

Abstract
The prototypic arenavirus lymphocytic choriomeningitis virus (LCMV) represents a powerful experimental model for the study of the basic virology and pathogenesis of arenaviruses. In the present study, we used the LCMV model to evaluate the anti-viral potential of phosphorothioate oligonucleotides against arenaviruses. Our findings indicate that amphipathic DNA polymers (APs) are potent inhibitors of infection with a series of LCMV isolates with IC50 in the low nanomolar range. APs target the surface glycoprotein (GP) of LCMV and block viral entry and cell-cell propagation of the virus, without affecting later steps in replication or release of progeny virus from infected cells. The anti-viral action of APs is sequence-independent but is critically dependent on their size and hydrophobicity. Mechanistically, we provide evidence that APs disrupt the interaction between LCMVGP and its cellular receptor, α-dystroglycan. Exposure of LCMV to APs does not affect the stability of the GP virion spike and has no effect on the conformation of a neutralizing antibody epitope, suggesting rather subtle changes in the conformation and/or conformational dynamics of the viral GP.
INTRODUCTION
Arenaviruses merit significant attention as powerful experimental models and important human pathogens. Infection of the prototypic Old World arenavirus lymphocytic choriomeningitis virus (LCMV) in its natural host, the mouse, illuminated fundamental concepts in immunology and virology that have been extended to other viruses, bacteria, and parasites (Oldstone, 2002). LCMV is also an important pathogen in human pediatric medicine (Jamieson, 2006) and has recently caused lethal infections in transplantation patients (Fischer et al., 2006). The closely related Lassa fever virus (LFV) is the causative agent of a severe hemorrhagic fever with high mortality in humans and represents the most important human pathogen among the arenaviruses, responsible for over 300, 000 infections and several thousand deaths per year (McCormick and Fisher-Hoch, 2002; Geisbert and Jahrling, 2004).
Arenaviruses are enveloped single-strand RNA viruses with a bisegmented genome in ambisense organization that consists of two single-stranded RNA species: the larger segment encodes the virus polymerase (L) and a small zinc finger motif protein (Z), the smaller RNA segment encodes the virus nucleoprotein (NP) and glycoprotein precursor (GPC). GPC is processed into the peripheral glycoprotein GP1 and the transmembrane glycoprotein GP2. GP1 is implicated in receptor binding (Parekh and Buchmeier, 1986, Borrow and Oldstone, 1992) and GP2 is structurally similar to the fusion active membrane proximal portions of other enveloped viruses (Gallaher et al., 2001; Eschli et al., 2006).
The initial step of LCMV and LFV infection is the attachment of the virus to specific glycan structures on the cellular receptor α-dystroglycan (α-DG) (Cao et al., 1998; Kunz et al., 2001; Imperiali et al., 2005; Kunz et al., 2005a/b, Rojek et al., 2007). Upon attachment, LCMV virions are taken up in smooth vesicles that enter the endocytic pathway and deliver the virus to endosomes where pH-dependent membrane fusion occurs (Borrow and Oldstone, 1994).
Studies in human Lassa fever patients and experimental LFV and LCMV infection in animals showed that the host's control of viral replication is primarily mediated by the anti-viral T-cell response with limited roles of antibodies (Oldstone et al., 2002; McCormick and Fisher-Hoch, 2002). The strong predictive value of virus concentration in blood for a disastrous disease outcome in human Lassa fever indicates further a close competition between virus spread and the patient's anti-viral immune response. Since rapid viral dissemination critically depends on efficient attachment of the viruses to host cells and subsequent entry, drugs targeting these steps will give the host's immune system an advantage by providing a wider window of opportunity for the generation of an efficient anti-viral immune response. Targeting virus entry also has the advantage that the numbers of virus particles present at early stages of infection tend to be smaller, which allows maximization of the inhibitory effect of an anti-viral drug.
Over the past two decades, phosphorothioate oligonucleotides (PS-ONs) have emerged as potent anti-viral substances that can target early steps of virus infection as illustrated by efficient blocking of receptor binding and fusion of human immunodeficiency virus (HIV) (Matsukura et al., 1987; Stein et al., 1989, Stein et al., 1991; Yamaguchi et al., 1997; Este et al., 1998). These effects were known to be unrelated to the specific nucleotide sequence of PS-ONs but were not clearly elucidated until Vaillant et al. (2006) described the antiviral activity of PS-ONs as being derived from their sequence-independent activity as amphipathic polymers (APs). This unique chemical property allowed the interaction of APs with the alpha-helices in HIV-1 gp41 and neutralized their fusion activity. While HIV and arenaviruses clearly differ in virion structure, cellular receptor use, and mechanism of membrane fusion, recent studies revealed that the cell surface GP of LCMV shares some key features with other fusion-active class I viral glycoproteins of enveloped viruses (Gallagher et al., 2001; Eschli et al., 2006). In the present study, we used the LCMV model to evaluate the anti-viral potential of APs against arenaviruses. Our findings indicate that APs are potent inhibitors of LCMV infection that target the viral GP and block viral entry and cell-cell propagation of the virus, without affecting later steps in replication. The anti-viral action of APs is sequence-independent but depends on size and the combination of hydrophobic and hydrophilic properties (amphipathicity). We provide evidence that APs perturb the interaction between LCMVGP and its cellular receptor, α-dystroglycan, without disrupting the overall structure of the GP virion spike.
RESULTS
Amphipathic DNA polymers (APs) efficiently block LCMV infection and cell-to-cell propagation
To evaluate the anti-viral potential of APs against arenaviruses, we tested a series of LCMV isolates including the prototypic isolate ARM53b and its immunosuppressive variant clone-13 (cl-13). While infection of adult mice with LCMV ARM53b is efficiently cleared by the host's anti-viral CD8+ cytotoxic T lymphocyte response, LCMV cl-13 causes a persistent infection accompanied by a generalized immunosuppression (Ahmed et al., 1984). As such, LCMV cl-13 differs from ARM53b only by two amino acids, an F260L change in GP1 and K1079Q change in the viral polymerase (Salvato et al., 1988). The immunosuppressive phenotype of LCMV cl-13 is linked to the F260L mutation in GP1, which causes a >400-fold increase in binding affinity to the cellular receptor α-DG (Sevilla et al., 2000). LCMV cl-13 has receptor binding characteristics and cellular tropism similar to LFV (Kunz et al., 2005a/b; Rojek et al., 2007). In addition, we tested the LCMV isolates WE54, E350, and Traub, whose disease potential and receptor binding characteristics have been described previously (Sevilla et al., 2000; Smelt et al., 2001). As a control, we used the rhabdovirus vesicular stomatitis virus (VSV) that is unrelated to arenaviruses and uses different cellular receptors (Rose and Whitt, 2001).
As a first candidate AP, we chose the 40-base completely degenerate (randomer) REP 9, which had previously shown anti-viral activity against HIV (Vaillant et al., 2006). LCMV isolates were pre-incubated with increasing concentrations of drug and then added to monolayers of VeroE6 cells. Viral infection was determined after 16 hours by detection of LCMV nucleoprotein (NP), which represents the earliest viral gene product using immunofluorescence. Pre-incubation with REP 9 resulted in a marked reduction of infection with all LCMV isolates tested (IC50 = 200 nM), but did not affect infection with a recombinant VSV expressing green fluorescent protein (VSV-GFP) (Fig. 1A). Since both LCMV and VSV enter cells by endocytosis followed by pH-dependent membrane fusion in an endosomal compartment (Borrow and Oldstone, 1994; Rose and Whitt, 2001), the lack of inhibition of VSV infection excludes unspecific drug effects including general perturbation of vesicular trafficking or depletion of endosomal proton gradients.
Figure 1. Blocking of LCMV infection with REP 9.

