

Abstract
We tested the hypotheses that (1) nitric oxide (NO) contributes to augmented skeletal muscle vasodilatation during hypoxic exercise and (2) the combined inhibition of NO production and adenosine receptor activation would attenuate the augmented vasodilatation during hypoxic exercise more than NO inhibition alone. In separate protocols subjects performed forearm exercise (10% and 20% of maximum) during normoxia and normocapnic hypoxia (80% arterial O2 saturation). In protocol 1 (n= 12), subjects received intra-arterial administration of saline (control) and the NO synthase inhibitor NG-monomethyl-l-arginine (l-NMMA). In protocol 2 (n= 10), subjects received intra-arterial saline (control) and combined l-NMMA–aminophylline (adenosine receptor antagonist) administration. Forearm vascular conductance (FVC; ml min−1 (100 mmHg)−1) was calculated from forearm blood flow (ml min−1) and blood pressure (mmHg). In protocol 1, the change in FVC (Δ from normoxic baseline) due to hypoxia under resting conditions and during hypoxic exercise was substantially lower with l-NMMA administration compared to saline (control; P < 0.01). In protocol 2, administration of combined l-NMMA–aminophylline reduced the ΔFVC due to hypoxic exercise compared to saline (control; P < 0.01). However, the relative reduction in ΔFVC compared to the respective control (saline) conditions was similar between l-NMMA only (protocol 1) and combined l-NMMA–aminophylline (protocol 2) at 10% (−17.5 ± 3.7 vs.−21.4 ± 5.2%; P= 0.28) and 20% (−13.4 ± 3.5 vs.−18.8 ± 4.5%; P= 0.18) hypoxic exercise. These findings suggest that NO contributes to the augmented vasodilatation observed during hypoxic exercise independent of adenosine.
Introduction
The combination of submaximal exercise and hypoxia produces a ‘compensatory’ vasodilatation relative to the same level of exercise under normoxic conditions (Rowell et al. 1986; Roach et al. 1999; Calbet et al. 2003; Wilkins et al. 2006, 2008; Casey et al. 2009). This augmented vasodilatation exceeds that predicted by a simple sum of the individual dilator responses to hypoxia alone and normoxic exercise. The compensatory vasodilation during hypoxic exercise is essential to ensure the maintenance of oxygen delivery to the active muscles. However, the mechanisms responsible for the compensatory vasodilatation during hypoxic exercise, as well as their interactions, remain unclear. We reported that, in the absence of overlying sympathetic vasoconstriction, β-adrenergic receptor activation contributes to the augmented hypoxic vasodilatation during mild forearm exercise. However, the β-adrenergic component decreases with increased exercise intensity (Wilkins et al. 2008). In addition, our recent report demonstrated that adenosine is not obligatory for the augmented hyperaemia during hypoxic exercise in humans (Casey et al. 2009).
Taken together, these findings suggest that additional vasoactive substances are playing a role in the compensatory vasodilatation during hypoxic exercise, especially as exercise intensity increases. In this context, NO release from an endothelial source and/or desaturation of haemoglobin in erythrocytes (Pohl & Busse, 1989; Stamler et al. 1997; Singel & Stamler, 2005; Leuenberger et al. 2008) has been proposed during hypoxia. Additionally, both β-adrenergic receptor activation (Weisbrod et al. 2001) and adenosine receptor stimulation (Bryan & Marshall, 1999b) have been implicated as potential sources for the endothelial release of NO during hypoxia at rest. Finally, muscle contraction per se may also stimulate endothelial NO release via flow-mediated mechanisms or mechanical vessel distortion (Tschakovsky & Joyner, 2008). Thus, the aim of the current study was to investigate if the compensatory vasodilatation due to combined hypoxia and exercise includes an NO-dependent component. Since the potential exists for a strong NO-dependent component in adenosine mediated vasodilatation, a secondary aim of the study was to examine the potential for an adenosine–NO interaction during hypoxic exercise (Smits et al. 1995; Martin et al. 2006a; Mortensen et al. 2009b). Although our previous results suggest that adenosine is not obligatory for the compensatory hypoxic vasodilatation (Casey et al. 2009), ∼40–60% of exogenous adenosine mediated vasodilatation is NO independent. Thus, we wanted to examine the potential role of adenosine in hypoxic exercise hyperaemia in the absence of NO.
With this information as a background, we tested the hypothesis that NO contributes to the compensatory vasodilatation observed during hypoxic forearm exercise. Additionally, we tested the secondary hypothesis that adenosine receptor mediated vasodilatation independent of NO contributes to the compensatory vasodilatation seen during hypoxic exercise.
Methods
Subjects
A total of 22 young healthy subjects (12 female and 10 male) volunteered to participate in two separate protocols (12 subjects in protocol 1; 10 subjects in protocol 2). Subjects completed written informed consent and underwent a standard screening, and were healthy, non-obese, non-smokers, and were not taking any medications (except for oral contraceptives in some women). Studies were performed after an overnight fast and refraining from exercise and caffeine for at least 24 h. Female subjects were studied during the early follicular phase of the menstrual cycle or the placebo phase of oral contraceptives. All study protocols were approved by the Institutional Review Board and were performed according to the Declaration of Helsinki.
Forearm exercise
Subjects performed rhythmic forearm exercise with a handgrip device by the non-dominant arm at 10% and 20% of each subject's maximal voluntary contraction (MVC, mean 37 ± 3 kg, range 19–56 kg), determined at the beginning of each experiment. The weight was lifted 4–5 cm over a pulley at a duty cycle of 1 s contraction and 2 s relaxation (20 contractions per minute) using a metronome to ensure correct timing. The average weight used for forearm exercise in protocol 1 was 3.8 ± 0.3 and 7.6 ± 0.7 kg for 10 and 20% MVC, respectively. The average weight used for forearm exercise in protocol 2 was 3.5 ± 0.3 and 7.1 ± 0.6 kg for 10 and 20% MVC, respectively.
Arterial and venous catheterization
A 20 gauge, 5 cm (Model RA-04020, Arrow International, Reading, PA, USA) catheter was placed in the brachial artery of the exercising arm under aseptic conditions after local anaesthesia (2% lidocaine) for administration of study drugs and to obtain arterial blood samples. The catheter was connected to a three-port connector in series, as previously described in detail (Dietz et al. 1994). One port was linked to a pressure transducer positioned at heart level (Model PX600F, Edwards Lifescience, Irvine, CA, USA) to allow measurement of arterial pressure and was continuously flushed (3 ml h−1) with heparinized saline with a stop-cock system to enable arterial blood sampling. The remaining two ports allowed arterial drug administration. Deep venous blood was sampled via an 18 gauge, 3 cm catheter inserted retrograde in an antecubital vein (Joyner et al. 1992).
Forearm blood flow
Brachial artery mean blood velocity and brachial artery diameter were determined with a 12 MHz linear-array Doppler probe (Model M12L, Vivid 7, General Electric, Milwaukee, WI, USA). Brachial artery blood velocity was measured throughout each condition with a probe insonation angle previously calibrated to 60 deg. Brachial artery diameter measurements were obtained at end diastole between contractions during steady-state conditions. Forearm blood flow (FBF) was calculated as the product of mean blood velocity (cm s−1) and brachial artery cross-sectional area (cm2) and expressed as millilitres per minute (ml min−1).
Systemic hypoxia
The hypoxic conditions involved a self-regulating partial rebreathe system that effectively clamps end-tidal CO2 at baseline levels despite large changes in minute ventilation during hypoxia (Banzett et al. 2000; Weisbrod et al. 2001; Wilkins et al. 2006, 2008). The level of inspired O2 was titrated to achieve an arterial O2 saturation (assessed via pulse oximetry) of ∼80%. Carbon dioxide concentrations were monitored (Cardiocap/5, Datex-Ohmeda, Louisville, CO, USA) and ventilation was assessed via a turbine (Model VMM-2a, Interface Associates, Laguna Nigel, CA, USA).
