

Abstract
Artemisinin the sesquiterpene endoperoxide lactone extracted from the herb Artemisia annua, remains the basis for the current preferred treatment against the malaria parasite Plasmodium falciparum. In addition, artemisinin and its derivatives show additional anti-parasite, anti-cancer, and anti-viral properties. Widespread use of this valuable secondary metabolite has been hampered by low production in vivo and high cost of chemical synthesis in vitro. Novel production methods are required to accommodate the ever-growing need for this important drug. Past work has focused on increasing production through traditional breeding approaches, with limited success, and on engineering cultured plants for high production in bioreactors. New research is focusing on heterologous expression systems for this unique biochemical pathway. Recently discovered genes, including a cytochrome P450 and its associated reductase, have been shown to catalyze multiple steps in the biochemical pathway leading to artemisinin. This has the potential to make a semi-synthetic approach to production both possible and cost effective. Artemisinin precursor production in engineered Saccharomyces cerevisiae is about two orders of magnitude higher than from field-grown A. annua. Efforts to increase flux through engineered pathways are on-going in both E. coli and S. cerevisiae through combinations of engineering precursor pathways and downstream optimization of gene expression. This review will compare older approaches to overproduction of this important drug, and then focus on the results from the newer approaches using heterologous expression systems and how they might meet the demands for treating malaria and other diseases.
Keywords: Amorpha diene, CYP71 AV1, terpenoid, genetic engineering, malaria
ARTEMISININ: EFFECTIVE AGAINST MALARIA AND OTHER DISEASES
In low income and developing nations, malaria is the fifth most prevalent infectious disease and the tenth overall cause of death and is projected to remain at that level until at least 2030 [1, 2]. The UN World Health Organization (WHO) estimates that more than 380 million cases of malaria occur each year and account for more than 1 million deaths. Furthermore, while not always fatal, the malaria burden has been estimated at more than 46 million disease adjusted life years (DALY) [3]. Though virtually eradicated in temperate climates and developed nations, developing nations still suffer a significant malaria burden.
Artemisinin and its more potent derivatives (Table 1) all contain an endoperoxide bridge that is required for therapeutic activity and these drugs provide an effective cure of even severe cerebral malaria. Unfortunately, treatment courses, while effective, remain costly and thus are often unavailable to the people that need them most. Current treatment centers on artemisinin combination therapy (ACT) in which artemisinin is used in conjunction with other drugs, or a variety of chloroquinone and sulfadoxine-pyramethamine combination courses. ACT is preferred because it has been shown to minimize the development of resistance by the parasite when compared to a single artemisinic drug [4]. Artemisinin has a relatively Short half-life thus requiring significant quantities in prescribed doses and thus there is a large and increasing need for this important compound. Although ACT remains the preferred treatment for high risk patients, its use is hampered by cost; it does, however, remain the most cost effective approach in terms of DALYs averted [4]. The cost of a single course of ACT treatment for uncomplicated malaria is approximately 10 times that of monotherapy using one of the more traditional drugs [5].
Table 1.
Potential Non-Malarial Therapeutic Uses of Artemisinin and its Derivatives
| Compound | Therapeutic target | Structure |
|---|---|---|
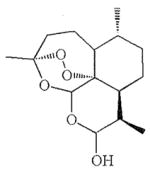
| Artemisinin | Toxoplasma gondii [8]; P. Carinii [11] |  |
| Dihydroartemisinin | Ovarian Cancer [18] Oral Cancer [13]; Antiangiogenesis [19] |
 |
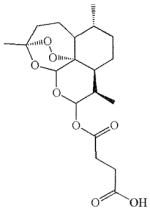
| Artesunate | Leukemia [22]; Uveal Melanoma [23] Prostate Cancer [97] Hepatitis B [7] Cytomegalovirus [6] |
 |
| Artemether | Schistosomiasis [9] | 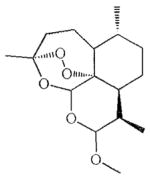 |
| Arteether | C. neoformans [98] | 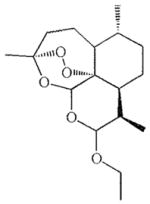 |
Besides malaria, artemisinin and its derivatives (Table 1) have been shown to be effective against a number of viruses including human cytomegaloviruses, herpes simplex, and hepatitis B and C [6, 7]. The drug is also effective against other prokaryotic and eukaryotic agents such as Toxoplasma gondii [8], Schistosoma [9, 10], and Pneumocystis carinii [11] (Table 1).
Artemisinin has further been shown to be useful in the treatment of some cancers (Table 1). Various artemisinin derivatives have been tested for their ability to induce apoptosis in cultured human cancer cell lines [12–14]. Efficacy apparently stems from the anti-angiogenesis activities of artemisinin along with its possible role in protein alkylation in high iron environments [15–19]. Cancer cells maintain a higher iron content than do non-cancerous cells, with the exception of erythrocytes, making this a viable working hypothesis for the specific activity of artemisinin. In addition, dihydroartemisinin has been shown to induce cell cycle arrest and apoptosis in certain cancer cell lines [20–22]. Limited clinical trials are in progress using artesunate in conjunction with standard chemotherapies as a treatment for uveal melanoma with some positive results [23]. Artemisinin and its derivatives therefore, may have wide applicability in the treatment of various cancers.