(A) The PS-ON REP 9 blocks infection of cells with LCMV but not VSV: The indicated LCMV isolates and VSV-GFP were pre-incubated with REP 9 for 1 hour at 4 °C and added to monolayers of VeroE6 cells (MOI = 3). After 24 hours, LCMV infection was detected by intracellular staining for LCMVNP using flow cytometry. Infection with VSV-GFP was detected by direct fluorescence excitation of GFP. Percentages of NP or GFP positive cells are given (n = 3 ± SD). (B) REP 9 blocks viral entry and cell-to-cell propagation: Monolayers of VeroE6 cells were infected with LCMV at the indicated MOI. REP 9 was added at the indicated time points and kept throughout the experiment. After total 48 hours, infection was determined as in (A). In histograms, the y-axis represents cell numbers and the x-axis fluorescence intensity for NP. The dotted line represents uninfected controls and the solid line infected samples. Percentages of NP positive cells are given. (C) Detection of virus infection and propagation by immunofluorescence: Monolayers of VeroE6 cells were infected with LCMV ARM53b at MOI of 0.1 and REP 9 added at the indicated time points. After 48 hours, cells were fixed, permeabilized and stained with mAb 113 anti-LCMVNP using a FITC-conjugated secondary antibody and phalloidin-rhodamine for counterstaining of actin filaments. Bar = 100 μM
Next, we addressed the effect of REP 9 on cell-to-cell propagation of LCMV, by adding the drug at different time points of infection. The presence of REP 9 during infection efficiently blocked viral entry, as indicated by negligible numbers of NP positive cells after 48 hours (Fig. 1B, C). Addition of the drug 6 or 24 hours post-infection resulted in a marked decrease in the number of infected cells, indicating efficient blocking of cell-to-cell spread of the virus. Assessment of cell viability at different time points of the experiment by Cell Titer Glo® assay did not reveal significant cytotoxicity of the drug (data not shown). The similar levels of NP expression in infected cells in all specimens, as illustrated by the comparable mean immunofluorescence intensities for NP detected in flow cytometry analysis (Fig. 1B), indicate that the drug does not affect NP expression once infection has occurred. In contrast to the effects on virus-to-cell infection and cell-to-cell spread of virus, pre-treatment of cells with REP 9 for up to 6 hours, followed by wash out of the drug, did not affect subsequent infection (data not shown).
APs target the GP of LCMV
Infection and cell-to-cell propagation of arenaviruses critically depend on the viral GP, which therefore appears to be a likely target for the drug. Since REP 9 blocks infection with LCMV but not VSV (Fig. 1A), we generated a recombinant VSV containing the GP of LCMV in its envelope to directly address the role of LCMVGP as a drug target. For this purpose, the GP derived from LCMV cl-13 was incorporated into the recombinant VSV variant VSVΔG*, in which the VSV G gene is replaced by a GFP reporter gene, by pseudotyping as described previously (Perez et al., 2001; Kunz et al., 2005a). Infection of cells by the VSVΔG*-LCMVGP pseudotype was specifically blocked by inactivated LCMV cl-13, but not by inactivated VSV (Fig. 2A), confirming that the recombinant LCMVGP displayed by VSVΔG*-LCMVGP shares the binding characteristics of the GP present in LCMV cl-13. As shown in Figure 2B, the VSVΔG*-LCMVGP chimera was efficiently neutralized with REP 9, with an efficacy of the drug comparable to the one observed against LCMV cl-13 (Fig. 1A). Since REP 9 did not affect infection with VSV (Fig. 1A, Fig. 2B), the susceptibility of VSVΔG*-LCMVGP strongly suggested that LCMVGP was a main target of the drug.
Figure 2. REP 9 targets the GP of LCMV.
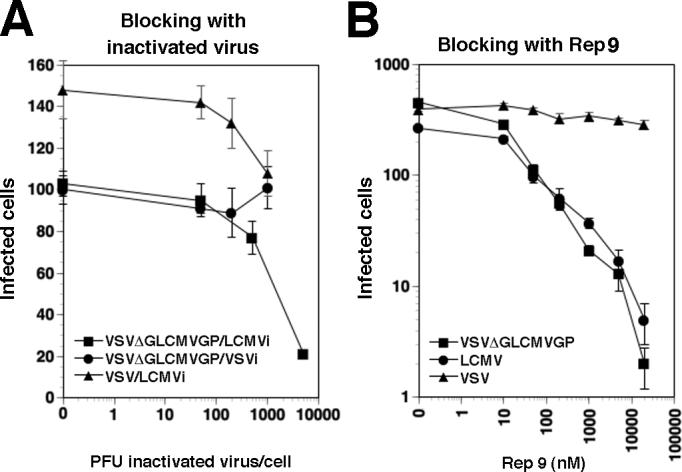
(A) Blocking of VSVΔG*-LCMVGP infection by inactivated LCMV cl-13: VeroE6 cells were blocked with inactivated LCMV cl-13 (LCMVi) or VSV (VSVi) at the indicated ratios of virus particles per cell followed by infection with 200 PFU of the indicated viruses. Infection of LCMV was assessed by immunofluorescence staining for LCMVNP and VSVΔG*-LCMVGP and VSV-GFP were detected using the GFP reporter. Infected cells per well were scored (n = 3, ± SD). (B) Inhibition of infection of VSVΔG*-LCMVGP with REP 9: LCMV, VSVΔG*-LCMVGP, and VSV-GFP (500 PFU) were pre-incubated with increasing concentrations of REP 9 and added to monolayers of VeroE6 cells. Infection was determined as in (A). Data are positive cells per well, n = 3, ± SD.
APs do not affect virion assembly and release of virus from infected cells
The efficient blocking of cell-to-cell propagation of LCMV may involve effects of REP 9 on viral entry as well as on release of progeny virus from infected cells, e.g. by targeting the viral GP present in budding zones at the cell surface. To address a possible impact of REP 9 on the assembly and/or release of virions from infected cells, VeroE6 cells were infected with LCMV at a multiplicity of infection (MOI) of 1. After 24 hours, medium was removed, cells washed extensively and fresh medium added with or without REP 9. After another 24 hours of incubation total cell lysates were prepared and virions isolated from supernatants by ultracentrifugation through sucrose cushions. Detection of NP and GP in total cell lysates by Western-blot revealed reduced levels of viral proteins in cells cultured in the presence of REP 9, likely due to inhibition of cell-to-cell spread of the virus in the presence of drug (Fig. 3). Similar amounts of viral proteins were detected in virions isolated from culture supernatants in the presence or absence of drug (Fig. 3), indicating that the drug does not inhibit release of virions from infected cells. No viral infectivity was detected in cell culture supernatants containing drug (data not shown), which is consistent with the efficient neutralization of free virus observed previously (Fig. 1A).
Figure 3. REP 9 does not interfere with LCMV virus production.
