The hydrogen bond formed with sulfoxide oxygen is shorter in 9-R (1.81 Å) than in 8-R (1.97 Å), but longer in 9-TS (2.08 Å) than in 8-TS (1.83 Å). The stabilization effect by this hydrogen bond is stronger in 9-R than in 8-R, but weaker in 9-TS than in 8-TS. The energy of 9-R is 4.1 kcal/mol lower than 8-R, while the energy of 9-TS is 1.1 kcal/mol higher than 8-TS. Consequently, the ring opening reaction barrier in complex 9 is 5.2 higher than in complex 8.
The two lowest vibrational frequencies calculated for 9-TS were 550.4i cm−1 and 11.9 cm−1, confirming that there is only one imaginary frequency. The full vibrational frequency analysis for the partial model also found only one imaginary frequency (554.8i cm−1). The lower imaginary frequency in 9-TS is due to a larger reduced mass and a lower force constant, both of which may be attributable to the structural differences between 8-TS and 9-TS. Animations of the imaginary frequency mode and the IRC path are available in the supporting information.
In the active site of MMP2, the barriers for ring opening for the sulfoxide analogues of SB-3CT (23.3 kcal/mol for 4 and 28.5 kcal/mol for 6) are higher than for SB-3CT (19.9 kcal/mol), and the overall reaction energetics for 4 and 6 (−17.2 and −17.3 kcal/mol, respectively) are less exothermic than for SB-3CT (−21.0 kcal/mol). The ring opening reactions of sulfoxide analogues of SB-3CT at MMP2 active site are less favorable than SB-3CT both kinetically and thermodynamically. While the sulfoxide analogue of SB-3CT is observed to be linear competitive inhibitor, SB-3CT is found to be slow binding inhibitor.25 This suggests that the sulfoxide analogue may bind at the MMP2 active site without the ring opening, whereas SB-3CT undergoes ring opening after binding. The higher barriers calculated for ring opening of the sulfoxide analogues than for SB-3CT agree with this observation.
Conclusions
In this study, the energetics of the deprotonation-induced ring opening of the sulfoxide analogues of SB-3CT (4 and 6) at the active site of MMP2 were examined by DFT and QM/MM calculations. The mechanism involves deprotonation of the inhibitor by a glutamate in the active site, opening of the three-membered ring and binding of the product thiolate to the zinc ion in the active site. The reaction barriers for a model system, 2-(methylsulfinylmethyl)thiirane (3 and 5), were studied in methanol. Two configurations of sulfoxide group were investigated for the effect on reaction barriers. Overall, the reaction barriers range from 17 to 23 kcal/mol due to steric and stereoelectronic effects associated with the rotation about the C–C and C–S bonds. The lowest barriers were 17 kcal/mol for both the R and S configurations of sulfoxide group in the model system.
QM/MM methods with an accurate partial charge fitting procedure were used to study the two diastereomers (4 and 6) in the active site of MMP2. One has a hydrogen bond between the sulfoxide oxygen and the backbone NH of Leu191 (8), while the other has a hydrogen bond between the sulfoxide oxygen and a water molecule (9). The barrier for 9 (28.5 kcal/mol) is higher than 8 (23.3 kcal/mol). Both TSs were confirmed by vibrational frequency and intrinsic reaction coordinate calculations. In addition to small differences in the geometries of 8-TS and 9-TS, differences in the hydrogen bonds in both reactant and TS complexes contribute to the differences between the two barriers. The ring opening reactions of the two diastereomers have similar reaction energetics, and both are exothermic from the reactant to the thiolate ring opening product. The protonation of the thiolate by an adjacent water molecule is endothermic in both cases. The ring opening of 4 at the active site of MMP2 is thermodynamically favorable, and the ring-opened product is stable. However, the methylene group adjacent to sulfoxide has lower acidity than the one adjacent to sulfone group. This lower acidity makes the ring opening reaction of the sulfoxide analogue of SB-3CT in the MMP2 active site more difficult than SB-3CT, accounting for the fact that it is a linear competitive inhibitor rather than a slow binding inhibitor.
Supplementary Material
Acknowledgments
This work is supported at Wayne State University by the National Science Foundation (CHE0910858) and at the University of Notre Dame by the National Institutes of Health (CA122417). We thank Wayne State University for generous allocations of computer time on its computational grid.