The primary cost of ACT comes from the high production costs of the artemisinin component relative to the primary effector in other treatments. The WHO estimates that the cost of ACT courses must be brought within the $ 0.10 threshold before widespread adoption and policy change will be feasible. Current levels have stabilized at between $0.50 and $1.00 per treatment course. These costs are due primarily to the necessity of harvesting large amounts of the annual plant, Artemisia annua, with relatively small yields of active compounds per acre [24]. In addition, the extraction and concentration of active components is both time and labor intensive and requires the use of a considerable volume of solvents and the associated infrastructure to carry out the procedures. Numerous in vivo semi-synthetic and synthetic production strategies, outlined here, are being pursued to significantly increase overall artemisinin production while reducing cost. While production of precursors to artemisinin in heterologous systems have been promising, one or more steps in the biosynthetic pathway have yet to be elucidated, thereby limiting production capability and presenting only a partial solution to the shortage of this very important drug. As these final enzymes or processes are identified, cost-effective treatments move closer to reality [25, 26]. Indeed if artemisinin is ever to be as broadly useful as its therapeutic power portends, then considerably greater amounts of this valuable drug must be produced. Here we review the current status of efforts, beyond some other recent reviews [25, 27, 28], to produce artemisinin, focusing mainly on transgenic approaches.
ARTEMISIA ANNUA: A PLANT WITH LOW LEVELS OF A VALUABLE DRUG
The annual herb, Artemisia annua L. Fig. (1), cultivated in much of Southeast Asia and which has featured prominently in traditional Chinese medicine since 168 BCE [21, 29–31] is the primary source of artemisinin. Artemisinin is localized to the trichomes, protuberances that are located on the leaves and flowers [32] and harvested from the shoots. The drug is present in exceedingly small amounts, usually < 1% of dry mass, thereby hindering sufficient production for treating malaria let alone any of the other diseases against which it is effective. Seasonal and cultivar specific variation in artemisinin content is also high and thus makes biotechnological approaches to production very attractive for maintaining a steady and reliable supply [33].
Fig. 1.

a) Greenhouse grown A. annua, b) hairy root culture, c) In vitro propagated shoot cultures.
ARTEMISINlN BIOSYNTHETIC PATHWAY
The artemisinin molecule is a sesquiterpene endoperoxide lactone derived from the common sesquiterpene precursor, farnesyl diphosphate (FPP), which itself is formed by the condensation of three isopentenyl diphosphate (IPP) molecules synthesized by either the mevalonic acid dependent (MVA) or independent (MEP) pathways present in the cytosol and plastid, respectively [34–36] Fig. (2). We recently showed that similar to other plants within the Asteraceae, the IPP used in the biosynthesis of FPP leading to artemisinin originates from both the MVA and the MEP pathways [37]. FPP is then converted by the artemisinin specific sesquiterpene cyclase, amorpha diene synthase (ADS), to yield amorpha-4, 11-diene (amorpha diene) Fig. (3) [38–40]. Following this reaction, a cytochrome P450, CYP71AV1, catalyzes multiple steps yielding artemisinic acid in vitro [41, 42]. Beyond this first P450 step, many possible intermediates have been identified through metabolic fingerprinting of A. annua, and indeed the primary biosynthetic pathway is not fully known [39,43]. For example, earlier Nair and Basile (44) proposed that artemisinic acid was a precursor based on the observation of its conversion in cell free extracts to arteannuin B and subsequently to artemisinin. However, Wallaart et al. [45] and Sy and Brown [46] both showed evidence that the primary route is through dihydroartemisinic acid, suggesting that the route through artemisinic acid is merely a side pathway. This would further suggest that there is a possible competition between the identified cytochrome P450 and a double bond reductase that would create the balance between artemisinic acid and dihydroartemisinic acid Fig. (3).
Fig. 2.

Isoprenoids synthesized by the cytosolic MVA pathway and the plastidal MEP pathway lead to diverse metabolites.
Fig. 3.

Artemisinin biosynthetic pathway and potential branching points.
This pre-artemisinic acid split is bolstered by the work of Brown and Sy [47] who fed 13C labeled artemisinic acid to intact plantlets through cut shoots and observed no conversion to artemisinin. However, a large enrichment of arteannuin B was observed suggesting this may be the end product in the artemisinic acid pathway and not artemisinin as had been previously hypothesized [48]. Additionally, Brown and Sy saw no conversion to dihydroartemisinic acid which suggested that the chemotypes proposed by Wallaart et al. [33] are in fact differences in the balance between these two pathways. Although recent work by Zhang et al. [49] has shown the existence of a double bond reductase (DBR 2, Fig. 3) seemingly capable of the conversion of artemisinic aldehyde to dihydroartemisinic aldehyde, it is still not clear at which point the reduction to the 11,13-dihydro metabolites occurs. This would suggest the presence of a final aldehyde dehydrogenase enzyme capable of the final conversion to dihydroartemisinic acid. It should also be noted that while this split in the pathway between the reduced and non-reduced forms likely exists in planta, the concentrations of certain precursors, notably artemisinic acid and dihydroartemisinic acid, are often an order of magnitude higher than that of artemisinin itself and seem to vary depending on environmental conditions, genetic, or epigenetic factors [33, 43].