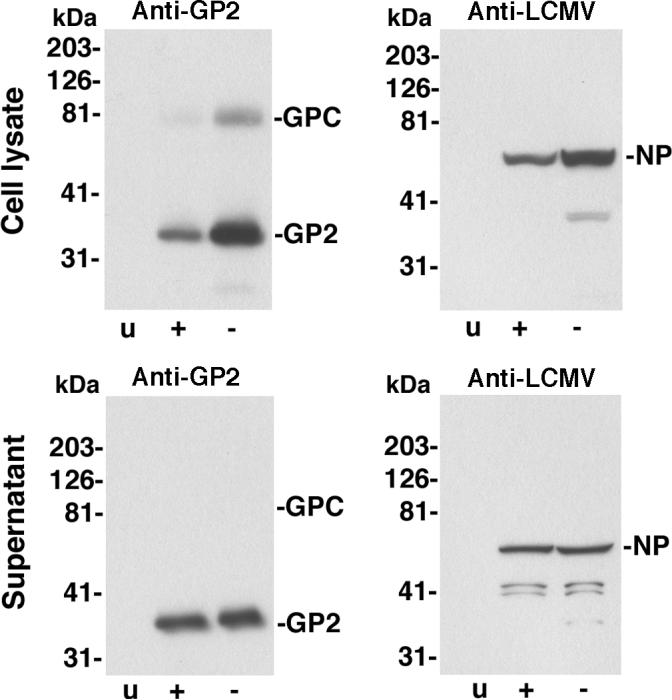
VeroE6 cells were infected with LCMV ARM53b at an MOI of 1 or mock infected (u). After 24 hours, medium was removed, cells washed extensively and new medium added with (+) or without (-) 2 μM REP 9. After 24 hours, total cell lysates were prepared. Supernatants were subjected to ultracentrifugation through a sucrose cushion and pellets solubilized in SDS-PAGE sample buffer. Total proteins of cell lysates (cell lysate) and pellets recovered from supernatants by ultracentrifugation (supernatant) were separated by SDS-PAGE, blotted to nitrocellulose and probed with mAb 83.6 to LCMVGP2 (anti-GP) and polyclonal guinea pig serum anti-LCMV. Primary antibodies were detected with HRP-conjugated secondary antibodies using enhanced chemiluminescence (ECL). Molecular masses and the positions of GPC, GP2, and NP are indicated.
Anti-viral activity of APs depends on size and hydrophobicity, but is sequence independent
In a next step, we wanted to determine if the physicochemical characteristics of APs required for activity against LCMV were similar to those necessary for interaction with the core alpha-helices of HIV-1 gp41. In order to screen a comprehensive set of oligonucleotide AP derivatives in a cell-based infection assay, we made use of a firmly established retroviral pseudotype system. The GPs of LCMV ARM53b and cl-13 were inserted into recombinant Moloney leukemia virus (MLV) containing a luciferase reporter gene, which allows rapid and reliable quantification of infection in a high-throughput assay format (Rojek et al., 2006; Rojek et al., 2007). Generation and characterization of the retroviral pseudotypes of LCMV ARM53b and cl-13 are described in Supplementary Materials and Fig. S1. Pre-treatment of retroviral LCMV pseudotypes with REP 9 blocked subsequent infection with an IC50 of approximately 200 nM, similar to that observed with live LCMV isolates (Fig. 4). In contrast to the potent anti-viral activity of REP 9, the anionic polymers dextran sulfate and heparin showed only limited and no anti-viral activity, respectively, against retroviral pseudotypes of LCMV ARM53b and cl-13 (Fig. 4) and the corresponding live viruses (not shown). Consistent with the data obtained with LCMV, REP 9 blocked pseudotype infection only when added to the virus prior to or during infection, but not when added at later time points post-infection (Fig. 5A) or when added to cells prior to infection (Fig. 5B). In contrast to the experiments shown in Fig. 1, the pseudotypes used here are replication deficient, and as such no cell-cell spread of the virus is observed.
Figure 4. Blocking of infection of cells with LCMV pseudotypes by anionic polymers.

Retroviral pseudotypes containing the GPs of LCMV ARM53b and cl-13 (MOI = 1) were pre-incubated with the indicated concentrations of REP 9, dextran sulfate, or heparin for one hour on ice. Pseudotypes were then added to monolayers of VeroE6 cells. After 48 h, infection was quantified by luciferase reporter gene assay. Luminescence is expressed as fold-increase over uninfected controls (n = 3 ± SD).
Figure 5. Effect of REP 9 on retroviral LCMV pseudotypes.
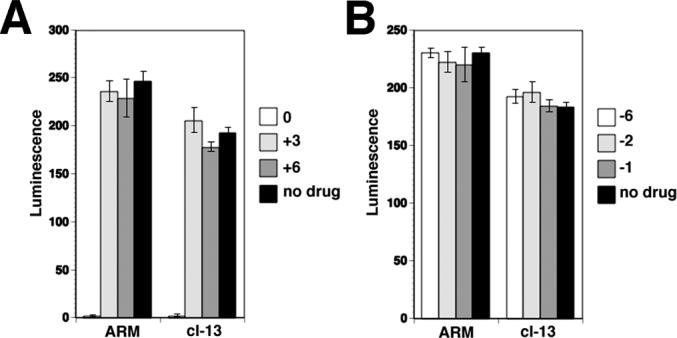
(A) Addition of drug at or post infection: VeroE6 cells were treated with 2 μM REP 9 during infection (0 h) and 3 or 6 hours post infection (+ 3 h, +6 h) with replication-deficient pseudotypes of LCMV (MOI = 1). Infection was assessed after 48 hours as in Fig. 4 (mean ± SD; n = 3). (B) Effect of the pre-treatment of cells with REP 9 on pseudotype infection: Cells were pre-treated with 2 μM REP 9 for 6, 2, and 1 hour, washed extensively and then infected with replication-deficient pseudotypes of LCMV (MOI = 1). Infection was assessed after 48 hours as in (A).
As a first step to elucidate the physicochemical requirements for the anti-viral activity of APs, we addressed size-dependence. REP 9 analogs of different lengths were synthesized and tested for their activity against retroviral pseudotypes of LCMV ARM53b and cl-13. As shown in Table 1, REP 9 size analogs became more active with increasing length, with the maximal activity at a length of 30 − 60 bases.
Table 1. REP 9 anti-LCMV activity depends on size and hydrophobicity.
LCMV pseudotypes (MOI = 1) were pre-incubated with increasing concentrations of the indicated polymers for one hour on ice and added to monolayers of VeroE6 cells in 96 well-plates. After 48 h, infection was quantified using the Steady-Glo® luciferase reporter gene assay as in Fig. 3B and IC90 calculated as the concentration of drug required to reduce the above background luminescence signal by 90% (n = 3 ± SD).
| Chemistry | Size (bases) | IC90 (μM, mean + SD) | |
|---|---|---|---|
| LCMV ARM53b | LCMV cl-13 | ||
| Phosphorothioate (PS) | 6 | >50 | >50 |
| 10 | 21 ± 4.5 | 36 ± 8.9 | |
| 20 | 0.945 ± 0.154 | 1.24 ± 0.32 | |
| 30 | 0.523 ± 0.141 | 0.556 ± 0.159 | |
| 40 (REP 9) | 0.454 ± 0.067 | 0.623 ± 0.187 | |
| 50 | 0.396 ± 0.075 | 0.503 ± 0.091 | |
| 60 | 0.488 ± 0.062 | 0.605 ± 0.175 | |
| PS + 2'-O-methyl | 40 | 1.41 ± 0.36 | 1.88 ± 0.31 |
| 2'-O-methyl | 40 | >50 | >50 |
| Propane PS | 40 | >50 | >50 |
| Abasic PS | 40 | >50 | >50 |
Next, we addressed the impact of altering the chemical properties of the phosphorothioate backbone on anti-viral activity. Comparison of the phosphorothioated 40-base randomer (REP 9) with a 2’-O-methylated analog revealed an approximately three-fold reduction in anti-viral activity (Table 1). In contrast, 2’-O-methylated, non-phosphorothioated 40-base randomer oligonucleotide showed significantly reduced activity, demonstrating that the phosphorothioate moiety, and thus hydrophobicity, is critical for the anti-viral effect. In addition, we tested the anti-viral activity of two additional polymers: propane-phosphorothioate (propane-PS) containing only a three-carbon linker between the phosphorothioate groups as well as abasic-phosphorothioate (abasic-PS), which consists of only the ribose sugars with phosphorothioate linkage. In contrast to normal phosphororthioate oligonuceotides, propane-PS and abasic-PS lack the hydrophilic activity that resides in the base paring moieties of the nucleobases, and neither of these analogs showed significant inhibition of LCMV pseudotype infection (Table 1). Together, these data suggest that the anti-LCMV activity of phosphorothioate oligonucleotides critically depends on their size and the presence of both hydrophobic and hydrophilic properties and thus the amphipathic nature of the oligonucleotide.