Footnotes
Supporting Information Available Energetics of model reactions for 1, 3 and 5, conformers of 5, reactant and TS complexes of 3 and 5, ring open products of 3 and 5, QM/MM geometries for ring opening reaction of 4 and 6 in the active site of MMP2 (8 and 9, respectively), partial QM/MM model, animations of the imaginary frequency modes and intrinsic reaction paths for 8-TS and 9-TS, Cartesian coordinates of all the conformers, reactant and TS complexes of 3 and 5, active site coordinates for MMP2 complexes 8 and 9. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Shi Y-B, Fu L, Hasebe T, Ishizuya-Oka A. Pharmacol Ther. 2007;116:391. doi: 10.1016/j.pharmthera.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith MF, Ricke WA, Bakke LJ, Dow MP, Smith GW. Mol Cell Endocrinol. 2002;191:45. doi: 10.1016/s0303-7207(02)00054-0. [DOI] [PubMed] [Google Scholar]
- 3.Kawasaki Y, Xu Z-Z, Wang X, Park JY, Zhuang Z-Y, Tan P-H, Gao Y-J, Roy K, Corfas G, Lo EH, Ji R-R. Nat Med. 2008;14:331. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Egeblad M, Werb Z. Nat Rev Cancer. 2002;2:161. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 5.Noël A, Jost M, Maquoi E. Semin Cell Dev Biol. 2008;19:52. doi: 10.1016/j.semcdb.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 6.Dollery CM, McEwan JR, Henney AM. Circ Res. 1995;77:863. doi: 10.1161/01.res.77.5.863. [DOI] [PubMed] [Google Scholar]
- 7.Luft FC. J Mol Med. 2004;82:781. doi: 10.1007/s00109-004-0609-1. [DOI] [PubMed] [Google Scholar]
- 8.Janssens S, Lijnen HR. Cardiovasc Res. 2006;69:585. doi: 10.1016/j.cardiores.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 9.Chow AK, Cena J, Schulz R. Br J Pharmacol. 2007;152:189. doi: 10.1038/sj.bjp.0707344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liotta LA, Tryggvason K, Garbisa S, Robey PG, Abe S. Biochemistry. 1981;20:100. doi: 10.1021/bi00504a017. [DOI] [PubMed] [Google Scholar]
- 11.Morgunova E, Tuuttila A, Bergmann U, Isupov M, Lindqvist Y, Schneider G, Tryggvason K. Science. 1999;284:1667. doi: 10.1126/science.284.5420.1667. [DOI] [PubMed] [Google Scholar]
- 12.Briknarova K, Gehrmann M, Banyai L, Tordai H, Patthy L, Llinas M. J Biol Chem. 2001;276:27613. doi: 10.1074/jbc.M101105200. [DOI] [PubMed] [Google Scholar]
- 13.Feng Y, Likos JJ, Zhu L, Woodward H, Munie G, McDonald JJ, Stevens AM, Howard CP, De Crescenzo GA, Welsch D, Shieh H-S, Stallings WC. Biochim Biophys Acta, Proteins Proteomics. 2002;1598:10. doi: 10.1016/s0167-4838(02)00307-2. [DOI] [PubMed] [Google Scholar]
- 14.Lukacova V, Zhang Y, Mackov M, Baricic P, Raha S, Calvo J, Balaz S. J Biol Chem. 2004;279:14194. doi: 10.1074/jbc.M313474200. [DOI] [PubMed] [Google Scholar]
- 15.Diaz N, Suarez D, Sordo T. J Phys Chem B. 2006;110:24222. doi: 10.1021/jp0656882. [DOI] [PubMed] [Google Scholar]
- 16.Tochowicz A, Maskos K, Huber R, Oltenfreiter R, Dive V, Yiotakis A, Zanda M, Bode W, Goettig P. J Mol Biol. 2007;371:989. doi: 10.1016/j.jmb.2007.05.068. [DOI] [PubMed] [Google Scholar]
- 17.Diaz N, Suarez D. Proteins: Struct Funct Bioinf. 2008;72:50. doi: 10.1002/prot.21894. [DOI] [PubMed] [Google Scholar]
- 18.Ikejiri M, Bernardo MM, Bonfil RD, Toth M, Chang M, Fridman R, Mobashery S. J Biol Chem. 2005;280:33992. doi: 10.1074/jbc.M504303200. [DOI] [PubMed] [Google Scholar]
- 19.Fisher JF, Mobashery S. Cancer Metastasis Rev. 2006;25:115. doi: 10.1007/s10555-006-7894-9. [DOI] [PubMed] [Google Scholar]
- 20.Khandelwal A, Balaz S. J Comput-Aided Mol Des. 2007;21:131. doi: 10.1007/s10822-007-9104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khandelwal A, Balaz S. Proteins: Struct Funct Bioinf. 2007;69:326. doi: 10.1002/prot.21500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gupta SP. Chem Rev. 2007;107:3042. doi: 10.1021/cr030448t. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Lukacova V, Bartus V, Nie X, Sun G, Manivannan E, Ghorpade S, Jin X, Manyem S, Sibi M, Cook G, Balaz S. Chem Biol Drug Des. 2008;72:237. doi: 10.1111/j.1747-0285.2008.00710.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown S, Bernardo MM, Li Z-H, Kotra LP, Tanaka Y, Fridman R, Mobashery S. J Am Chem Soc. 2000;122:6799. [Google Scholar]