Others later offered evidence suggesting that certain late steps in the pathway may be non-enzymatic and might instead be spontaneous photo-oxidative reactions Fig. (3). For example, Wallaart et al. [45] demonstrated that dihydroartemisinic acid incubated in the presence of chlorophyll a was slowly converted to artemisinin. They further proposed that the reaction was a combination of photo- and air oxidation reactions that served to limit reactive oxygen species (ROS) through sequestration. This hypothesis was supported by Wallaart et al. [50] who observed that the cDNA for ADS could not be amplified using RT-PCR unless tissues were drought and photo stressed prior to harvest, thereby increasing levels of ROS. Contrary to these results, the Covello lab and ours have been readily able to isolate, from unstressed greenhouse and in vitro cultured plants, full length cDNAs of ADS using RT-PCR [41] (Unpublished results). This may, however, be due to a difference in chemotypes, the result of selective breeding of high yielding cultivars, or plants that no longer induce ADS in response to ROS. Brown and Sy [51] reported that feeding isotope-labeled dihydroartemisinic acid to intact A. annua plants via the roots resulted in labeled sesquiterpene metabolites present in shoot tissue. However, they observed no enrichment of labeled artemisinin suggesting that artemisinin is not produced from dihydroartemisinic acid. However, they also observed that a variety of other labeled sesquiterpenes were present, though, interestingly, no conversion to artemisininc acid was observed. Included in these metabolites was a carboxyenol derivative of dihydroartemisinic acid that when incubated in chloroform will convert spontaneously to artemisinin. Brown and Sy [51] hypothesized that this conversion may only happen in a lipophillic environment capable of stabilizing the hydroxyl groups and that in planta this could explain the observed enrichment of artemisinin in the lipophillic glandular trichomes and would offer the most complete picture of artemisinin biosynthesis.
Although artemisinic acid lacks the key endoperoxide bridge that has been shown to be important for anti-plasmodial action in vivo, semi-synthetic processing can produce either artemisinin or a derivative that contains this important moiety [52]. Unfortunately the semi-synthetic processing of artemisinin precursors leads to environmental and economic costs that contribute heavily to the cost per treatment for ACT [42]. The semi-synthetic approach may never be entirely avoided, however, because some artemisinin derivatives are more efficacious than their parent molecules [14]. Notably compared to artemisinin, dihydroartemisinin has been shown to have elevated anti-plasmodial activity, and artesunate is more effective against certain forms of cancer (Table 1) [13, 14].
PLASTID VS. CYTOSOL AND COMPARTMENTALIZATION OF ARTEMISININ BIOSYNTHESIS
To further complicate artemisinin biosynthesis, IPP is a product of two distinct biochemical pathways located in two separate cellular compartments Fig. (2). These compartments have been shown to give rise to discrete subsets of terpenoids often with some evidence of IPP flow or cross-talk, between plastid and cytosol [34, 53}. Additionally, the MVA pathway leading to IPP also plays a role in sterol biosynthesis. A major regulatory step in this pathway is at the enzyme HMG-CoA Reductase (HMGR). Molecular studies and sequence analyses have shown that plant HMGRs show high levels of similarity between species [54] and exist in small gene families ranging from 2 to > 12 isoforms in, for example, A. thaliana and potato, respectively [55] These isoforms exhibit complex developmental and environmental regulation for altering carbon shunting towards sterols. For example, in tobacco transformed with hmg1 from Arabidopsis, phytosterols were increased in seeds, but not in leaves [56]. This carbon shunting can be an advantage for increasing production levels, although the necessity of carefully balancing production of other products and the after effects of reduced carbon flux elsewhere must be considered. Although there is no significant evidence for “cross-talk” in A. annua, it can be either limited or extensive in other higher plants including Arabidopsis where cross-talk seems bi-directional and extensive [57] or as in carrot and snapdragon where cross-talk is uni-directional with plastidal IPP from the MEP pathway supporting the production of both plastidal monoterpenes and cytosolic sesquiterpenes [35, 58].
STRATEGIES FOR INCREASING PRODUCTION OF ARTEMISININ
At least three different approaches have been used to increase production of artemisinin: nontransgenic, transgenic A. annua, and heterologous transgenic systems. Nontransgenic approaches include selective breeding, alteration of nutrient and environmental conditions, use of in vitro cultures, and exploitation of a plant’s natural defense system through elicitation. Transgenic A. annua plants have also been produced using a variety of different genes. The most recent approach for increasing artemisinin is via heterologous systems, insertion of key artemisinin biosynthetic genes into organisms other than A. annua.
Nontransgenic Efforts
Much of the nontransgenic work to increase artemisinin content in A. annua has focused primarily on selective breeding and in vitro cell culture approaches including the formation and testing of calli, suspension cells, hairy root and shoot cultures [59–61]. This section will focus mainly on the in vitro work.
With the exception of certain hairy root cultures and recent work with elicited suspension cells, very little, if any, artemisinin has been observed using these in vitro approaches. Interestingly, although A. annua shoot cultures produce small amounts of artemisinin, rooted shoots produce considerably more [62]. Indeed we have confirmed that rooted shoots produce almost 10 fold more artemisinin than unrooted shoots (Weathers, unpublished data). Other attempts at optimizing culture conditions have been marginally successful at inducing higher levels of artemisinin, usually focusing on effects of added plant hormones [63, 64], chitosan [65], nutrient cues [66], light [67, 68], or other abiotic elicitors [59, 69]. Most attempts at stimulating artemisinin production, however, have not been encouraging. For example, although glucose was shown to increase artemisinin production compared to sucrose, yields were at best doubled at the studies used 2–3 week old seedlings [60, 70]. In another case, addition of chitosan to the medium increased yields in A. annua hairy roots 6 fold, but the final concentration was only 1.8 mg/g DW [65].
There are clearly numerous points of control for artemisinin. One control point occurs after IPP condensation at the decision to synthesize sterols or sesquiterpenes. These two pathways are under coordinate control as demonstrated in both yeast and plants through either genetic engineering or inhibition of specific enzymes [42, 71]. For example, when miconazole, an inhibitor of squalene synthase, was fed to A. annua shoots [64, 72] and seedlings [37] artemisinin concentrations rose significantly compared to uninhibited controls. These studies indicate that inducible shunting of carbon may be a reasonable approach for engineering increased levels of artemisinin in planta.