To address the sequence-dependence of the anti-viral activity, a series of 40-base phosphorothioate oligonucelotides, including poly(A), −(T), −(C), −(G), and −(I) as well as co-polymers of (A + C), (A + G), (T + C), and (T + G), were tested. While most sequences had an IC50 for LCMV pseudotypes within the same order of magnitude, we observed significantly reduced activity with poly(G), and, to a lesser degree, with poly(A) (Table 2). The reduced activity of the poly(G) PS-ON was likely due to the propensity of poly(G) sequences to form stable secondary structures (Kim et al., 1991). A similar phenomenon may also underlie the higher IC50 observed with the polypurine poly(A).
Table 2. Anti-LCMV activity of APs is sequence independent.
LCMV pseudotypes (MOI = 1) were pre-incubated with increasing concentrations of 40-mers phosphorothioate oligonucleotides with the indicated sequences for one hour on ice and added to monolayers of VeroE6 cells in 96 well plates. After 48 h, infection was quantified and IC90 calculated as in Table 2 (n = 3 ± SD).
| Sequence | IC90 (μM, mean + SD) | |
|---|---|---|
| LCMV ARM53b | LCMV cl-13 | |
| G40 PS | 18.2 ± 4.1 | 16.7 ± 2.9 |
| A40 PS | 4.12 ± 0.92 | 3.29 ± 1.27 |
| T40 PS | 0.311 ± 0.043 | 0.795 ± 0.121 |
| C40 PS | 2.13 ± 0.42 | 1.91 ± 0.38 |
| I40 PS | 0.284 ± 0.031 | 0.567 ± 0.161 |
| TG20 PS | 0.218 ± 0.081 | 0.367 ± 0.143 |
| AC20 PS | 0.812 ± 0.196 | 1.34 ± 0.048 |
| TC20 PS | 0.789 ± 0.221 | 0.923 ± 0.316 |
| AG20 PS | 0.534 ± 0.163 | 0.845 ± 0.164 |
| C20 PS | 11.6 ± 2.0 | 13.6 ± 4.5 |
| C30 PS | 1.84 ± 0.54 | 2.26 ± 1.01 |
| C50 PS | 0.946 ± 0.207 | 1.12 ± 0.25 |
| C60 PS | 0.981 ± 0.197 | 1.26 ± 0.32 |
Thus the chemical determinants for antiviral activity of APs against LCMV were remarkably similar to those required for optimal antiviral activity against HIV-1 (Vaillant et al., 2006), suggesting that domains analogous to the HIV-1 gp41 core alpha helices formed the target domain of LCMVGP.
APs disrupt the binding of LCMVGP to its receptor α-DG
The first step of LCMV infection is the attachment of the virus to its cellular receptor α-DG. To test the impact of APs on virus receptor binding, we tested REP 9 in two in vitro binding assays, a virus overlay protein binding assay (VOPBA) and an ELISA-format solid phase virus-binding assay. Purified LCMV cl-13 was pre-incubated with increasing concentrations of REP 9 and then added to immobilized α-DG derived from human HEK293 cells. As a control, we tested the effect of REP 9 on the binding of laminin-1, which is a natural high affinity ligand for α-DG (Ervasti and Campbell, 1993). In both assay formats, treatment with REP 9 blocked the binding of LCMV cl-13 to α-DG in a dose-dependent manner, but did not affect laminin binding (Fig. 6A, B). To test the ability of REP 9 to dissociate bound virus from α-DG, LCMV cl-13 was pre-incubated with immobilized α-DG to allow the formation of a virus-receptor complex. Addition of drug to the pre-formed virus-receptor complex caused a significant dissociation of the virus (Fig. 6C). However, addition of the same concentrations of laminin-1 did not result in significant dissociation of the virus-receptor complex (Fig. 6C), in line with previous reports (Kunz et al, 2001).
Figure 6. REP 9 disrupts virus-receptor binding.

(A) REP 9 blocks α-DG binding of LCMV but not laminin: LCMV cl-13 (107 PFU/ml) (top panel) and mouse EHS laminin (10 μg/ml) were pre-incubated with the indicated concentrations of REP 9 and then added to nitrocellulose blots of immobilized α-DG. After 4 hours of incubation, bound virus was detected using mAb 83.6 anti-LCMVGP2 and an HRP-coupled secondary antibody using ECL. Bound laminin was detected with a polyclonal Ab anti-laminin-1 and an HRP-coupled secondary antibody (B) Blocking of LCMV and laminin binding to immobilized α-DG in ELISA: LCMV cl-13 (107 PFU/ml) and EHS laminin (10 μg/ml) were pre-incubated with the indicated concentrations of REP 9 and then added to microtiter plates containing immobilized α-DG from rabbit skeletal muscle. Bound virus and laminin were detected using the antibody concentrations in (A) in a color reaction with ABTS substrate. OD (405) was measured in an ELISA reader and background binding to BSA subtracted (n = 3 ± SD). (C) REP 9 dissociates the LCMVGP/receptor complex. LCMV cl-13 (107 PFU/ml) was bound to immobilized α-DG for 16 hours at 6 °C. After brief washing, the indicated concentrations of REP 9 or laminin-1 were added for 2 hours at room temperature and bound virus was detected as in (B).
APs do not dissociate GP subunits or affect a conformation-sensitive antibody epitope
Within the LCMVGP virion spike, GP1 and GP2 are associated by non-covalent, predominantly ionic interactions (Burns and Buchmeier, 1991). To test a possible effect of REP 9 interaction on the association of GP1 with GP2, purified LCMV was exposed to either REP 9 or increasing concentrations of NaCl. After incubation for one hour at 4 °C, virus particles were subjected to ultracentrifugation through a sucrose cushion. Recovered virus was immobilized in microtiter plates and probed with monoclonal antibody (mAb) 36.1, which recognizes a conformation-sensitive neutralizing epitope on GP1 (Wright et al., 1989) and mAb 83.6 that binds to a linear epitope in GP2 (Weber and Buchmeier, 1988). While exposure to 1M NaCl resulted in significant dissociation of GP1 and a concomitant drop in viral infectivity, no change in the GP1 signals was observed after treatment with REP 9 (Fig. 7A), excluding dissociation of GP1. In a direct competition assay, REP 9 did not interfere with binding of mAbs 36.1 and 83.6, but caused a slight increase in binding of mAb 36.1, indicating that the polymer does not disrupt but rather stabilizes the conformation-sensitive epitope of this antibody.
Figure 7. REP 9 inactivates LCMV without dissociation of GP1 from GP2.

(A) Purified LCMV cl-13 (106 PFU/ml) was incubated with the indicated concentrations of NaCl or REP 9 in PBS for one hour at 4 °C. After addition of a 10-fold volume of PBS, samples were subjected to ultracentrifugation through a sucrose cushion. Supernatants were removed and pellets re-suspended. Virus was immobilized in microtiter plates and the presence of GP1 and GP2 detected in ELISA using mAbs 36.1 and 83.6, respectively. Primary antibodies were detected with POD-conjugated anti-mouse IgG in a color reaction using ABTS substrate. OD (405) was measured using an ELISA reader (n = 3 ± SD). Inactivation of LCMV was verified by plaque assay on monolayers of VeroE6 cells. (B) REP 9 does not compete with the binding of mAbs 36.1 and 83.6 to LCMVGP: Purified LCMV ARM53b was immobilized in microtiter plates and incubated with the indicated concentrations of REP 9 for one hour at room temperature. Monoclonal antibodies 36.1 and 83.6 were added in the presence of REP 9 for one hour and bound antibody detected as in (A).