Pharmacological infusions
NG-Monomethyl-l-arginine (l-NMMA; NOS inhibitor; Bachem, Switzerland) was infused at a loading dose of 5 mg min−1 for 5 min and then at a maintenance dose of 1 mg min−1 for the remainder of the study. The efficacy of the NOS inhibition with l-NMMA was confirmed via intra-arterial acetylcholine (ACh; a non-specific muscarinic agonist) infusion at 2.0, 4.0 and 8.0 μg (dl forearm volume)−1 min−1 for 2 min each before and after l-NMMA administration (protocol 1 only). Aminophylline (adenosine receptor antagonist) was administered to the forearm via the brachial artery catheter at a dose of 200 μg (dl forearm volume)−1 min−1. This dose of aminophylline has been shown to effectively attenuate the forearm vasodilator response to exogenous adenosine administration (Martin et al. 2006b; Casey et al. 2009).
Blood gas and catecholamine analysis
Brachial artery and deep venous blood samples were analysed with a clinical blood gas analyser (Bayer 855 Automatic Blood Gas System, Boston, MA, USA) for partial pressures of O2 and CO2 (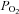 and
and 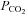 ), pH, and O2 saturation (
), pH, and O2 saturation (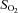 ). Arterial and venous O2 content was calculated using the measured
). Arterial and venous O2 content was calculated using the measured 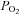 and
and 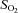 values. Arterial and venous plasma catecholamine (adrenaline and noradrenaline) levels were determined by HPLC with electrochemical detection.
values. Arterial and venous plasma catecholamine (adrenaline and noradrenaline) levels were determined by HPLC with electrochemical detection.
Experimental protocol
A schematic diagram of the general experimental design is illustrated in Fig. 1. Each subject completed a resting baseline condition followed by rhythmic forearm exercise at 10% MVC, which was immediately increased to 20% MVC during normoxia and normocapnic hypoxia. Exposure to normoxia or hypoxia was alternated and randomized. Protocol 1: resting baseline and forearm exercise (normoxia and hypoxia) were performed during a control (saline) infusion, followed by l-NMMA infusion. Due to the long half-life of l-NMMA, study drugs were always administered in the same order. A rest period of at least 20 min was allowed between conditions under each drug infusion. Protocol 2: resting baseline and forearm exercise (normoxia and hypoxia) were performed during a control (saline) infusion, followed by infusion of combined l-NMMA and aminophylline. As in protocol 1, a rest period of at least 20 min was allowed between conditions under each drug infusion. During each infusion (saline or l-NMMA in protocol 1 and saline or l-NMMA–aminophylline in protocol 2) and each condition (normoxia and hypoxia), arterial and venous blood was sampled at rest and at steady-state exercise for blood gas analysis and plasma catecholamine determination.
Figure 1. Schematic diagram of experimental protocol.

Measurements were obtained at baseline and incremental exercise (10% and 20% of maximum) under normoxic and hypoxic conditions. Protocol 1 was performed during control (saline) and l-NMMA infusions. Protocol 2 was performed during control (saline) and combined l-NMMA–aminophylline (Aph). Ach, acetylcholine.
Data analysis and statistics
Data were collected at 200 Hz, stored on a computer and analysed off-line with signal processing software (WinDaq, DATAQ Instruments, Akron, OH, USA). Mean arterial pressure (MAP) was determined from the brachial artery pressure waveform and HR was determined from the electrocardiogram. Values for minute ventilation, end-tidal CO2, and O2 saturation (pulse oximetry) were determined by averaging minutes 4 and 5 at rest and at each exercise intensity. Forearm blood flow and arterial pressure were determined by averaging values from the 4th minute at rest and each exercise bout. Forearm vascular conductance (FVC) was calculated as (FBF/arterial pressure) × 100 and expressed as ml min−1 (100 mmHg)−1. The change (Δ) in FBF and FVC due to hypoxia at rest and due to hypoxic exercise (10% and 20%) was calculated by subtracting resting FBF and FVC during normoxia at each drug infusion (saline or l-NMMA in protocol 1 and saline or combined l-NMMA–aminophylline in protocol 2) from FBF and FVC values obtained during hypoxia (at rest and during exercise) within each drug infusion. Blood gas and catecholamine values were determined from blood samples obtained during normoxia and hypoxia with each drug infusion. Arteriovenous oxygen difference during forearm exercise was calculated by the difference between arterial and venous O2 content. Venous–arterial (v-a) noradrenaline difference was calculated as the difference between venous and arterial noradrenaline concentrations.
All values are expressed as means ±s.e.m. To determine the effect of hypoxia with each pharmacological treatment, differences in absolute FBF and FVC at rest (normoxia and hypoxia) and differences in ΔFBF and ΔFVC at rest and during each exercise intensity (normoxia and hypoxia) were determined via repeated measures analysis of variance (ANOVA). Haemodynamic, respiratory blood gases and catecholamine variables were compared via repeated measures ANOVA to detect differences between responses during hypoxia at rest and during exercise across pharmacological infusions. Appropriate post hoc analysis determined where statistical differences occurred. Statistical difference was set a priori at P < 0.05. To compare the effects of single blockade (l-NMMA only in protocol 1) to double blockade (l-NMMA–aminophylline in protocol 2) on the vasodilator response to hypoxic exercise, differences in ΔFBF and ΔFVC (percentage reduction compared to respective control (saline) conditions) during hypoxia at each exercise intensity were determined via independent t tests.
Results
Twelve (6 male; 6 female) subjects completed protocol 1. The subjects were 26 ± 2 years of age, 172 ± 3 cm in height, and weighed 69 ± 4 kg (BMI: 23 ± 1 kg m−2). Ten subjects (4 male, 6 female) completed protocol 2. The subjects were 26 ± 2 years of age, 173 ± 2 cm in height, and weighed 74 ± 4 kg (BMI: 24 ± 1 kg m−2).
Systemic haemodynamic and respiratory responses (Table 1)
Table 1.
Systemic haemodynamic and respiratory responses at rest and with incremental exercise during normoxia and hypoxia under each drug infusion
| Normoxia |
Hypoxia |
|||||
|---|---|---|---|---|---|---|
| Rest | 10% | 20% | Rest | 10% | 20% | |
| Protocol 1 (n= 12) | ||||||
| Control (saline) | ||||||
| Mean arterial pressure (mmHg) | 89 ± 2 | 90 ± 2 | 93 ± 2 | 88 ± 2 | 90 ± 2 | 91 ± 3 |
| Heart rate (beats min−1)* | 62 ± 3 | 66 ± 3† | 68 ± 3 | 73 ± 4 | 77 ± 5† | 80 ± 5 |
| Minute ventilation (l min−1; BTPS)* | 6.3 ± 0.7 | 8.7 ± 0.7 | 9.1 ± 0.8 | 10.1 ± 1.7 | 13.7 ± 2.7† | 14.7 ± 2.1 |
| End-tidal CO2 (mmHg) | 37 ± 1 | 36 ± 1 | 37 ± 1 | 36 ± 1 | 36 ± 1 | 37 ± 1 |
| l-NMMA | ||||||
| Mean arterial pressure (mmHg) | 91 ± 2 | 92 ± 2 | 95 ± 2 | 92 ± 2 | 92 ± 2 | 93 ± 2 |
| Heart rate (beats min−1)* | 62 ± 3 | 65 ± 3 | 68 ± 3† | 76 ± 5 | 81 ± 5† | 82 ± 5 |
| Minute ventilation (l min−1; BTPS)* | 8.3 ± 1.0 | 10.2 ± 1.3 | 11.9 ± 1.5† | 14.4 ± 3.1‡ | 16.5 ± 2.9 | 19.0 ± 3.0†‡ |
| End-tidal CO2 (mmHg) | 37 ± 1 | 37 ± 1 | 37 ± 1 | 36 ± 1 | 37 ± 1 | 37 ± 1 |
| Protocol 2 (n= 10) | ||||||
| Control (saline) | ||||||
| Mean arterial pressure (mmHg) | 88 ± 2 | 90 ± 3 | 92 ± 3 | 91 ± 3 | 90 ± 3 | 91 ± 3 |
| Heart rate (beats min−1)* | 60 ± 3 | 65 ± 3† | 68 ± 4 | 75 ± 3 | 79 ± 3† | 81 ± 3 |
| Minute ventilation (l min−1; BTPS)* | 7.2 ± 0.8 | 8.7 ± 1.0 | 10.8 ± 1.4 | 14.9 ± 3.1 | 17.0 ± 2.3 | 20.0 ± 3.1 |
| End-tidal CO2 (mmHg) | 35 ± 1 | 35 ± 1 | 36 ± 1 | 35 ± 1 | 35 ± 1 | 36 ± 1 |
| l-NMMA–aminophylline | ||||||
| Mean arterial pressure (mmHg) | 91 ± 3 | 92 ± 3 | 93 ± 3 | 91 ± 2 | 90 ± 3 | 90 ± 2 |
| Heart rate (beats min−1)* | 60 ± 3 | 66 ± 3† | 70 ± 4 | 77 ± 2 | 80 ± 3† | 84 ± 4† |
| Minute ventilation (l min−1; BTPS)* | 10.7 ± 1.4‡ | 15.2 ± 2.0†‡ | 17.4 ± 2.2‡ | 18.8 ± 3.3 | 22.5 ± 4.4‡ | 27.3 ± 4.8†‡ |
| End-tidal CO2 (mmHg) | 35 ± 1 | 35 ± 1 | 36 ± 1 | 35 ± 1 | 36 ± 1 | 36 ± 1 |
Values are means ±s.e.m.