Production of Artemisinin via Transgenic A. annua
Early attempts at using transgenic approaches for increasing artemisinin levels in A. annua have included the insertion of genes affecting flowering [73, 74], phytohormone levels [75], and farnesyl diphosphate synthesis [76]. Since artemisinin levels have been reported as highest during flowering, genes stimulating flowering were transferred into A. annua. The flowering promoting factor 1 gene (fpf1) and CONSTANS (CO), an early flowering gene, both from Arabidopsis, were transferred into A. annua using Agrobacterium tumefaciens. Although fpf1 plants flowered 20 d earlier than nontransgenic plants, no significant difference in artemisinin levels was observed upon flowering [73]. Similar results were observed for CO transformed plants; flowering occurred earlier than in the controls with no change in artemisinin concentration leading Wang et al. [74] to conclude that there appears to be no direct link between artemisinin production and flowering.
Artemisinin is produced in the shoots of A. annua so Sa et al. [75] considered that increasing cytokinin, the phytohormone that stimulates shoot production, may also increase artemisinin productivity. After using A. tumefaciens containing the isopentenyl transferase gene (ipt) to transform A. annua plants, they observed that some of the transformed lines constitutively produced 2–3 times more cytokinins than nontransgenic plants. Although chlorophyll and artemisinin also increased as much as 60 and 70%, respectively, above controls, these levels were still disappointingly low.
FPP is the starting molecule for the committed step in sesquiterpene biosynthesis. Chen et al. [77] hypothesized that increasing FPP by over expression of FPS in A. annua should result in enhanced yields of artemisinin. They first transformed A. annua with an A. rhizogenes containing a Gossypium (cotton) FPS cDNA construct to obtain hairy roots that produced 2–3 mg/g DW artemisinin, 3–4 times greater than controls. Later Chen et al. [76] transfected A. annua plants with A. tumefaciens containing the same FPS gene and obtained five lines showing artemisinin levels at 8–10 mg/g DW which were 2–3 times greater than controls. These results would suggest, to some degree, that FPP synthesis is a rate-limiting step in artemisinin biosynthesis. This result, however, contradicts metabolic profiles showing dihydroartemisinic acid in very high quantities suggesting a bottleneck instead at that much later step [43]. Unfortunately long term productivity of many of these more successful transformations has not, to our knowledge, been reported and little is known about the clearly complex mechanisms of control.
Heterologous Production of Artemisinin
Because in planta production of artemisinin has thus far been somewhat disappointing, heterologous systems have been identified as potentially more productive platforms for pathway engineering. A number of terpenoid and plant secondary metabolite pathways have been engineered into a variety of hosts including, E. coli, Saccharomyces cerevisiae, and other microbial organisms [71, 78, 79]. In addition, several plant species have been used to heterologously express enzymes from a variety of sources, including tomato, potato, and tobacco [80–82]. For example, various carotenoid pigments have been engineered in novel systems for increased nutritional benefit Most notably, genes encoding the β-carotene biosynthetic pathway have been introduced into rice endosperm [83]. Production of valuable compounds not intended for direct human consumption, like artemisinin or artemisinic acid, can be made using microbial systems or nonfood crops.
Two enzymes specific to artemisinin biosynthesis, ADS and CYP71AV1, have also been targeted for heterologous expression and genetic engineering mainly into microbes (Table 2) [41, 42, 84, 85]. Unfortunately, the genes encoding the catalytic steps beyond CYP7lAV1 have not yet been identified. Most of these efforts have been focused in the labs of Jay Keasling (University of California, Berkeley, CA) and Patrick Covello (plant Biotechnology Institute, Saskatoon, Canada).
Table 2.
Transgenic Approaches Used for Amorpha 4,11- Diene Production
| Species | Heterologous target | Yield |
|---|---|---|
| N. tabacum | Cytosolic Avian FPS A. annua ADS [79] | 0.3–1 ng/g FW |
| Plastidal Avian FPS. A. annua ADS [79] | 10–25 ug/g FW | |
| E. coli | 1. Codon optimized ADS [85] | 0.086 g/L/OD600 at 10h. |
| 2. As in “1” + SOE4 operon of DXS pathway [85] | 0.3133 g/L OD600 at 5h. | |
| 3. As in “1” With engineered mevalonate pathway and 40mM mevalonate [85] | 3.1 mg/L/OD600 at 9h.a | |
| 4. As in “3” with addition of 0.8% glycerol [85] | 24 mg/L/OD600 at 12h.b | |
| 5. As in “3” using dodecane trap bioreactor (6L) and additional carbon [82] | >450 mg/L at 60h. (OD600 approx. 25) | |
| S. cerevisiae | 1. Plasmid encoded ADS gene alone [86] | 0.6 mg/L at 384h. |
| 2. As in “1” with plasmid encoded ADS under inducible promoter [42] | 4.4 mg/L at 144h | |
| 3. As in “2” with truncated soluble tHMGR [42] | 20 mg/L at 144h. | |
| 4. As in “3” with downregulation of squalene synthase (erg9) [42] | 40 mg/L at 144h | |
| 5. As in “4” with additional chromosomal tHMGR, upregulation of transcription factor upc2-1 and overexpression of FPS [42] | 153 mg/L at 144h. |
Estimated as high as 22.6 mg/L when accounting for effect, of air-stripping
Estimated as high as 112.2 mg/L when accounting for effects of air-stripping
Promising work done with carotenoids has shown that E coli can potentially be a viable platform for isoprenoid biosynthesis [86]. Martin et al. [87] engineered a mevalonate pathway for IPP production from yeast into E coli in anticipation of creating a microbial factory for terpenoids (Table 2) [87]. This group made an important improvement in codon optimization of the ADS gene co-expressed with an engineered MVA pathway. Their efforts yielded a 10–36 fold improvement in amorpha diene production compared to wild type ADS mRNA sequences; maximum yield was approximately 112.2 mg/L when 0.8% glycerol was added as an additional feedstock [87]. Amorpha diene is volatile, so they further optimized its extraction by using a two phase bioreactor incorporating a dodecane layer to trap it. Further improvements were seen altering culture conditions to allow production of compounds well into stationary phase [84]. Use of dodecane trapping and media supplementation subsequently resulted in total yields of more than 450 mg/L after a 60 h fermentation in a 6 L bioreactor (Table 2) [84]. These are the highest reported levels to date and represent at least a 4-fold improvement over previous work [84]. ADS has also been a target for expression in S. cerevisiae. The work done by the Brodelius group [88] showed that a plasmid encoded ADS gene yielded 0.6mg/L of amorpha diene over a 16 day fermentation. Further work by the Keasling group has shown that by mutating a squalene synthese, introduction of a truncated HMGR reductase, and overexpressing a transcription factor controlling sterol biosynthesis, they were able to obtain yields of amorpha diene on the order of 153 mg/L (Table 2) [42]. While this is not as high as the levels obtained in optimized E. coli bioreactors, the ability to engineer bacteria to produce terpenoids has been limited due to difficulty in expressing various plant cytochrome P450s for isoprene anabolism.