DISCUSSION
In the present study, we used the prototypic arenavirus LCMV as a model to evaluate amphipathic DNA polymers (APs) as anti-arenaviral drugs and to investigate their mechanism of action. We found that the prototypic AP, REP 9, blocked LCMV entry and cell-to-cell propagation without affecting subsequent steps of replication and formation of progeny virus. When tested against a series of LCMV isolates, REP 9 efficiently blocked infection with IC50 in the low nanomolar range and prevented cell-to-cell propagation of virus, without affecting expression of viral protein in infected cells. The anti-viral activity of REP 9 against LCMV critically depended on size, hydrophobicity, and an amphipathic nature of the oligonucleotide, but was sequence independent. Mechanistically, we found that REP 9 targeted the viral GP and perturbed virus-receptor binding without disrupting the overall composition of the GP virion spike.
We provide evidence that the PS-ON targets the viral GP and blocks virus entry, but does not affect virion assembly and release of progeny virus from infected cells. In the time window of acute LCMV infection in the cell lines tested, we found no evidence for the ability of the drug to clear virus from infected cells.
Using retroviral pseudotypes of LCMV in a cellular high-throughput infection assay, we performed initial structure-function studies to determine the physicochemical features of APs required for anti-viral activity. We found optimal activity with phosphorothioated oligonucleotide randomers of 30−60 bases, a size range similar to PS-ONs that inhibit receptor binding and fusion of HIV (Vaillant et al., 2006). The importance of the hydrophobic and hydrophilic characters of phosphorothioated oligonucleoties was illustrated by markedly reduced anti-viral activity in the absence of the phosphothioate groups (lacking hydrophobicity) or low anti-viral activity of abasic or propane phopshorothioates (lacking hydrophilicity). This indicates that the amphiphatic character of phosphorothioated oligonucleotides and not their polyanionic nature is the underlying chemistry conferring anti-viral activity to these compounds. This is in line with our finding that anionic polymers of similar size like dextran sulfate and heparin show little if any anti-viral activity. As reported earlier for HIV (Stein et al., 1989; Vaillant et al., 2006), anti-viral activity of PS-ONs against LCMV was essentially sequence-independent. The physicochemical requirements for activity of APs against LCMV are strikingly similar to the ones recently identified for HIV (Vaillant et al., 2006). This suggests that, despite obvious differences in structure, receptor specificity, as well as site and pH-requirements of fusion, the envelope GPs of arenaviruses and retroviruses share common features, supporting the idea that arenavirus GPs belong to the class I viral fusion active membrane proteins (Gallagher et al., 2001; Eschli et al., 2006).
Since the first step of every virus infection is the attachment of virus particles to their cellular receptors on the target cell, we investigated the impact of REP 9 on the interaction of LCMV with its primary receptor α-DG. Old World arenaviruses and host-derived binding partners like laminin recognize specific, highly conserved O-glycan structures present on α-DG (Imperiali et al., 2005; Kunz et al., 2005b, Rojek et al., 2007). As a consequence, the viruses and laminin compete for α-DG binding (Kunz et al., 2001). Using in vitro binding assays, we found that REP 9 efficiently blocked the high affinity binding of LCMV cl-13 to α-DG with an IC50 similar to the one observed for virus neutralization (approximately 200 nM). In contrast, REP 9 had no effect on the interaction between α-DG and laminin-1, indicating specificity of the drug. REP 9 behaved exactly opposite to heparin, which had no effect on LCMV receptor binding or infection (Kunz et al., 2001; this study), but efficiently blocked the interaction between laminin and α-DG (Ervasti and Campbell, 1993). Moreover, REP 9 was able to disrupt pre-formed virus-receptor complexes. This observation is rather striking since previous studies indicated virtually irreversible binding of LCMV cl-13 to α-DG (Kunz et al., 2001) and no displacement of bound virus from its receptor by competition with laminin (Kunz et al., 2001; Kunz et al., 2005a). Interestingly, REP 9 IC50 values within the same order of magnitude were found against LCMV isolates with high α-DG binding affinity such as LCMV cl-13, WE54, and Traub (low nanomolar Kd) and low affinity binders like ARM53b and E350 (micromolar Kd). This lack of correlation between IC50 of REP 9 and the receptor binding affinity of the LCMV isolate makes a mechanism involving competitive inhibition unlikely. A non-competitive mechanism involving allosteric effects might also explain the ability of the PS-ON to disrupt pre-formed virus-receptor complexes that are inert against high concentrations of the competitive inhibitor laminin.
Despite its drastic impact on receptor binding, we found no evidence for significant dissociation of the receptor-binding GP1 from the transmembrane GP2 in LCMVGP present on virions upon REP 9 treatment. Likewise, exposure to REP 9 did not affect binding of a neutralizing monoclonal antibody (mAb 36.1) recognizing a conformational epitope on GP1. In direct competition assays, the presence of REP 9 resulted even in a slight increase of antibody binding, suggesting a stabilizing effect of the drug on the neutralizing epitope. Together, these findings exclude mechanisms involving disruption of the hetero-oligomeric GP spike and suggest rather subtle changes in the conformation and/or conformational dynamics of the viral GP induced by the drug. Based on the large size (>30 nucleotides) and amphipathic nature required for anti-viral activity, REP 9 may undergo at least partially amphipathic interactions with the GP involving a large molecular target domain. REP 9 may bind to such a target on GP1, or, alternatively may interact with GP2 inducing conformational changes in GP1 via allosteric effects. Given the involvement of rather extended molecular surfaces on the target domain in the viral GP, it would be reasonable to assume that single point mutations in GP would not result in drug resistance, making the emergence of viral escape variants unlikely. This is perhaps one of the more striking features of the mechanism of action of APs in general as the development of drug resistance is a critical issue for the treatment of most viral infections. As Old World arenavirus infection is a complex, multi-step process involving possible unknown co-receptors and endocytosis followed by pH-dependent membrane fusion, REP 9 could target additional steps in viral entry, a possibility we are currently investigating.
Based on the striking correlation between initial virus titers in blood and disease outcome of human infections with Old World arenaviruses like LFV, drugs able to block viral entry appear promising. However, efforts to specifically target Old World arenavirus entry have thus far been limited to attempts to neutralize free virus with antibody, using immune plasma from convalescent patients. This resulted in improvement in some cases of human LFV infection (Leifer et al., 1970; Monath et al., 1974; Frame et al., 1984), but not in others (McCormick et al., 1986). Subsequent proof-of-principle studies in animal models showed significant protection by sera with sufficient titers of neutralizing antibodies (Jahrling et al., 1983; Jahrling et al., 1984; Jahrling and Peters, 1984; Jahrling et al., 1985). However, due to the unusually low titers of neutralizing antibody in convalescent human plasma from Lassa patients (Jahrling et al., 1985) and logistic problems associated with collection, storage, and processing of immune plasma in endemic areas, the therapeutic use of neutralizing antibodies does not appear feasible at this point. Novel, synthetic drugs able to target Old World arenavirus entry are therefore needed.
Based on experimental and clinical experience with phosphorothioate oligonucleotides as antisense agents, this class of compounds appear to be tolerated well at therapeutic doses in humans and in animal models (Stein et al., 2005) and novel, improved strategies for drug delivery are currently being evaluated (Zhang et al., 2007). Together with their reported anti-viral activity in other viral systems, these properties made REP 9 and other APs promising candidates as potential anti-arena virus drugs that could attack arenaviruses early in infection, before they can gain control over the host cell machinery for replication.