Main effect of hypoxia, P < 0.05 vs. normoxia;
P < 0.05 vs. previous intensity;
P < 0.05 vs. Control.
The group data (means ±s.e.m.) for haemodynamic and respiratory responses due to combined forearm exercise and hypoxia with each drug infusion (protocol 1 and 2) are presented in Table 1. As expected, during both protocols heart rate and minute ventilation increased as a consequence of systemic hypoxia and incremental forearm exercise. Despite the elevated heart rate, mean arterial pressure was similar during normoxia and hypoxia independent of drug infusion at rest and incremental exercise. By design, end-tidal CO2 was maintained throughout rest and incremental exercise under both normoxic and hypoxic conditions for both protocols.
Forearm exercise (Table 2)
Table 2.
Forearm haemodynamics at rest and with incremental exercise during normoxia and hypoxia with each drug infusion
| Δ from normoxia rest |
|||||
|---|---|---|---|---|---|
| Rest | 10% | 20% | 10% | 20% | |
| Protocol 1 (n= 12) | |||||
| Forearm blood flow (ml min−1) | |||||
| Control (saline) | |||||
| Normoxia | 55 ± 5 | 205 ± 22† | 329 ± 35† | 150 ± 17 | 274 ±29† |
| Hypoxia | 68 ± 8* | 245 ± 27*† | 388 ± 42*† | 190 ± 22* | 333 ± 37*† |
| l-NMMA | |||||
| Normoxia | 41 ± 4‡ | 174 ± 23† | 334 ± 44† | 131 ± 18 | 291 ± 38† |
| Hypoxia | 44 ± 5‡ | 202 ± 24†‡ | 341 ± 38†‡ | 161 ± 21*‡ | 300 ± 35†‡ |
| Forearm vascular conductance (ml min−1 100 mmHg−1) | |||||
| Control (saline) | |||||
| Normoxia | 62 ± 6 | 225 ± 22† | 354 ± 34† | 163 ± 17 | 292 ± 28† |
| Hypoxia | 76 ± 9* | 273 ± 28*† | 429 ± 39*† | 211 ± 23* | 367 ± 34*† |
| l-NMMA | |||||
| Normoxia | 44 ± 5‡ | 187 ± 23† | 349 ± 41† | 140 ± 17 | 302 ± 35† |
| Hypoxia | 49 ± 5‡ | 219 ± 24†‡ | 362 ± 37†‡ | 175 ± 20*‡ | 318 ± 32†‡ |
| Protocol 2 (n= 10) | |||||
| Forearm blood flow (ml min−1) | |||||
| Control (saline) | |||||
| Normoxia | 43 ± 7 | 164 ± 23† | 304 ± 56† | 121 ± 16 | 261 ± 46† |
| Hypoxia | 63 ± 11* | 211 ± 28*† | 362 ± 58*† | 168 ± 21* | 320 ± 49*† |
| l-NMMA–aminophylline | |||||
| Normoxia | 61 ± 9‡ | 159 ± 23† | 288 ± 55† | 98 ± 15# | 227 ± 45† |
| Hypoxia | 69 ± 11* | 194 ± 26*† | 314 ± 50*†‡ | 133 ± 17*‡ | 253 ± 39*†‡ |
| Forearm vascular conductance (ml min−1 100 mmHg−1) | |||||
| Control (saline) | |||||
| Normoxia | 47 ± 7 | 180 ± 23† | 325 ± 54† | 133 ± 17 | 277 ± 45† |
| Hypoxia | 68 ± 11* | 231 ± 27*† | 393 ± 58*† | 183 ± 20* | 346 ± 48*† |
| l-NMMA–aminophylline | |||||
| Normoxia | 67 ± 8‡ | 171 ± 22† | 306 ± 52† | 104 ± 14# | 239 ± 42† |
| Hypoxia | 76 ± 12* | 213 ± 26*† | 344 ± 51*†‡ | 145 ± 17*‡ | 278 ± 39*†‡ |
Values are means ±s.e.m.
Main effect of hypoxia, P < 0.05;
P < 0.05 (vs. previous exercise intensity);
P < 0.01 vs. Control. #P < 0.05 vs. Control.
Presented in Table 2 are group data (means ±s.e.m.) forearm haemodynamics at rest and with increasing exercise intensity during control saline and l-NMMA infusion (protocol 1) and saline and combined l-NMMA–aminophylline (protocol 2).
Protocol 1
Systemic hypoxia increased resting FBF and FVC during the control saline infusion (P < 0.05). The absolute FBF and FVC as well as the change (Δ) in FBF and FVC (relative to normoxic baseline values) to incremental hypoxic exercise were higher compared to normoxic exercise of the same intensity during the control saline infusion (main effect of hypoxia; P < 0.05). Intra-arterial administration of l-NMMA decreased normoxic baseline (resting) blood flow below values observed during control saline infusion (P < 0.05). l-NMMA infusion did not reduce absolute FBF or FVC (Table 2) or the ΔFBF or ΔFVC during normoxic exercise at 10% and 20% MVC compared to control saline (Fig. 3A–D). l-NMMA infusion reduced absolute FBF and FVC (Table 2) and the change (Δ) in FBF and FVC due to acute systemic hypoxia at rest (Fig. 2A and B). Furthermore, infusion of l-NMMA decreased absolute FBF and FVC (Table 2) as well as the ΔFBF and ΔFVC with hypoxic exercise at 10% MVC (P < 0.05, Fig. 3A and B) and at 20% MVC (P < 0.05, Fig. 3C and D) compared to control saline infusions. In contrast to exercise at 10% MVC, the ΔFBF and ΔFVC during hypoxic exercise at 20% MVC was not statistically different from normoxic exercise observed during l-NMMA infusion (P= 0.54 for ΔFBF; P= 0.27 for ΔFVC; Fig. 3C and D). That is, the administration of l-NMMA seemed to abolish the elevated FBF and FVC response due to hypoxia during forearm exercise.
Figure 3. Change (Δ) in forearm blood flow (FBF) and forearm vascular conductance (FVC) due to hypoxic exercise during saline (control) and l-NMMA administration (n= 12).

At 10% or 20% forearm exercise, NO synthase inhibition (l-NMMA) reduced forearm blood flow (A and C) and vascular conductance (B and D) compared to control (saline) during hypoxic exercise. †Main effect of hypoxia, P < 0.01 vs. normoxia. *P < 0.05 vs. control (saline).
Figure 2. Change (Δ) in forearm blood flow (FBF) and forearm vascular conductance (FVC) due to hypoxia at rest during saline and l-NMMA administration (Protocol 1; n= 12) and during saline and combined l-NMMA–aminophylline administration (Protocol 2; n= 10).

NO synthase inhibition (l-NMMA) reduced hypoxic vasodilatation compared to control (saline) (Protocol 1). Combined NO synthase inhibition (l-NMMA) and adenosine receptor antagonist (aminophylline) reduced hypoxic vasodilatation compared to control (saline) (Protocol 2). *P < 0.05 vs. control (saline).