Further work has been done to express the final known gene in the pathway, CYF71AV1, in S. cerevisiae in combination with FPS and ADS genes as described above. This work focused on engineering both a precursor pathway optimized for FPP production and also down regulating sterol biosynthesis through alteration of the erg9 gene of yeast in an attempt to shunt carbon towards the engineered pathway [42,71]. The genes for ADS and also CYP71AVI with its associated NADPH oxido-reductase were all introduced into an engineered yeast strain. The products obtained were artemisinic alcohol, artemisinic aldehyde, and most prominently artemisinic acid [41]. Additional work, using an E. coli strain with a myriad of optimizations including codon usage, N-terminal engineering, and isolation improvements yielded a strain able to produce 105 mg/L of artemisinic acid compared to 115 mg/L in the engineered yeast strain from the same group [42, 78]. Together these approaches hold great promise; the synthetic conversion of artemisinic acid to artemisinin and other derivatives is well understood and production, with scale up, has the potential to be far less expensive than harvesting field-grown A. annua and subsequent extraction and purification of artemisinin. Even after including the costs of carbon feed stocks and the capital costs of large scale fermenters, this microbial approach may yield more cost-effective artemisinin than current field-based production methods.
The strength of these microbial approaches lies in the yield of precursor obtained per day of growth. However, maximizing expression of these genes has been slow and metabolic flux analysis is on-going [89]. Given the high amounts of artemisinic acid present in planta relative to downstream products, it is possible that the steps directly preceding its production may represent a bottleneck in biosynthesis [49]. Accurate analytical standards for many of the precursors to artemisinin have been difficult to produce and are not commercially available, thereby making large scale flux analysis in either heterologous or natural systems difficult at best. Further work with heterologous systems will likely focus on carbon flux through the pathways and how that flux is controlled. Is flux regulated mainly by differences in enzymatic efficiency, by pre- or post-translational controls, or by some yet to be described mechanism? Eventually these microorganisms will likely be engineered for high level production that would far exceed the natural production potential of A. annua, though the final steps in the pathway for complete biosynthetic production of artemisinin must still be identified for that to occur.
Currently all artemisinin is obtained from field-grown plants and production relies heavily on a semi-synthetic approach using harvested artemisinin precursors, costly organic solvents and production facilities. If additional steps in biosynthesis can be performed in a higher yield in genetically engineered systems than in field-grown plants, then the overall cost per treatment course should similarly decrease. Efforts at engineering other terpenoid products have shown that a variety of strategies from engineered control loops, in vitro evolution, and combinatorial biosynthesis have been able to increase yield of desired or novel terpenoid products in engineered systems [90, 91]. Increased yield per liter of culture would also serve to decrease costs through the elimination of costly feed stocks for growing cultures.
Recompartmentalizing Artemisinin Biosynthesis
Engineering of higher plants for small molecule production has also yielded some promising results. Specifically efforts at engineering a tobacco plant to produce amorpha diene yielded some surprising data. A construct containing the genes for both an avian farnesyl diphosphate synthase and the gene for ADS was introduced through Agrobacterium-mediated transformation into tobacco [81]. Each gene was also introduced with a transit peptide to mark the enzymes for transport into chloroplasts. This shifted normally cytosolic biosynthetic steps into a different cell compartment, the plastid, where the enzymes are not normally found. Results showed that levels of sesquiterpene production were markedly higher in transgenic plants with the plastid targeted proteins. Labeled carbon was used to further show that pools of IPP were relatively isolated in the production of amorpha diene and that very little “cross-talk” was occurring during biosynthesis. Furthermore, similar results were obtained for plants engineered using the same approach but to produce a different sesquiterpene, patchoulol. The enhanced yields for both products suggested that the effects of the subcellular compartmental shift were not isolated to one particular set of enzymes. Indeed it appeared that the plant “lost control” of a normally cytosolic enzyme once it was moved into the plastid. Also, through the use of a mutant background with a truncated HMGR that accumulated tenfold higher levels of sterols, they Saw as much as a 13 fold increase in production of patcholul over wild type transformed plants [81]. Together these results suggest that pathways may be engineered for high level production in plants in a similar fashion to microbial systems. Transformed tobacco plants expressing amorpha diene synthase targeted to plastids yielded approximately 10–25 μg/g FW of amorpha diene compared to the 0.1–1 ng/g FW observed with non-targeted enzymes (Table 2) [81].