Amphipathic DNA polymers (APs) represent a new class of nucleic-acid based potent, broad-spectrum anti-viral substances able to target entry of enveloped viruses including HIV (Vaillant et al., 2006), arenaviruses (this study), other enveloped viruses with type 1 fusion proteins including influenza viruses and respiratory syncytial virus as well as non type 1 entry viruses including HCV, HBV, dengue virus, and yellow fever virus (A. Vaillant, personal communication and manuscripts in preparation). Since these compounds act primarily as “gatekeeper” drugs they may be applied in combination with established inhibitors of later stages of arenavirus replication like ribavirin (Parker, 2005). Thus REP 9 and other APs represent promising new candidate anti-virals for combinatorial therapy to improve the anti-viral treatments available at present.
MATERIALS AND METHODS
Reagents
Alpha-DG was purified from human HEK293 cells as reported (Michele et al, 2002) and natural mouse laminin-1 was obtained from Invitrogen. Monoclonal antibodies (mAbs) 113 (anti-LCMVNP), 36.1 and 83.6 (anti-LCMVGP) as well as guinea pig hyperimmune serum against LCMV have been described (Buchmeier et al, 1981; Weber and Buchmeier, 1988). Rabbit polyclonal anti-laminin antibody and actin-Phalloidin were purchased from Sigma (St. Louis, MO). Fluorescein isothiocyanate (FITC)- and phycoerthrin (PE)-conjugated secondary antibodies were from Jackson Immuno-Research (West Grove, PA) and horseradish peroxidase (HRP)-conjugated secondary antibodies from Pierce (Rockford, IL). Heparin (average Mr 4,000 − 6, 000 Da) and dextrane sulfate (average Mr 10, 000 Da) were from Sigma. Oligonucleotides used in this study were synthesized as reported previously (Vaillant et al., 2006).
Cells and viruses
African green monkey kidney (VeroE6) cells were maintained in MEM (Gibco BRL, NY) containing 7% fetal bovine serum (FBS) and supplemented with glutamine and penicillin/streptomycin. HEK293 cells were maintained in DMEM (Gibco BRL, NY) containing 10% FBS and supplemented with non-essential amino acids, glutamine, and penicillin/streptomycin.
Origin, passage and characteristics of LCMV ARM53b, cl-13, WE54, E350, and Traub have been described elsewhere (Dutko and Oldstone, 1983; Ahmed et al., 1984; Teng et al., 1996; Smelt et al., 2001). Seed stocks of all LCMV isolates were prepared by growth in BHK-21 cells and titers determined as reported (Dutko and Oldstone, 1983). For virus purification, LCMV virions were precipitated with polyethylene glycol, and purified by ultracentrifugation on a renografin-gradient as described (Dutko and Oldstone, 1983). VSV containing a green fluorescent protein (GFP) reporter (rVSV-GFP) was provided by Dr. Juan-Carlos de la Torre (TSRI) and stocks grown in VeroE6 cells and titers determined by plaque assay on VeroE6 cells. Recombinant VSV pseudotyped with LCMV cl-13 GP (VSVΔG*-LCMVGP) was generated as reported (Kunz et al., 2005a). Virus titers were determined by infection of VeroE6 cell monolayers and detection of GFP positive cells by fluorescence microscopy.
Virus infection of cells
For infection of cells with LCMV isolates, VSVΔG*-LCMVGP, and rVSV-GFP, virus stocks were diluted to the indicated multiplicity of infection (MOI) and added to cells for 1 h at 37°C. After one hour incubation at the indicated temperature, inoculum was removed, cells washed twice with medium without serum, and incubated for the indicated time periods at 37 °C, 5% CO2. Infection was quantified by immunofluorescence detection of LCMVNP using mAbs 113 (anti-LCMVNP) combined with fluorescence-labeled secondary antibodies (Cao et al., 1998). Quantification of LCMV infection by flow cytometry was performed as reported previously (Rojek et al. 2007). Infection of VSVΔG*-LCMVGP and rVSV-GFP was assessed by detection of the GFP reporter by direct fluorescence.
For infection with retroviral pseudotypes, cells were plated in 96-well plates in a density of 104 cells/well. After 24 hours, retroviral pseudotypes were added at the indicated MOI and incubated for 1 hour at 37°C. Unbound viral particles were removed, cells washed twice with DMEM, and fresh medium added. Luciferase activity was determined after 48 hours by Steady-Glo® luciferase assay (Promega).
For blocking experiments with inactivated virus LCMV cl-13 and VSV were inactivated by UV irradiation (Rojek et al., 2007). VeroE6 cells cultured in 96 well plates (2 × 103 cells/well), were incubated with the indicated concentration of inactivated viruses in a total volume of 100 μl/well in 50% OPTIMEM/PBS for two hours on ice. Then, 200 PFU of VSVΔG*-LCMVGP or VSV were mixed with the same concentrations of inactivated viruses in a total volume of 100 μl OPTIMEM/PBS and the inoculum added to the cells. After 45 minutes, supernatants were removed, cells washed three times with medium and incubated for 16 and infection quantified by detection of GFP.
Blocking with PS-ONs, heparin, and dextran sulfate were performed as follows. Viruses were incubated with the indicated concentrations of drugs in complete medium containing 10 mM Hepes, pH 7.5 on ice for 45 minutes, followed by addition to cells for one hour. If not indicated otherwise, cells were washed three times with serum-free medium and infection assessed as described above.
For Western-blot analysis of virus produced in VeroE6 cells, 2 × 106 cells were plated in poly-L-lysine coated T75 tissue culture flasks and 24 hours later infected with LCMV ARM53b (MOI = 1). After 24 hours, 2 μM REP 9 were added. After 24 h, supernatnants were harvested and centrifuged at 1,200 rpm for 20 min at 4° C. Cleared supernatants were layered on a 20% sucrose-cushion (equal volume of 20% (wt/vol) sucrose in PBS) and subjected to ultracentrifugation in a SW41 rotor at 35, 000 rpm for 2 h at 4 °C. Supernatants were carefully removed and pellets were solubilized by boiling for 10 min in SDS-PAGE sample buffer.
Immunoblotting, laminin overlay assay, virus overlay assay
Standard immunoblotting involved proteins being separated by SDS-PAGE gel electrophoresis and transferred to nitrocellulose. After blocking in 5% (wt/vol) skim milk in PBS, membranes were incubated with the primary antibodies mAb 83.6 anti-LCMVGP2 (20 μg/ml purified IgG) and guinea pig hyperimmune serum against LCMV (1: 500) in 2% (wt/vol) skim milk, PBS overnight at 6 °C. After several washes in PBS, 0.1 % (wt/vol) Tween-20 (PBST), secondary antibodies coupled to HRP were applied 1: 5 ,000 in PBST for 1 h at room temperature. Blots were developed using Super Signal West Pico ECL Substrate (Pierce). Laminin overlay assay was performed as described in Michele et al. (2002) and virus overlay protein binding assay (VOPBA) as described in Cao et al. (1998). Solid-phase virus binding and laminin binding assays were performed as reported (Kunz et al., 2005b).
Detection of arenavirus GP incorporated into retroviral pseudotypes
For the detection of GP in LCMV virions and retroviral pseudotypes, viruses at 106 −107 PFU/ml in PBS were coated in triplicate wells in 96-well EIA/RIA high-bond microtiter plates (Corning) for 2 hours at 6 °C and non-specific binding blocked with 1% (wt/vol) BSA/PBS. For the detection of GP, mAbs 36.1 (anti-LCMVGP1) and 83.6 (anti-LCMVGP2) were applied at 20 μg/ml for 2 hours at 6 °C and detected with peroxidase-conjugated anti-mouse IgG (1: 1000) in a color reaction using ABTS (2,2’azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) substrate. OD (405) was measured with an ELISA reader. For the determination of specific binding, background binding to BSA was subtracted.