The vasodilator response (change in FVC from baseline) to ACh infusion was substantially lower at all three doses (2.0, 4.0 and 8.0 μg (dl forearm volume)−1 min−1) in the presence of l-NMMA (153 ± 33, 174 ± 41 and 219 ± 47 ml min−1 (100 mmHg)−1, respectively) compared to no drug (255 ± 50, 267 ± 58, and 364 ± 65 ml min−1 (100 mmHg)−1, respectively; P < 0.01), thus confirming effective NOS inhibition.
During control saline infusion, forearm oxygen consumption increased (P < 0.05) to 22 ± 3 ml min−1 and to 34 ± 4 ml min−1 with normoxic exercise at 10% and 20% MVC, respectively. This was similar to oxygen consumption values obtained during hypoxic exercise (22 ± 3 ml min−1 and 35 ± 4 ml min−1 for 10% and 20% respectively, P < 0.05 for exercise intensity). During l-NMMA administration, forearm oxygen consumption increased (P < 0.05) to 18 ± 2 ml min−1 and 32 ± 4 ml min−1 with normoxic exercise at 10% and 20% MVC, respectively. Values obtained during normoxic exercise with l-NMMA infusion were similar to those obtained during hypoxic exercise (18 ± 3 ml min−1 and 29 ± 4 ml min−1 for 10% and 20% respectively, P < 0.05 for exercise intensity). Forearm oxygen consumption during hypoxic exercise was greater during control saline infusion compared to l-NMMA infusion (P < 0.05).
Protocol 2
Systemic hypoxia increased resting FBF and FVC during control saline infusion (P < 0.05). The absolute FBF and FVC as well as the ΔFBF and ΔFVC during incremental hypoxic exercise were higher compared to normoxic exercise of the same intensity during the control saline infusion (main effect of hypoxia; P < 0.05; Table 2). Combined l-NMMA–aminophylline administration increased baseline resting blood flow compared to values observed during saline alone (P < 0.05; Table 2). However, ΔFBF and ΔFVC due to acute systemic hypoxia at rest were substantially lower during combined l-NMMA–aminophylline infusion (Fig. 2C and D).
Although the combined infusion of l-NMMA–aminophylline did not alter the absolute FBF and FVC, it decreased the ΔFBF and ΔFVC during normoxic exercise at 10% MVC (P < 0.05, Fig. 4A and B). However, the ΔFBF or ΔFVC during normoxic exercise at 20% MVC was not statistically different between control saline infusion and l-NMMA–aminophylline infusion (P= 0.13 for ΔFBF; P= 0.11 for ΔFVC; Fig. 4C and D). During hypoxic exercise, the absolute FBF and FVC as well as the ΔFBF (Fig. 4A and C) and ΔFVC (Fig. 4B and D) were greater at 10% and 20% MVC compared to normoxic exercise of the same intensity (main effect of hypoxia, P < 0.01). Of primary importance to this study, the combination of l-NMMA–aminophylline decreased ΔFBF (Fig. 4A and C) and ΔFVC (Fig. 4B and D) due to incremental hypoxic exercise at 10% and 20% MVC (P < 0.01). The relative (%) reduction in ΔFVC compared to the respective control (saline) conditions was statistically identical with combined l-NMMA–aminophylline (protocol 2) and l-NMMA only (protocol 1) at 10% (−21.4 ± 5.2 vs.−17.5 ± 3.7%; P= 0.28) and at 20% (−18.8 ± 4.5 vs.−13.4 ± 3.5%; P= 0.18) hypoxic forearm exercise (Fig. 5).
Figure 4. Change (Δ) in forearm blood flow (FBF) and forearm vascular conductance (FVC) due to hypoxic exercise during saline and combined l-NMMA–aminophylline administration (n= 10).

At 10% or 20% forearm exercise, combined NO synthase inhibition (l-NMMA) and adenosine receptor antagonist (aminophylline) reduced forearm blood flow (A and C) or vascular conductance (B and D) compared to control (saline) during hypoxic exercise. †Main effect of hypoxia, P < 0.01 vs. normoxia. *P < 0.05 vs. control (saline).
Figure 5. Percentage reduction (compared to respective control (saline) condition) in the change (Δ) in forearm vascular conductance (FVC) due to hypoxic exercise following l-NMMA administration (Protocol 1; n= 12) and l-NMMA plus aminophylline administration (Protocol 2; n= 10).

At 10% and 20% forearm exercise, both single blockade (NO synthase inhibition; l-NMMA) and double blockade (NO synthase inhibition and adenosine receptor antagonist; l-NMMA–aminophylline) revealed a substantial reduction in hypoxic exercise vasodilatation. However, there was no difference in the percentage reduction in ΔFVC between the two protocols.
Forearm oxygen consumption increased (P < 0.05) to 15 ± 2 ml min−1 and to 25 ± 5 ml min−1 with normoxic exercise at 10% and 20% MVC, respectively, during control saline. This was similar to oxygen consumption values obtained during hypoxic exercise (15 ± 3 ml min−1 and 28 ± 5 ml min−1 for 10% and 20%, respectively, P < 0.05 for exercise intensity). During combined l-NMMA–aminophylline administration, forearm oxygen consumption increased (P < 0.05) to 13 ± 2 ml min−1 and 24 ± 4 ml min−1 with normoxic exercise at 10% and 20% MVC respectively. Values obtained during normoxic exercise with combined l-NMMA–aminophylline infusion were similar to those obtained during hypoxic exercise (14 ± 2 ml min−1 and 24 ± 5 ml min−1 for 10% and 20%, respectively, P < 0.05 for exercise intensity). There was no statistical difference in oxygen consumption between saline and combined l-NMMA–aminophylline infusions.
Blood gases (Table 3)
Table 3.
Arterial and venous blood gas responses at rest and with incremental exercise during normoxia and hypoxia with each drug infusion
| Normoxia |
Hypoxia |
|||||
|---|---|---|---|---|---|---|
| Rest | 10% | 20% | Rest | 10% | 20% | |
| Protocol 1 (n= 12) | ||||||
| Control (saline) | ||||||
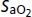 (%)* (%)*
|
97 ± 0 | 97 ± 0 | 97 ± 0 | 82 ± 0 | 82 ± 0 | 83 ± 0 |
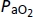 (Torr)* (Torr)*
|
107 ± 3 | 107 ± 4 | 109 ± 3 | 48 ± 1 | 48 ± 1 | 49 ± 1 |
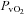 (Torr)* (Torr)*
|
38 ± 2 | 26 ± 1† | 27 ± 1 | 30 ± 1 | 22 ± 1† | 23 ± 1 |
| Arterial O2 content (ml l−1)* | 171 ± 5 | 173 ± 5 | 172 ± 5 | 144 ± 4 | 147 ± 4 | 149 ± 4 |
| Venous O2 content (ml l−1)* | 118 ± 8 | 68 ± 4† | 72 ± 4 | 95 ± 7 | 58 ± 4† | 59 ± 3 |
| a-v O2 (ml l−1)* | 53 ± 6 | 105 ± 4† | 100 ± 3 | 49 ± 6 | 88 ± 4† | 90 ± 3 |
| l-NMMA | ||||||
|
(%)*
|
97 ± 0 | 97 ± 0 | 97 ± 0 | 82 ± 0 | 81 ± 1 | 82 ± 1 |
|
(Torr)*
|
107 ± 3 | 106 ± 3 | 109 ± 3 | 47 ± 1 | 46 ± 2 | 49 ± 1 |
|
(Torr)*
|
34 ± 3 | 24 ± 1† | 28 ± 1† | 25 ± 1‡ | 20 ± 0†‡ | 23 ± 0† |
| Arterial O2 content (ml l−1)* | 173 ± 5 | 173 ± 5 | 176 ± 5 | 143 ± 4 | 141 ± 5 | 148 ± 4 |
| Venous O2 content (ml l−1)* | 105 ± 10 | 68 ± 7† | 80 ± 7† | 67 ± 5‡ | 51 ± 2† | 61 ± 3† |
| a-v O2 (ml l−1) | 68 ± 9 | 106 ± 7† | 95 ± 6 | 76 ± 4‡ | 91 ± 5*† | 87 ± 4 |
| Protocol 2 (n= 10) | ||||||
| Control (saline) | ||||||
|
(%)*
|
97 ± 0 | 97 ± 0 | 97 ± 0 | 82 ± 1 | 83 ± 1 | 82 ± 1 |
|
(Torr)*
|
106 ± 3 | 110 ± 2 | 110 ± 3 | 49 ± 1 | 50 ± 1 | 50 ± 1 |
|
(Torr) |
43 ± 3 | 28 ± 2† | 29 ± 2† | 30 ± 2 | 27 ± 2† | 26 ± 1 |
| Arterial O2 content (ml l−1)* | 161 ± 3 | 162 ± 3 | 163 ± 3 | 136 ± 3 | 140 ± 2 | 143 ± 2† |
| Venous O2 content (ml l−1)* | 119 ± 10 | 72 ± 10† | 78 ± 8† | 83 ± 8 | 70 ± 8† | 65 ± 7 |
| a-v O2 (ml l−1)* | 41 ± 8 | 90 ± 8† | 85 ± 7† | 54 ± 7 | 70 ± 9† | 77 ± 6† |
| l-NMMA–aminophylline | ||||||
|
(%)*
|
97 ± 0 | 97 ± 0 | 97 ± 0 | 82 ± 0 | 82 ± 1 | 82 ± 0 |
|
(Torr)*
|
108 ± 2 | 110 ± 2 | 111± 2 | 48 ± 1 | 48 ± 1 | 50 ± 1 |
|
(Torr) |
43 ± 2 | 28 ± 2† | 28 ± 2 | 35 ± 1‡ | 25 ± 1† | 25 ± 1 |
| Arterial O2 content (ml l−1)* | 157 ± 2 | 159 ± 2 | 159 ± 3 | 132 ± 2 | 134 ± 3 | 138 ± 3 |
| Venous O2 content (ml l−1) | 127 ± 5 | 74 ± 10† | 72 ± 9 | 107 ± 3 | 61 ± 6† | 63 ± 5 |
| a-v O2 (ml l−1) | 29 ± 4 | 85 ± 9† | 87 ± 7 | 26 ± 3‡ | 74 ± 4† | 75 ± 4 |
Values are means ±s.e.m.