The reason behind the observed 4–5 orders of magnitude increase in sesquiterpene production when genes are targeted to alternative compartments is not entirely understood but does hold promise for future engineering of metabolic pathways. This approach, bolstered by careful control of metabolic flux through regulation of other genes, may yield a plethora of biosynthetic factories not constrained by the need for costly equipment, media, or feed stock as required by engineered microbial systems [91, 92]. While not suffering from the high initial capital costs of microbial systems, it remains to be seen if productivity (mg/g DW), or absolute production (mg/g DW/ha) levels in plants can be cost-effective. The high producing tobacco lines produced by Wu et el. [81] still produced only 25 μg/g FW of amorpha diene, far less than its engineered microbial counterparts. However, FPS transgenic A. annua have yields of artemisinin of 8–10 mg/gDW. Furthermore, transformed hairy root clones of A annua have been able to produce as much as 1.8 mg/g DW in bioreactor set-ups [76, 65]. The possibility of introducing other genes with far less pre-engineering and optimization than required with the microbial systems makes heterologous plants systems still an attractive platform and one worthy of further study.
Current work in artemisinin production focuses both on elucidating the last steps in the biosynthesis of artemisinin and its precursors, and finding those final genes. Although the yields of artemisinin precursors from engineered yeast are promising, [41, 42], the problem of semi-synthesis is still an important economic consideration. Artemisinic acid lacks the endoperoxide bridge, the key feature needed to make artemisinin and its derivatives therapeutically effective. Artemisinic acid produced by yeast cultures still must be extracted and then synthetically modified to produce a useful form of the drug and this also results in increased cost.
NON BIOLOGICAL PROSPECTS
To be therapeutically active, artemisinin requires both the endo-peroxide bridge and the tri-oxane ring. These are cleaved in an iron-dependant Fenton reaction to yield an active molecule. There are two competing mechanisms for artemisinin’s therapeutic activity. Artemisinin has been shown to inactivate a SERCA protein (sarco/endoplasmic reticulum Ca2+-ATPase) which is eventually fatal to the parasite. Other data suggest artemisinin inhibits haemozoin formation as part of the mechanism for limiting the effects of reactive oxygen species [93, 94]. In support of the first hypothesized mechanism, homology modeling has shown that artemisinin may bind similar to thapsigargin, another sesquiterpene lactone that has been shown to bind and inhibit SERCA proteins. Single point mutations in the SERCA thapsigargin binding cleft also confer resistance to artemisinin as predicted. These results suggested that the two molecules act similarly to inhibit SERCA activity and show the importance of the sesquiterpene backbone for proper docking of active molecules [95, 96]. It is still not entirely clear, however, by which mechanism artemisinin acts in vivo.
Currently, work is being done on cheaper synthetic tri-oxane molecules that may be able to replicate the effects of artemisinin on Plasmodium [97]. Although combinations of chloroquinine-like motifs with moieties from artemisinin have shown some positive results, most results have shown only limited efficacy of non-artemisinin derived tri-oxanes [98]. This less than desirable response may be due to the lack of a sesquiterpene backbone known to be an important factor in the efficacy of artemisinin [93]. Transport of artemisinin intra-cellularly also affects Plasmodium and it is unlikely that the significantly smaller tri-oxane derivatives will be able to match that property in vivo [93]. Despite some of the advances in developing these synthetic trioxane drugs, it appears that biologically produced artemisinin still offers significant therapeutic advantages over use of synthetic versions of the drug.
CONCLUSIONS AND NEW DIRECTIONS
Thus far, work in heterologous systems has made some progress in the goal to produce enough artemisinin to meet world demand. Microbial systems offer good prospects, but process optimization along with more precise engineering of metabolic pathways are needed in order to maximize artemisinin yields. A complete understanding of all the steps in the biosynthetic pathway will provide new opportunities as well as challenges in metabolically engineering the production of this important drug. It remains to be seen, however, if this will be a truly cost-effective strategy. Although transgenic A. annua systems have also shown some recent progress, they are still less productive than microbial counterparts. Production in heterologous plant systems is in its infancy, and yields are still far below that of microbial systems and transgenic A. annua. Further efforts are needed to optimize their production potential. Improvements in synthetic derivatives may yield not only less expensive production methodologies but new molecules with greater variety of potential applications; however, it is not clear they will provide the same therapeutic benefit as the natural products they are intended to replace.
Acknowledgments
The authors thank Dr. Keat H. (Thomas) Teoh, Arkansas Bioscience Institute (ABI), for a critical review of the manuscript. Funding from WPI, and NIH 2R15GM069562-02 provided partial support to P. Arsenault. Support was also provided in part to P. Arsenault by the Arkansas Biosciences Institute, the major research component of the Arkansas Tobacco Settlement Proceeds Act of 2000.