ACKNOWLEDGEMENTS
This is publication 18989 from the Molecular and Integrative Neurosciences Department (MIND) of the Scripps Research Institute. The authors thank Michael B.A. Oldstone (TSRI) for his generous support and insightful discussions. Dr. Juan-Carlos de la Torre (TSRI) and Dr. Michael Buchmeier (TSRI) are acknowledged for materials. This research was supported by US PHS grant AI55540 and grant 1U54 AI065359. A.T.G. performed part of this work in fulfillment of the requirements for her Master Thesis at the University of Oslo, Norway.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
REFERENCES
- Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med. 1984;160(2):521–40. doi: 10.1084/jem.160.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow P, Oldstone MB. Mechanism of lymphocytic choriomeningitis virus entry into cells. Virology. 1994;198(1):1–9. doi: 10.1006/viro.1994.1001. [DOI] [PubMed] [Google Scholar]
- Buchmeier MJ, Lewicki HA, Tomori O, Oldstone MB. Monoclonal antibodies to lymphocytic choriomeningitis and pichinde viruses: generation, characterization, and cross-reactivity with other arenaviruses. Virology. 1981;113(1):73–85. doi: 10.1016/0042-6822(81)90137-9. [DOI] [PubMed] [Google Scholar]
- Burns JW, Buchmeier MJ. Protein-protein interactions in lymphocytic choriomeningitis virus. Virology. 1991;183(2):620–9. doi: 10.1016/0042-6822(91)90991-j. [DOI] [PubMed] [Google Scholar]
- Cao W, Henry MD, Borrow P, Yamada H, Elder JH, Ravkov EV, Nichol ST, Compans RW, Campbell KP, Oldstone MB. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus [see comments]. Science. 1998;282(5396):2079–81. doi: 10.1126/science.282.5396.2079. [DOI] [PubMed] [Google Scholar]
- Dutko FJ, Oldstone MB. Genomic and biological variation among commonly used lymphocytic choriomeningitis virus strains. J Gen Virol. 1983;64(Pt 8):1689–98. doi: 10.1099/0022-1317-64-8-1689. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122(4):809–23. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschli B, Quirin K, Wepf A, Weber J, Zinkernagel R, Hengartner H. Identification of an N-terminal trimeric coiled-coil core within arenavirus glycoprotein 2 permits assignment to class I viral fusion proteins. J Virol. 2006;80(12):5897–907. doi: 10.1128/JVI.00008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Este JA, Cabrera C, Schols D, Cherepanov P, Gutierrez A, Witvrouw M, Pannecouque C, Debyser Z, Rando RF, Clotet B, Desmyter J, De Clercq E. Human immunodeficiency virus glycoprotein gp120 as the primary target for the antiviral action of AR177 (Zintevir). Mol Pharmacol. 1998;53(2):340–5. doi: 10.1124/mol.53.2.340. [DOI] [PubMed] [Google Scholar]
- Fischer SA, Graham MB, Kuehnert MJ, Kotton CN, Srinivasan A, Marty FM, Comer JA, Guarner J, Paddock CD, DeMeo DL, Shieh WJ, Erickson BR, Bandy U, DeMaria A, Jr., Davis JP, Delmonico FL, Pavlin B, Likos A, Vincent MJ, Sealy TK, Goldsmith CS, Jernigan DB, Rollin PE, Packard MM, Patel M, Rowland C, Helfand RF, Nichol ST, Fishman JA, Ksiazek T, Zaki SR. Transmission of lymphocytic choriomeningitis virus by organ transplantation. N Engl J Med. 2006;354(21):2235–49. doi: 10.1056/NEJMoa053240. [DOI] [PubMed] [Google Scholar]
- Frame JD, Verbrugge GP, Gill RG, Pinneo L. The use of Lassa fever convalescent plasma in Nigeria. Trans R Soc Trop Med Hyg. 1984;78(3):319–24. doi: 10.1016/0035-9203(84)90107-x. [DOI] [PubMed] [Google Scholar]
- Gallaher WR, DiSimone C, Buchmeier MJ. The viral transmembrane superfamily: possible divergence of Arenavirus and Filovirus glycoproteins from a common RNA virus ancestor. BMC Microbiol. 2001;1(1):1. doi: 10.1186/1471-2180-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbert TW, Jahrling PB. Exotic emerging viral diseases: progress and challenges. Nat Med. 2004;10(12 Suppl):S110–21. doi: 10.1038/nm1142. [DOI] [PubMed] [Google Scholar]
- Imperiali M, Thoma C, Pavoni E, Brancaccio A, Callewaert N, Oxenius A. O Mannosylation of alpha-dystroglycan is essential for lymphocytic choriomeningitis virus receptor function. J Virol. 2005;79(22):14297–308. doi: 10.1128/JVI.79.22.14297-14308.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahrling PB. Protection of Lassa virus-infected guinea pigs with Lassa-immune plasma of guinea pig, primate, and human origin. J Med Virol. 1983;12(2):93–102. doi: 10.1002/jmv.1890120203. [DOI] [PubMed] [Google Scholar]
- Jahrling PB, Frame JD, Rhoderick JB, Monson MH. Endemic Lassa fever in Liberia. IV. Selection of optimally effective plasma for treatment by passive immunization. Trans R Soc Trop Med Hyg. 1985;79(3):380–4. doi: 10.1016/0035-9203(85)90388-8. [DOI] [PubMed] [Google Scholar]
- Jahrling PB, Peters CJ. Passive antibody therapy of Lassa fever in cynomolgus monkeys: importance of neutralizing antibody and Lassa virus strain. Infect Immun. 1984;44(2):528–33. doi: 10.1128/iai.44.2.528-533.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahrling PB, Peters CJ, Stephen EL. Enhanced treatment of Lassa fever by immune plasma combined with ribavirin in cynomolgus monkeys. J Infect Dis. 1984;149(3):420–7. doi: 10.1093/infdis/149.3.420. [DOI] [PubMed] [Google Scholar]
- Jamieson DJ, Kourtis AP, Bell M, Rasmussen SA. Lymphocytic choriomeningitis virus: an emerging obstetric pathogen? Am J Obstet Gynecol. 2006;194(6):1532–6. doi: 10.1016/j.ajog.2005.11.040. Epub 2006 Apr 21. [DOI] [PubMed] [Google Scholar]
- Kim J, Cheong C, Moore PB. Tetramerization of an RNA oligonucleotide containing a GGGG sequence. Nature. 1991;351(6324):331–2. doi: 10.1038/351331a0. [DOI] [PubMed] [Google Scholar]
- Kunz S, Rojek J, Perez M, Spiropoulou C, MB O. Characterization of the interaction of Lassa fever virus with its cellular receptor α-dystroglycan. J. Virol. 2005a;79:5979–5987. doi: 10.1128/JVI.79.10.5979-5987.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz S, Rojek J, Spiropoulou C, Barresi R, Campbell KP, MB O. Post-translational modification of alpha-dystroglycan, the cellular receptor for arenaviruses by the glycosyltransferase LARGE is critical for virus binding. J. Virol. 2005b;79:14282–14296. doi: 10.1128/JVI.79.22.14282-14296.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz S, Sevilla N, McGavern DB, Campbell KP, Oldstone MB. Molecular analysis of the interaction of LCMV with its cellular receptor [alpha]-dystroglycan. J Cell Biol. 2001;155(2):301–10. doi: 10.1083/jcb.200104103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leifer E, Gocke DJ, Bourne H. Lassa fever, a new virus disease of man from West Africa. II. Report of a laboratory-acquired infection treated with plasma from a person recently recovered from the disease. Am J Trop Med Hyg. 1970;19(4):677–9. doi: 10.4269/ajtmh.1970.19.677. [DOI] [PubMed] [Google Scholar]