Main effect of hypoxia, P < 0.05 vs. normoxia;
P < 0.05 vs. previous intensity;
P < 0.05 vs. control.
Protocol 1
Systemic hypoxia reduced arterial 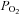 (P < 0.01) and arterial oxygen content (P < 0.01) at rest and with incremental forearm exercise during both drug trials (saline or l-NMMA). Acute hypoxia also decreased venous oxygen content during each drug infusion (P < 0.05). Under control (saline) conditions the lower arterial and venous oxygen content during hypoxic exercise led to a lower a-v oxygen difference compared to normoxic exercise (P < 0.05). However, during hypoxic exercise with l-NMMA the a-v oxygen difference was only lower at 10% MVC compared to normoxic exercise (P < 0.05).
(P < 0.01) and arterial oxygen content (P < 0.01) at rest and with incremental forearm exercise during both drug trials (saline or l-NMMA). Acute hypoxia also decreased venous oxygen content during each drug infusion (P < 0.05). Under control (saline) conditions the lower arterial and venous oxygen content during hypoxic exercise led to a lower a-v oxygen difference compared to normoxic exercise (P < 0.05). However, during hypoxic exercise with l-NMMA the a-v oxygen difference was only lower at 10% MVC compared to normoxic exercise (P < 0.05).
Protocol 2
Systemic hypoxia reduced arterial 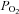 (P < 0.01) and arterial O2 content (P < 0.01) at rest and with increasing exercise intensity during both drug trials (saline or l-NMMA–aminophylline). Similar to protocol 1, acute hypoxia also decreased venous oxygen content during each control saline infusion (P < 0.05). Under control (saline) conditions the lower arterial and venous oxygen content during hypoxic exercise led to a lower a-v oxygen difference compared to normoxic exercise (P < 0.05). Contrary to control saline infusion, extraction of oxygen was similar during normoxic and hypoxic exercise during l-NMMA–aminophylline.
(P < 0.01) and arterial O2 content (P < 0.01) at rest and with increasing exercise intensity during both drug trials (saline or l-NMMA–aminophylline). Similar to protocol 1, acute hypoxia also decreased venous oxygen content during each control saline infusion (P < 0.05). Under control (saline) conditions the lower arterial and venous oxygen content during hypoxic exercise led to a lower a-v oxygen difference compared to normoxic exercise (P < 0.05). Contrary to control saline infusion, extraction of oxygen was similar during normoxic and hypoxic exercise during l-NMMA–aminophylline.
Catecholamines (Table 4)
Table 4.
Adrenaline and noradrenaline at rest and incremental exercise during normoxia and hypoxia with each drug infusion
| Normoxia |
Hypoxia |
|||||
|---|---|---|---|---|---|---|
| Rest | 10% | 20% | Rest | 10% | 20% | |
| Protocol 1 (n= 12) | ||||||
| Control (saline) | ||||||
| Venous noradrenaline (pg ml−1) | 161 ± 12 | 164 ± 15 | 182 ± 20 | 193 ± 9 | 176 ± 9 | 176 ± 11 |
| v-a noradrenaline difference (pg ml−1) | 45 ± 8 | 36 ± 10 | 36 ± 17 | 64 ± 10 | 40 ± 5 | 30 ± 6 |
| Arterial adrenaline (pg ml−1)* | 31 ± 4 | 35 ± 7 | 47 ± 7 | 50 ± 8 | 64 ± 8 | 79 ± 12 |
| l-NMMA | ||||||
| Venous noradrenaline (pg ml−1) | 174 ± 16 | 158 ± 17 | 178 ± 18 | 182 ± 11 | 177 ± 12 | 190 ± 15 |
| v-a noradrenaline difference (pg ml−1) | 43 ± 10 | 24 ± 5† | 25 ± 4 | 54 ± 11 | 31 ± 6† | 28 ± 5 |
| Arterial adrenaline (pg ml−1)* | 31 ± 4 | 40 ± 6 | 47 ± 7 | 51 ± 9 | 76 ± 13 | 86 ± 12 |
| Protocol 2 (n= 10) | ||||||
| Control (saline) | ||||||
| Venous noradrenaline (pg ml−1)* | 198 ± 37 | 177 ± 35† | 178 ± 36 | 246 ± 38 | 214 ± 34† | 211 ± 32 |
| v-a noradrenaline difference (pg ml−1) | 65 ± 10 | 36 ± 4† | 30 ± 5 | 93 ± 17 | 68 ± 8*† | 49 ± 10*† |
| Arterial adrenaline (pg ml−1)* | 34 ± 4 | 36 ± 7 | 44 ± 7 | 59 ± 7 | 69 ± 11 | 81 ± 13† |
| l-NMMA–aminophylline | ||||||
| Venous noradrenaline (pg ml−1) | 205 ± 29 | 186 ± 30 | 199 ± 33 | 212 ± 31‡ | 220 ± 36 | 228 ± 35 |
| v-a noradrenaline difference (pg ml−1) | 78 ± 13‡ | 61 ± 7‡ | 70 ± 10‡ | 79 ± 15 | 74 ± 15 | 68 ± 12 |
| Arterial adrenaline (pg ml−1)* | 49 ± 9‡ | 51 ± 9‡ | 60 ± 10‡ | 91 ± 19 | 131 ± 33†‡ | 143 ± 37‡ |
Values are means ±s.e.m.
Main effect of hypoxia, P < 0.05 vs. normoxia;
P < 0.05 vs. previous intensity;
main effect of drug, P < 0.05 vs. control.
Protocol 1
Systemic hypoxia increased arterial adrenaline during control saline and l-NMMA infusions (P < 0.05). Arterial adrenaline did not increase with incremental exercise under normoxic or hypoxic conditions for either drug condition. Additionally, systemic hypoxia did not increase v-a noradrenaline difference (P= 0.44).
Protocol 2
Systemic hypoxia increased arterial adrenaline during both saline and combined l-NMMA–aminophylline infusions (P < 0.05). There was a statistical main effect of drug infusion on arterial adrenaline concentration during normoxic and hypoxic exercise. That is, combined l-NMMA–aminophylline infusion resulted in higher levels of arterial adrenaline at each level of exercise during normoxia and hypoxia (P < 0.05). There was a main effect of drug on v-a noradrenaline difference during normoxia (P < 0.05). v-a noradrenaline difference was higher during incremental normoxic exercise with combined l-NMMA–aminophylline infusion (P < 0.05).