References
- 1.Nahlen BL, Korenromp EL, Miller JM, Shibuya K. Nature. 2005;437:E3. doi: 10.1038/nature04178. discussion E4–5. [DOI] [PubMed] [Google Scholar]
- 2.Mathers CD, Loncar D. PLoS Med. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathers CD, Ezzati M, Lopez AD. PLoS Neglected Trop Dis. 2007;1:e114. doi: 10.1371/journal.pntd.0000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhattarai A, Ali AS, Kachur SP, Martensson A, Abbas AK, Khatib R, Al-Mafazy AW, Ramsan M, Rotllant G, Gerstenmaier JF, Molteni F, Abdulla S, Montgomery SM, Kaneko A, Bjorkman A. PLoS Med. 2007;4:e309. doi: 10.1371/journal.pmed.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiseman V, Kim M, Mutabingwa TK, Whitty CJ. PLoS Med. 2006;3:e373. doi: 10.1371/journal.pmed.0030373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Efferth T, Marschall M, Wang X, Huong SM, Hauber I, Olbrich A, Kronschnabl M, Stamminger T, Huang ES. J Mol Med (Berlin, Germany) 2002;80:233–242. doi: 10.1007/s00109-001-0300-8. [DOI] [PubMed] [Google Scholar]
- 7.Romero MR, Efferth T, Serrano MA, Castano B, Macias RI, Briz O, Marin JJ. Antiviral Res. 2005;68:75–83. doi: 10.1016/j.antiviral.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Jones-Brando L, D’Angelo J, Posner GH, Yolken R. Antimicrobial Agents Chemother. 2006;50:4206–4208. doi: 10.1128/AAC.00793-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Utzinger J, Xiao S, Keiser J, Chen M, Zheng J, Tanner M. Curr Med Chem. 2001;8:1841–1860. doi: 10.2174/0929867013371581. [DOI] [PubMed] [Google Scholar]
- 10.Utzinger J, Xiao S, N’Goran EK, Bergquist R, Tanner M. Int J Parasitol. 2001;31:1549–1562. doi: 10.1016/s0020-7519(01)00297-1. [DOI] [PubMed] [Google Scholar]
- 11.Merali S, Meshnick SR. Antimicrobial Agents Chemother. 1991;35:1225–1227. doi: 10.1128/aac.35.6.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh NP, Lai HC. Anticancer Res. 2004;24(4):2277–2280. [PubMed] [Google Scholar]
- 13.Nam W, Tak J, Ryu JK, Jung M, Yook JI, Kim HJ, Cha IH. Head Neck. 2007;29:335–340. doi: 10.1002/hed.20524. [DOI] [PubMed] [Google Scholar]
- 14.de Vries PJ, Dien TK. Drugs. 1996;52:818–836. doi: 10.2165/00003495-199652060-00004. [DOI] [PubMed] [Google Scholar]
- 15.Chen HH, Zhou HJ, Fang X. Pharmacol Res. 2003;48:231–236. doi: 10.1016/s1043-6618(03)00107-5. [DOI] [PubMed] [Google Scholar]
- 16.Wu XH, Zhou HJ, Lee J. Anti-Cancer Drugs. 2006;17:839–848. doi: 10.1097/01.cad.0000224443.85834.32. [DOI] [PubMed] [Google Scholar]
- 17.Yang YZ, Little B, Meshnick SR. Biochem Pharmacol. 1994;48:569–573. doi: 10.1016/0006-2952(94)90287-9. [DOI] [PubMed] [Google Scholar]
- 18.Messori L, Gabbiani C, Casini A, Siragusa M, Vincieri FF, Bilia AR. Bioorg Med Chem. 2006;14:2972–2977. doi: 10.1016/j.bmc.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 19.D’Alessandro S, Gelati M, Basilico N, Parati EA, Haynes RK, Taramelli D. Toxicology. 2007;241:66–74. doi: 10.1016/j.tox.2007.08.084. [DOI] [PubMed] [Google Scholar]
- 20.Jiao Y, Ge CM, Meng QH, Cao JP, Tong J, Fan SJ. Acta Pharmacol Sinica. 2007;28:1045–1056. doi: 10.1111/j.1745-7254.2007.00612.x. [DOI] [PubMed] [Google Scholar]
- 21.Efferth T. Curr Drug Targets. 2006;7:407–421. doi: 10.2174/138945006776359412. [DOI] [PubMed] [Google Scholar]
- 22.Efferth T. Planta Med. 2007;73:299–309. doi: 10.1055/s-2007-967138. [DOI] [PubMed] [Google Scholar]
- 23.Berger TG, Dieckmann D, Efferth T, Schultz ES, Funk JO, Baur A, Schuler G. Oncol Rep. 2005;14:1599–1603. [PubMed] [Google Scholar]
- 24.Kindermans JM, Pilloy J, Olliaro P, Gomes M. Malaria J. 2007;6:125. doi: 10.1186/1475-2875-6-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu C, Zhao Y, Wang Y. Appl Microbiol Biotechnol. 2006;72:11–20. doi: 10.1007/s00253-006-0452-0. [DOI] [PubMed] [Google Scholar]
- 26.Zeng Q, Qiu F, Yuan L. Biotechnol Lett. 2008;30(4):581–592. doi: 10.1007/s10529-007-9596-y. [DOI] [PubMed] [Google Scholar]
- 27.Covello PS, Teoh KH, Polichuk DR, Reed DW, Nowak G. Phytochemistry. 2007;68:1864–1871. doi: 10.1016/j.phytochem.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 28.Weathers PJ, Elkholy S, Wobbe KK. In Vitro Cell Dev Biol-Plant. 2006;42:309–317. [Google Scholar]
- 29.Hsu E. Transact Royal Soc Tropic Med Hyg. 2006;100:505–508. doi: 10.1016/j.trstmh.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 30.Acton N, Klayman DL. Planta Med. 1985;51:441–442. doi: 10.1055/s-2007-969543. [DOI] [PubMed] [Google Scholar]