- Matsukura M, Shinozuka K, Zon G, Mitsuya H, Reitz M, Cohen JS, Broder S. Phosphorothioate analogs of oligodeoxynucleotides: inhibitors of replication and cytopathic effects of human immunodeficiency virus. Proc Natl Acad Sci U S A. 1987;84(21):7706–10. doi: 10.1073/pnas.84.21.7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick JB, Fisher-Hoch SP. Lassa fever. Curr Top Microbiol Immunol. 2002;262:75–109. doi: 10.1007/978-3-642-56029-3_4. [DOI] [PubMed] [Google Scholar]
- McCormick JB, King IJ, Webb PA, Scribner CL, Craven RB, Johnson KM, Elliott LH, Belmont-Williams R. Lassa fever. Effective therapy with ribavirin. N Engl J Med. 1986;314(1):20–6. doi: 10.1056/NEJM198601023140104. [DOI] [PubMed] [Google Scholar]
- Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, Dollar J, Nishino I, Kelley RI, Somer H, Straub V, Mathews KD, Moore SA, Campbell KP. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418(6896):417–22. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- Monath TP, Maher M, Casals J, Kissling RE, Cacciapuoti A. Lassa fever in the Eastern Province of Sierra Leone, 1970−1972. II. Clinical observations and virological studies on selected hospital cases. Am J Trop Med Hyg. 1974;23(6):1140–9. doi: 10.4269/ajtmh.1974.23.1140. [DOI] [PubMed] [Google Scholar]
- Oldstone MB. Biology and pathogenesis of lymphocytic choriomeningitis virus infection. Curr Top Microbiol Immunol. 2002;263:83–117. doi: 10.1007/978-3-642-56055-2_6. [DOI] [PubMed] [Google Scholar]
- Parekh BS, Buchmeier MJ. Proteins of lymphocytic choriomeningitis virus: antigenic topography of the viral glycoproteins. Virology. 1986;153(2):168–78. doi: 10.1016/0042-6822(86)90020-6. [DOI] [PubMed] [Google Scholar]
- Parker WB. Metabolism and antiviral activity of ribavirin. Virus Res. 2005;107(2):165–71. doi: 10.1016/j.virusres.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Perez M, Watanabe M, Whitt MA, de la Torre JC. N-terminal domain of Borna disease virus G (p56) protein is sufficient for virus receptor recognition and cell entry. J Virol. 2001;75(15):7078–85. doi: 10.1128/JVI.75.15.7078-7085.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojek JM, Spiropoulou CF, Campbell KP, Kunz S. Old World and Clade C New World Arenaviruses Mimic the Molecular Mechanism of Receptor Recognition Used by {alpha}-Dystroglycan's Host-Derived Ligands. J Virol. 2007;81(11):5685–95. doi: 10.1128/JVI.02574-06. Epub 2007 Mar 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojek JM, Spiropoulou CF, Kunz S. Characterization of the cellular receptors for the South American hemorrhagic fever viruses Junin, Guanarito, and Machupo. Virology. 2006;349(2):476–91. doi: 10.1016/j.virol.2006.02.033. Epub 2006 Mar 30. [DOI] [PubMed] [Google Scholar]
- Rose JK, Whitt MA. Rhabdoviridae: the viruses and their replication. In: Fields BN, Knipe DL, Howley PM, editors. Fields Virology. Lippincott-Raven; Philadelphia: 2001. pp. 1221–1244. [Google Scholar]
- Salvato M, Borrow P, Shimomaye E, Oldstone MB. Molecular basis of viral persistence: a single amino acid change in the glycoprotein of lymphocytic choriomeningitis virus is associated with suppression of the antiviral cytotoxic T-lymphocyte response and establishment of persistence. J Virol. 1991;65(4):1863–9. doi: 10.1128/jvi.65.4.1863-1869.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvato M, Shimomaye E, Southern P, Oldstone MB. Virus-lymphocyte interactions. IV. Molecular characterization of LCMV Armstrong (CTL+) small genomic segment and that of its variant, Clone 13 (CTL-). Virology. 1988;164(2):517–22. doi: 10.1016/0042-6822(88)90566-1. [DOI] [PubMed] [Google Scholar]
- Sevilla N, Kunz S, Holz A, Lewicki H, Homann D, Yamada H, Campbell KP, de La Torre JC, Oldstone MB. Immunosuppression and resultant viral persistence by specific viral targeting of dendritic cells. J Exp Med. 2000;192(9):1249–60. doi: 10.1084/jem.192.9.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smelt SC, Borrow P, Kunz S, Cao W, Tishon A, Lewicki H, Campbell KP, Oldstone MB. Differences in affinity of binding of lymphocytic choriomeningitis virus strains to the cellular receptor alpha-dystroglycan correlate with viral tropism and disease kinetics. J Virol. 2001;75(1):448–57. doi: 10.1128/JVI.75.1.448-457.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein CA, Benimetskaya L, Mani S. Antisense strategies for oncogene inactivation. Semin Oncol. 2005;32(6):563–72. doi: 10.1053/j.seminoncol.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Stein CA, Matsukura M, Subasinghe C, Broder S, Cohen JS. Phosphorothioate oligodeoxynucleotides are potent sequence nonspecific inhibitors of de novo infection by HIV. AIDS Res Hum Retroviruses. 1989;5(6):639–46. doi: 10.1089/aid.1989.5.639. [DOI] [PubMed] [Google Scholar]
- Stein CA, Neckers LM, Nair BC, Mumbauer S, Hoke G, Pal R. Phosphorothioate oligodeoxycytidine interferes with binding of HIV-1 gp120 to CD4. J Acquir Immune Defic Syndr. 1991;4(7):686–93. [PubMed] [Google Scholar]
- Teng MN, Borrow P, Oldstone MB, de la Torre JC. A single amino acid change in the glycoprotein of lymphocytic choriomeningitis virus is associated with the ability to cause growth hormone deficiency syndrome. J Virol. 1996;70(12):8438–43. doi: 10.1128/jvi.70.12.8438-8443.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillant A, Juteau JM, Lu H, Liu S, Lackman-Smith C, Ptak R, Jiang S. Phosphorothioate oligonucleotides inhibit human immunodeficiency virus type 1 fusion by blocking gp41 core formation. Antimicrob Agents Chemother. 2006;50(4):1393–401. doi: 10.1128/AAC.50.4.1393-1401.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber EL, Buchmeier MJ. Fine mapping of a peptide sequence containing an antigenic site conserved among arenaviruses. Virology. 1988;164(1):30–8. doi: 10.1016/0042-6822(88)90616-2. [DOI] [PubMed] [Google Scholar]
- Wright KE, Salvato MS, Buchmeier MJ. Neutralizing epitopes of lymphocytic choriomeningitis virus are conformational and require both glycosylation and disulfide bonds for expression. Virology. 1989;171(2):417–26. doi: 10.1016/0042-6822(89)90610-7. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Papp B, Zhang D, Ali AN, Agrawal S, Byrn RA. The multiple inhibitory mechanisms of GEM 91, a gag antisense phosphorothioate oligonucleotide, for human immunodeficiency virus type 1. AIDS Res Hum Retroviruses. 1997;13(7):545–54. doi: 10.1089/aid.1997.13.545. [DOI] [PubMed] [Google Scholar]
- Zhang C, Pei J, Kumar D, Sakabe I, Boudreau HE, Gokhale PC, Kasid UN. Antisense oligonucleotides: target validation and development of systemically delivered therapeutic nanoparticles. Methods Mol Biol. 2007;361:163–85. doi: 10.1385/1-59745-208-4:163. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.