Discussion
The primary conclusions of the present study are (1) similar to resting conditions (Blitzer et al. 1996; Weisbrod et al. 2001) NO mediated mechanisms contribute to the compensatory vasodilatation during incremental hypoxic exercise and (2) there was no evidence for adenosine mediated vasodilatation after NOS inhibition with l-NMMA. These conclusions are supported by the significant blunting of the augmented vasodilatation with NOS inhibition during hypoxic exercise (Table 2 and Fig. 3A–D) and the lack of any further attenuation of the compensatory vasodilatation with combined NOS inhibition and adenosine receptor inhibition.
To our knowledge this study is the first report of a substantial NO mediated contribution to the augmented vasodilatation associated with hypoxic exercise. Nitric oxide is a potent vasodilator that contributes to the overall tone of the vasculature at rest (Dietz et al. 1994; Engelke et al. 1996), contributes to the regulation of blood pressure (Halliwill et al. 2000; Charkoudian et al. 2006; Gamboa et al. 2007), and has been shown to make a modest contribution to the rise in muscle blood flow during exercise (Radegran, 1999; Schrage et al. 2004). Data for both animals (Skinner & Marshall, 1996; Bryan & Marshall, 1999b; Edmunds & Marshall, 2001; Ray & Marshall, 2005) and humans (Blitzer et al. 1996; Weisbrod et al. 2001) demonstrate the important contribution of NO hypoxia induced vasodilatation at rest. Our findings confirm this previous work, and further describe the contribution of NO during hypoxic exercise. The conclusion that NO contributes to the compensatory vasodilatation during hypoxic exercise is evidenced by a reduction in absolute FBF and FVC (Table 2) as well as an attenuated Δ FBF and FVC (relative to normoxic baseline values; Fig. 3A–D). Furthermore, data from this study are consistent with our recent report (Casey et al. 2009) showing that adenosine mediated vasodilatation is not obligatory for compensatory vasodilatation during hypoxic exercise and demonstrates that adenosine does not act through a NO-independent pathway.
Stimulus for NO release during hypoxic exercise
Previous studies have demonstrated that β-adrenergic receptor activation is responsible for a substantial portion of the hypoxic vasodilatation observed at rest (Blauw et al. 1995; Weisbrod et al. 2001; Wilkins et al. 2008) and during mild forearm exercise (Wilkins et al. 2008). It appears, that at least a portion of the β-adrenergic receptor activation at rest is mediated through a NO pathway (Weisbrod et al. 2001). Combined with the findings from the current study, this suggests at least some contribution of β-adrenergic receptor mediated NO release contributing to the compensatory vasodilatation with hypoxic exercise. This β-adrenergic receptor stimulated NO component is yet to be examined directly during hypoxic exercise. At higher intensities of forearm exercise the contribution of β-adrenergic mechanisms to the augmented hypoxic vasodilatation decreases (Wilkins et al. 2008). In the present study there was no corresponding decrease in the NO mediated component of the augmented vasodilatation response. Therefore, the source of the NO may be less dependent on β-adrenergic mechanisms as exercise intensity increases.
In humans ∼40–60% of the vasodilator response to exogenous adenosine is mediated by NO (Smits et al. 1995; Martin et al. 2006a; Mortensen et al. 2009b), which suggests that the NO mediated vasodilatation during hypoxic exercise may be a result of stimulation of adenosine receptors. However, results from our previous study (Casey et al. 2009) and the current study suggest adenosine receptor activation is not a major source of NO production during hypoxic exercise (Fig. 5). Additionally, there is strong evidence that suggests an interaction between prostaglandins and NO in the regulation of skeletal muscle blood flow at rest and during exercise (Schrage et al. 2004; Saunders et al. 2005; Mortensen et al. 2007; Nicholson et al. 2009). However, it is currently unknown whether these interactions are operative under hypoxic conditions.
ATP release via oxygen sensitive mechanisms in erythrocytes or endothelial cells in response to hypoxia can elicit vasodilatation through binding to purinergic P2y receptors located on the vascular endothelial cells. In vitro studies suggest that ATP induced vasodilatation is mediated through an NO pathway (McCullough et al. 1997; Collins et al. 1998). A similar interaction between ATP and NO has been reported to exist in the human leg (Mortensen et al. 2009a) but not the forearm (Rongen et al. 1994). Of particular interest to this study, venous plasma levels of ATP tend to be elevated with hypoxic exercise compared to normoxic exercise (Gonzalez-Alonso et al. 2002). The findings highlighted above suggest that circulating ATP may have contributed to the NO-mediated hypoxic vasodilatation during exercise observed in the present study. However, the apparent differences in the contribution of NO to forearm vs. leg dilator responses to ATP indicate the need for caution in this interpretation.
Lastly, increases in plasma but not skeletal muscle interstitial NO during hypoxia in humans suggests an endovascular or endothelial NO source (Leuenberger et al. 2008). Therefore, it is possible that the augmented vasodilatation during hypoxic exercise is a result of the direct release of NO from endothelial cells and/or erythrocytes (independent of the aforementioned stimuli). In this context, luminal hypoxia can elicit the direct release of NO from the endothelium (Pohl & Busse, 1989) and from erythrocytes in the form S-nitrosohaemoglobin (Stamler et al. 1997). However, based on the location for intraluminal l-NMMA administration and that fact that NOS inhibition was effective in reducing forearm blood flow during hypoxic exercise, we would suspect that the NO responsible for the compensatory vasodilatation is from endothelial sources versus an erythrocyte source. Furthermore, the effectiveness of NOS inhibition in reducing the compensatory vasodilatation during hypoxic exercise also argues against the idea that nitrite is a key vasodilator in this response (Gladwin, 2008). In this context, if nitrite was responsible, either directly or indirectly (via reduction to NO), the augmented hyperaemia would have been maintained despite NOS inhibition.
Experimental considerations
Reductions in resting forearm blood flow and conductance due to l-NMMA may have overestimated the contribution of NO in the present study. However, the hypoxic vasodilatation was reduced to the same extent during combined infusion of l-NMMA and aminophylline (protocol 2) in which resting forearm blood flow was slightly elevated compared to control (saline) conditions. Therefore, we feel the attenuated vasodilator response to hypoxic exercise following l-NMMA administration was not an artifactual result of lower resting values.
The present study identified the contribution of NO to the vasodilator response without α-adrenergic blockade. Therefore, it can be argued that the contribution of NO to the augmented vasodilatation was underestimated in the present study due to the effects of sympathetic vasoconstrictor activity. Although this may be the case under resting conditions where the effect of sympathetic vasoconstrictor would be the greatest, it is likely that α-adrenergic influence would be minimized during exercise (via functional sympatholysis), especially with increasing exercise intensity (Tschakovsky et al. 2002). Along these lines, we have previously demonstrated that the difference (absolute and relative) between the hypoxic vasodilator response under control (saline) and α-adrenergic blockade (phentolamine) is greatest at rest and decreases with increasing exercise intensity (Wilkins et al. 2008).
In the present study the combined administration of l-NMMA and aminophylline had a minimal effect on absolute blood flow during normoxic exercise. These findings are in contrast to previous studies that have reported a ∼15–20% reduction in the hyperaemic response to leg exercise following adenosine receptor antagonism with theophylline (Radegran & Calbet, 2001; Martin et al. 2006b; Mortensen et al. 2009b). However, taking into account the change in baseline flow following administration of aminophylline, there was a significant reduction in the ΔFBF (−19%) at lower intensity exercise and a 13% reduction (P= 0.09) in ΔFBF at higher exercise workloads. In animal models, extravascular release of adenosine from active muscle is thought to be a signal for exercise hyperaemia per se (Ray & Marshall, 2009). Along these lines, the modest effect of intraarterial adenosine receptor blockade on exercise hyperaemia during normoxia (Radegran & Calbet, 2001; Martin et al. 2006b; Mortensen et al. 2009b) might reflect an inability to block extravascular adenosine receptors using intraarterial administration of aminophylline. However, intraluminal adenosine release from the endothelium appears to be responsible for the hypoxia-induced vasodilatation in rats (Bryan & Marshall, 1999a). Therefore, if the hyperaemia from the combination of hypoxia and exercise in the present study had an NO-independent adenosine component it should have been blunted to some degree with intravascular adenosine receptor blockade (Casey et al. 2009).