- 25.Forbes C, Shi QC, Fisher JF, Lee M, Hesek D, Llarrull LI, Toth M, Gossing M, Fridman R, Mobashery S. Chem Biol Drug Des. 2009;74:527. doi: 10.1111/j.1747-0285.2009.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bordwell FG, Branca JC, Johnson CR, Vanier NR. J Org Chem. 1980;45:3884. [Google Scholar]
- 27.Matthews WS, Bares JE, Bartmess JE, Bordwell FG, Cornforth FJ, Drucker GE, Margolin Z, McCallum RJ, McCollum GJ, Vanier NR. J Am Chem Soc. 1975;97:7006. [Google Scholar]
- 28.Tao P, Fisher JF, Mobashery S, Schlegel HB. Org Lett. 2009;11:2559. doi: 10.1021/ol9008393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tao P, Fisher JF, Shi QC, Vreven T, Mobashery S, Schlegel HB. Biochemistry. 2009;48:9839. doi: 10.1021/bi901118r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Scalmani G, Mennucci B, Barone V, Petersson GA, Caricato M, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Li X, Hratchian HP, Peralta JE, Izmaylov AF, Kudin KN, Heyd JJ, Brothers E, Staroverov V, Zheng G, Kobayashi R, Normand J, Sonnenberg JL, Iyengar SS, Tomasi J, Cossi M, Rega N, Burant JC, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Chen W, Wong MW, Pople JA. Gaussian DV; Revision F.02. Gaussian, Inc.; Wallingford, CT: 2007. [Google Scholar]
- 31.Lee C, Yang W, Parr RG. Phys Rev B: Condens Matter. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 32.Becke AD. Phys Rev A: Gen Phys. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 33.Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]
- 34.Montgomery JJA, Frisch MJ, Ochterski JW, Petersson GA. J Chem Phys. 1999;110:2822. [Google Scholar]
- 35.Montgomery JJA, Frisch MJ, Ochterski JW, Petersson GA. J Chem Phys. 2000;112:6532. [Google Scholar]
- 36.Tomasi J, Mennucci B, Cances E. THEOCHEM. 1999;464:211. [Google Scholar]
- 37.Maseras F, Morokuma K. J Comput Chem. 1995;16:1170. [Google Scholar]
- 38.Svensson M, Humbel S, Froese RDJ, Matsubara T, Sieber S, Morokuma K. J Phys Chem. 1996;100:19357. [Google Scholar]
- 39.Humbel S, Sieber S, Morokuma K. J Chem Phys. 1996;105:1959. [Google Scholar]
- 40.Dapprich S, Komaromi I, Byun KS, Morokuma K, Frisch MJ. THEOCHEM. 1999;461-462:1. [Google Scholar]
- 41.Vreven T, Morokuma K. J Comput Chem. 2000;21:1419. [Google Scholar]
- 42.Vreven T, Morokuma K, Farkas O, Schlegel HB, Frisch MJ. J Comput Chem. 2003;24:760. doi: 10.1002/jcc.10156. [DOI] [PubMed] [Google Scholar]
- 43.Vreven T, Frisch MJ, Kudin KN, Schlegel HB, Morokuma K. Mol Phys. 2006;104:701. [Google Scholar]
- 44.Vreven T, Byun KS, Komaromi I, Dapprich S, Montgomery JA, Jr, Morokuma K, Frisch MJ. J Chem Theory Comput. 2006;2:815. doi: 10.1021/ct050289g. [DOI] [PubMed] [Google Scholar]
- 45.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. J Am Chem Soc. 1995;117:5179. [Google Scholar]
- 46.Bayly CI, Cieplak P, Cornell W, Kollman PA. J Phys Chem. 1993;97:10269. [Google Scholar]
- 47.Cornell WD, Cieplak P, Bayly CI, Kollmann PA. J Am Chem Soc. 1993;115:9620. [Google Scholar]
- 48.Wolfe S, Rauk A, Csizmadia IG. J Am Chem Soc. 1967;89:5710. [Google Scholar]
- 49.Wolfe S, Stolow A, Lajohn LA. Tetrahedron Lett. 1983;24:4071. [Google Scholar]
- 50.Dhanaraj V, Williams MG, Ye QZ, Molina F, Johnson LL, Ortwine DF, Pavlovsky A, Rubin JR, Skeean RW, White AD, Humblet C, Hupe DJ, Blundell TL. Croat Chem Acta. 1999;72:575. [Google Scholar]
- 51.Rosenblum G, Meroueh SO, Kleifeld O, Brown S, Singson SP, Fridman R, Mobashery S, Sagi I. J Biol Chem. 2003;278:27009. doi: 10.1074/jbc.M301139200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.