Lastly, the lack of a sequential blockade study design (l-NMMA followed by aminophylline or vice versa) may limit our interpretation of the interactions between NO and adenosine in the regulation of vascular tone during hypoxic exercise. However, considering the results of the present study (protocols 1 and 2) and those of our previous study (Casey et al. 2009) together suggest that the contribution of NO to the augmented hyperaemia during hypoxic exercise in humans appears to be independent of adenosine.
Perspectives
We have previously demonstrated that the augmented vasodilatation during hypoxic exercise is not explained by an enhanced sympatholysis (Wilkins et al. 2006). Additionally, systemic adrenaline release, acting via β-adrenergic receptors, contributes to the augmented vasodilatation during hypoxic exercise at lower exercise intensities, but this β-adrenergic contribution decreases with increasing exercise intensity (Wilkins et al. 2008). Thus, an additional source of NO is likely to be engaged as exercise intensity increases during hypoxia. Based on the findings from our previous study (Casey et al. 2009) and the present study (protocol 2), this secondary source for NO is not adenosine. Additionally, our evidence suggests the major source of NO was endothelial vs. NO release from erythrocytes.
Conclusions
This study demonstrates that NO mediated vasodilatation plays an obligatory role in the compensatory vasodilatation at rest and during incremental hypoxic exercise (protocol 1). The present study also demonstrates that adenosine does not have an independent role in compensatory vasodilatation (protocol 2) during hypoxic exercise. The primary source of NO and its interaction with other potential vasodilator substances during hypoxic exercise remains an open question, but our data favour an endothelial source.
Acknowledgments
The authors are grateful to the study volunteers for their participation. We also thank Branton Walker, Rachel Elvebak, Christopher Johnson, Lakshmi (Madhuri) Somaraju, Pam Engrav, Karen Krucker, Jean Knutson and Shelly Roberts for their technical assistance. This research was supported by the National Institutes of Health research grants AR-55819 (to D.P.C.) and HL-46493 (to M.J.J.) and by CTSA RR-024150. The Caywood Professorship via the Mayo Foundation also supported this research.
Glossary
Abbreviations
- FBF
forearm blood flow
- FVC
forearm vascular conductance
- l-NMMA
NG-monomethyl-l-arginine
- MAP
mean arterial pressure
- MVC
maximal voluntary contraction
- NO
nitric oxide
- NOS
nitric oxide synthase
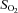
oxygen saturation
Author contributions
D.P.C.: conception and design of protocol, analysis and interpretation of data, drafting the article and revising it critically for important intellectual content. B.D.M.: analysis and interpretation of data, drafting the article. T.B.C.: analysis and interpretation of data, drafting the article. J.H.E.: analysis and interpretation of data, drafting the article. B.W.W.: conception and design of protocol, analysis and interpretation of data, drafting the article and revising it critically for important intellectual content. M.J.J.: conception and design of protocol, analysis and interpretation of data, drafting the article and revising it critically for important intellectual content. All authors gave final approval of the version to be published. All experiments were performed at the Mayo Clinic, Rochester, MN, USA.
References
- Banzett RB, Garcia RT, Moosavi SH. Simple contrivance ‘clamps’ end-tidal PCO2 and PO2 despite rapid changes in ventilation. J Appl Physiol. 2000;88:1597–1600. doi: 10.1152/jappl.2000.88.5.1597. [DOI] [PubMed] [Google Scholar]
- Blauw GJ, Westendorp RG, Simons M, Chang PC, Frolich M, Meinders AE. β-Adrenergic receptors contribute to hypoxaemia induced vasodilation in man. Br J Clin Pharmacol. 1995;40:453–458. [PMC free article] [PubMed] [Google Scholar]
- Blitzer ML, Lee SD, Creager MA. Endothelium-derived nitric oxide mediates hypoxic vasodilation of resistance vessels in humans. Am J Physiol Heart Circ Physiol. 1996;271:H1182–1185. doi: 10.1152/ajpheart.1996.271.3.H1182. [DOI] [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Adenosine receptor subtypes and vasodilatation in rat skeletal muscle during systemic hypoxia: a role for A1 receptors. J Physiol. 1999a;514:151–162. doi: 10.1111/j.1469-7793.1999.151af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Cellular mechanisms by which adenosine induces vasodilatation in rat skeletal muscle: significance for systemic hypoxia. J Physiol. 1999b;514:163–175. doi: 10.1111/j.1469-7793.1999.163af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calbet JA, Boushel R, Radegran G, Sondergaard H, Wagner PD, Saltin B. Determinants of maximal oxygen uptake in severe acute hypoxia. Am J Physiol Regul Integr Comp Physiol. 2003;284:R291–303. doi: 10.1152/ajpregu.00155.2002. [DOI] [PubMed] [Google Scholar]
- Casey DP, Madery BD, Pike TL, Eisenach JH, Dietz NM, Joyner MJ, Wilkins BW. Adenosine receptor antagonist and augmented vasodilation during hypoxic exercise. J Appl Physiol. 2009;107:1128–1137. doi: 10.1152/japplphysiol.00609.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charkoudian N, Joyner MJ, Barnes SA, Johnson CP, Eisenach JH, Dietz NM, Wallin BG. Relationship between muscle sympathetic nerve activity and systemic hemodynamics during nitric oxide synthase inhibition in humans. Am J Physiol Heart Circ Physiol. 2006;291:H1378–1383. doi: 10.1152/ajpheart.00234.2006. [DOI] [PubMed] [Google Scholar]
- Collins DM, McCullough WT, Ellsworth ML. Conducted vascular responses: communication across the capillary bed. Microvasc Res. 1998;56:43–53. doi: 10.1006/mvre.1998.2076. [DOI] [PubMed] [Google Scholar]
- Dietz NM, Rivera JM, Eggener SE, Fix RT, Warner DO, Joyner MJ. Nitric oxide contributes to the rise in forearm blood flow during mental stress in humans. J Physiol. 1994;480:361–368. doi: 10.1113/jphysiol.1994.sp020366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds NJ, Marshall JM. Vasodilatation, oxygen delivery and oxygen consumption in rat hindlimb during systemic hypoxia: roles of nitric oxide. J Physiol. 2001;532:251–259. doi: 10.1111/j.1469-7793.2001.0251g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelke KA, Halliwill JR, Proctor DN, Dietz NM, Joyner MJ. Contribution of nitric oxide and prostaglandins to reactive hyperemia in human forearm. J Appl Physiol. 1996;81:1807–1814. doi: 10.1152/jappl.1996.81.4.1807. [DOI] [PubMed] [Google Scholar]
- Gamboa A, Shibao C, Diedrich A, Choi L, Pohar B, Jordan J, Paranjape S, Farley G, Biaggioni I. Contribution of endothelial nitric oxide to blood pressure in humans. Hypertension. 2007;49:170–177. doi: 10.1161/01.HYP.0000252425.06216.26. [DOI] [PubMed] [Google Scholar]
- Gladwin MT. Evidence mounts that nitrite contributes to hypoxic vasodilation in the human circulation. Circulation. 2008;117:594–597. doi: 10.1161/CIRCULATIONAHA.107.753897. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Alonso J, Olsen DB, Saltin B. Erythrocyte and the regulation of human skeletal muscle blood flow and oxygen delivery: role of circulating ATP. Circ Res. 2002;91:1046–1055. doi: 10.1161/01.res.0000044939.73286.e2. [DOI] [PubMed] [Google Scholar]
- Halliwill JR, Minson CT, Joyner MJ. Effect of systemic nitric oxide synthase inhibition on postexercise hypotension in humans. J Appl Physiol. 2000;89:1830–1836. doi: 10.1152/jappl.2000.89.5.1830. [DOI] [PubMed] [Google Scholar]
- Joyner MJ, Nauss LA, Warner MA, Warner DO. Sympathetic modulation of blood flow and O2 uptake in rhythmically contracting human forearm muscles. Am J Physiol Heart Circ Physiol. 1992;263:H1078–1083. doi: 10.1152/ajpheart.1992.263.4.H1078. [DOI] [PubMed] [Google Scholar]
- Leuenberger UA, Johnson D, Loomis J, Gray KS, MacLean DA. Venous but not skeletal muscle interstitial nitric oxide is increased during hypobaric hypoxia. Eur J Appl Physiol. 2008;102:457–461. doi: 10.1007/s00421-007-0601-x. [DOI] [PubMed] [Google Scholar]
- Martin EA, Nicholson WT, Eisenach JH, Charkoudian N, Joyner MJ. Bimodal distribution of vasodilator responsiveness to adenosine due to difference in nitric oxide contribution: implications for exercise hyperemia. J Appl Physiol. 2006a;101:492–499. doi: 10.1152/japplphysiol.00684.2005. [DOI] [PubMed] [Google Scholar]
- Martin EA, Nicholson WT, Eisenach JH, Charkoudian N, Joyner MJ. Influences of adenosine receptor antagonism on vasodilator responses to adenosine and exercise in adenosine responders and nonresponders. J Appl Physiol. 2006b;101:1678–1684. doi: 10.1152/japplphysiol.00546.2006. [DOI] [PubMed] [Google Scholar]
- McCullough WT, Collins DM, Ellsworth ML. Arteriolar responses to extracellular ATP in striated muscle. Am J Physiol Heart Circ Physiol. 1997;272:H1886–1891. doi: 10.1152/ajpheart.1997.272.4.H1886. [DOI] [PubMed] [Google Scholar]
- Mortensen SP, Gonzalez-Alonso J, Bune LT, Saltin B, Pilegaard H, Hellsten Y. ATP-induced vasodilation and purinergic receptors in the human leg: roles of nitric oxide, prostaglandins, and adenosine. Am J Physiol Regul Integr Comp Physiol. 2009a;296:R1140–1148. doi: 10.1152/ajpregu.90822.2008. [DOI] [PubMed] [Google Scholar]
- Mortensen SP, Gonzalez-Alonso J, Damsgaard R, Saltin B, Hellsten Y. Inhibition of nitric oxide and prostaglandins, but not endothelial-derived hyperpolarizing factors, reduces blood flow and aerobic energy turnover in the exercising human leg. J Physiol. 2007;581:853–861. doi: 10.1113/jphysiol.2006.127423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen SP, Nyberg M, Thaning P, Saltin B, Hellsten Y. Adenosine contributes to blood flow regulation in the exercising human leg by increasing prostaglandin and nitric oxide formation. Hypertension. 2009b;53:993–999. doi: 10.1161/HYPERTENSIONAHA.109.130880. [DOI] [PubMed] [Google Scholar]
- Nicholson WT, Vaa B, Hesse C, Eisenach JH, Joyner MJ. Aging is associated with reduced prostacyclin-mediated dilation in the human forearm. Hypertension. 2009;53:973–978. doi: 10.1161/HYPERTENSIONAHA.108.121483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl U, Busse R. Hypoxia stimulates release of endothelium-derived relaxant factor. Am J Physiol Heart Circ Physiol. 1989;256:H1595–1600. doi: 10.1152/ajpheart.1989.256.6.H1595. [DOI] [PubMed] [Google Scholar]
- Radegran G. Limb and skeletal muscle blood flow measurements at rest and during exercise in human subjects. Proc Nutr Soc. 1999;58:887–898. doi: 10.1017/s0029665199001196. [DOI] [PubMed] [Google Scholar]
- Radegran G, Calbet JA. Role of adenosine in exercise-induced human skeletal muscle vasodilatation. Acta Physiol Scand. 2001;171:177–185. doi: 10.1046/j.1365-201x.2001.00796.x. [DOI] [PubMed] [Google Scholar]
- Ray CJ, Marshall JM. Measurement of nitric oxide release evoked by systemic hypoxia and adenosine from rat skeletal muscle in vivo. J Physiol. 2005;568:967–978. doi: 10.1113/jphysiol.2005.094854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray CJ, Marshall JM. Elucidation in the rat of the role of adenosine and A2A-receptors in the hyperaemia of twitch and tetanic contractions. J Physiol. 2009;587:1565–1578. doi: 10.1113/jphysiol.2008.163683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach RC, Koskolou MD, Calbet JA, Saltin B. Arterial O2 content and tension in regulation of cardiac output and leg blood flow during exercise in humans. Am J Physiol Heart Circ Physiol. 1999;276:H438–445. doi: 10.1152/ajpheart.1999.276.2.H438. [DOI] [PubMed] [Google Scholar]
- Rongen GA, Smits P, Thien T. Characterization of ATP-induced vasodilation in the human forearm vascular bed. Circulation. 1994;90:1891–1898. doi: 10.1161/01.cir.90.4.1891. [DOI] [PubMed] [Google Scholar]
- Rowell LB, Saltin B, Kiens B, Christensen NJ. Is peak quadriceps blood flow in humans even higher during exercise with hypoxemia? Am J Physiol Heart Circ Physiol. 1986;251:H1038–1044. doi: 10.1152/ajpheart.1986.251.5.H1038. [DOI] [PubMed] [Google Scholar]
- Saunders NR, Dinenno FA, Pyke KE, Rogers AM, Tschakovsky ME. Impact of combined NO and PG blockade on rapid vasodilation in a forearm mild-to-moderate exercise transition in humans. Am J Physiol Heart Circ Physiol. 2005;288:H214–220. doi: 10.1152/ajpheart.00762.2004. [DOI] [PubMed] [Google Scholar]
- Schrage WG, Joyner MJ, Dinenno FA. Local inhibition of nitric oxide and prostaglandins independently reduces forearm exercise hyperaemia in humans. J Physiol. 2004;557:599–611. doi: 10.1113/jphysiol.2004.061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singel DJ, Stamler JS. Chemical physiology of blood flow regulation by red blood cells: the role of nitric oxide and S-nitrosohemoglobin. Annu Rev Physiol. 2005;67:99–145. doi: 10.1146/annurev.physiol.67.060603.090918. [DOI] [PubMed] [Google Scholar]
- Skinner MR, Marshall JM. Studies on the roles of ATP, adenosine and nitric oxide in mediating muscle vasodilatation induced in the rat by acute systemic hypoxia. J Physiol. 1996;495:553–560. doi: 10.1113/jphysiol.1996.sp021615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits P, Williams SB, Lipson DE, Banitt P, Rongen GA, Creager MA. Endothelial release of nitric oxide contributes to the vasodilator effect of adenosine in humans. Circulation. 1995;92:2135–2141. doi: 10.1161/01.cir.92.8.2135. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Jia L, Eu JP, McMahon TJ, Demchenko IT, Bonaventura J, Gernert K, Piantadosi CA. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science. 1997;276:2034–2037. doi: 10.1126/science.276.5321.2034. [DOI] [PubMed] [Google Scholar]
- Tschakovsky ME, Joyner MJ. Nitric oxide and muscle blood flow in exercise. Appl Phys Nutr Metab. 2008;33:151–161. doi: 10.1139/H07-148. [DOI] [PubMed] [Google Scholar]
- Tschakovsky ME, Sujirattanawimol K, Ruble SB, Valic Z, Joyner MJ. Is sympathetic neural vasoconstriction blunted in the vascular bed of exercising human muscle? J Physiol. 2002;541:623–635. doi: 10.1113/jphysiol.2001.014431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisbrod CJ, Minson CT, Joyner MJ, Halliwill JR. Effects of regional phentolamine on hypoxic vasodilatation in healthy humans. J Physiol. 2001;537:613–621. doi: 10.1111/j.1469-7793.2001.00613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BW, Pike TL, Martin EA, Curry TB, Ceridon ML, Joyner MJ. Exercise intensity-dependent contribution of β-adrenergic receptor-mediated vasodilatation in hypoxic humans. J Physiol. 2008;586:1195–1205. doi: 10.1113/jphysiol.2007.144113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BW, Schrage WG, Liu Z, Hancock KC, Joyner MJ. Systemic hypoxia and vasoconstrictor responsiveness in exercising human muscle. J Appl Physiol. 2006;101:1343–1350. doi: 10.1152/japplphysiol.00487.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]