- 31.Klayman DL. Sci (New York, NY) 1985;228:1049–1055. doi: 10.1126/science.3887571. [DOI] [PubMed] [Google Scholar]
- 32.Lommen WJ, Schenk E, Bouwmeester HJ, Verstappen FW. Planta Med. 2006;72:336–345. doi: 10.1055/s-2005-916202. [DOI] [PubMed] [Google Scholar]
- 33.Wallaart TE, Pras N, Beekman AC, Quax WJ. Planta Med. 2000;66:57–62. doi: 10.1055/s-2000-11115. [DOI] [PubMed] [Google Scholar]
- 34.Newman JD, Chappell J. Crit Rev Biochem Mol Biol. 1999;34:95–106. doi: 10.1080/10409239991209228. [DOI] [PubMed] [Google Scholar]
- 35.Dudareva N, Andersson S, Orlova I, Gatto N, Reichelt M, Rhodes D, Boland W, Gershenzon J. Proc Natl Acad Sci USA. 2005;102:933–938. doi: 10.1073/pnas.0407360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thai L, Rush JS, Maul JE, Devarenne T, Rodgers DL, Chappell J, Waeehter CJ. Proc Natl Acad Sci USA. 1999;96:13080–13085. doi: 10.1073/pnas.96.23.13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Towler MJ, Weathers PJ. Plant Cell Reports. 2007;26:2129–2136. doi: 10.1007/s00299-007-0420-x. [DOI] [PubMed] [Google Scholar]
- 38.Bouwmeester HJ, Wallaart TE, Janssen MH, van Loo B, Jansen BJ, Posthumus MA, Schmidt CO, De Kraker JW, Konig WA, Franssen MC. Phytochemistry. 1999;52:843–854. doi: 10.1016/s0031-9422(99)00206-x. [DOI] [PubMed] [Google Scholar]
- 39.Mercke P, Bengtsson M, Bouwmeester HJ, Posthumus MA, Brodelius PE. Arch Biochem Biophys. 2000;381:173–180. doi: 10.1006/abbi.2000.1962. [DOI] [PubMed] [Google Scholar]
- 40.Picaud S, Olofsson L, Brodelius M, Brodelius PE. Arch Biochem Biophys. 2005;436:215–226. doi: 10.1016/j.abb.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 41.Teoh KH, Polichuk DR, Reed DW, Nowak G, Covello PS. FEBS Lett. 2006;580:1411–1416. doi: 10.1016/j.febslet.2006.01.065. [DOI] [PubMed] [Google Scholar]
- 42.Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, Chang MC, Withers ST, Shiba Y, Sarpong R, Keasling JD. Nature. 2006;440:940–943. doi: 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]
- 43.Bertea CM, Freije JR, van der Woude H, Verstappen FW, Perk L, Marquez V, De Kraker JW, Posthumus MA, Jansen BJ, de Groot A, Franssen MC, Bouwmeester HJ. Planta Med. 2005;71:40–47. doi: 10.1055/s-2005-837749. [DOI] [PubMed] [Google Scholar]
- 44.Nair MS, Basile DV. J Nat Prod. 1993;56:1559–1566. doi: 10.1021/np50099a015. [DOI] [PubMed] [Google Scholar]
- 45.Wallaart TE, Pras N, Quax WJ. J Nat Prod. 1999;62:1160–1162. doi: 10.1021/np9900122. [DOI] [PubMed] [Google Scholar]
- 46.Sy LK, Brown GD. Tetrahedron. 2002;58:897–908. [Google Scholar]
- 47.Brown GD, Sy LK. Tetrahedron. 2007;63:9548–9566. [Google Scholar]
- 48.Sangwan RS, Agrawal K, Luthra R, Thakur RS, Singh-Sangwan N. Phytochemistry. 1993;34(5):1301–1302. [Google Scholar]
- 49.Zhang Y, Teoh KH, Reed DW, Maes L, Goossens A, Olson DJH, Ross ARS, Covello PS. JBC. 2008;283:21501–21508. doi: 10.1074/jbc.M803090200. [DOI] [PubMed] [Google Scholar]
- 50.Wallaart TE, Bouwmeester HJ, Hille J, Poppinga L, Maijers NC. Planta. 2001;212:460–465. doi: 10.1007/s004250000428. [DOI] [PubMed] [Google Scholar]
- 51.Brown GD, Sy LK. Tetrahedron. 2004;60:1139–1159. [Google Scholar]
- 52.Roth RJ, Acton N. J Nat Prod. 1989;52:1183–1185. doi: 10.1021/np50065a050. [DOI] [PubMed] [Google Scholar]
- 53.Lange BM, Croteau R. Proc Natl Acad Sci USA. 1999;96:13714–13719. doi: 10.1073/pnas.96.24.13714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lange BM, Wildung MR, McCaskill D, Croteau R. Proc Natl Acad Sci USA. 1998;95:2100–2104. doi: 10.1073/pnas.95.5.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCaskill D, Croteau R. Tibtech. 1998;16:349–354. [Google Scholar]
- 56.Hey SJ, Powers SJ, Beale MH, Hawkins ND, Ward JL, Halford NG. Plant Biotechnol J. 2000;4:219–229. doi: 10.1111/j.1467-7652.2005.00174.x. [DOI] [PubMed] [Google Scholar]
- 57.Laule O, Furholz A, Chang HS, Zhu T, Wang X, Heifetz PB, Gruissem W, Lange M. Proc Natl Acad Sci USA. 2003;100:6866–6871. doi: 10.1073/pnas.1031755100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hampel D, Mosandl A, Wust M. Phytochemistry. 2005;66:305–311. doi: 10.1016/j.phytochem.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 59.Baldi A, Dixit VK. Bioresour Technol. 2008;99(11):4609–14. doi: 10.1016/j.biortech.2007.06.061. [DOI] [PubMed] [Google Scholar]
- 60.Weathers PJ, DeJesus-Gonzalez L, Kim YJ, Souret FF, Towler MJ. Plant Cell Reports. 2004;23:414–418. doi: 10.1007/s00299-004-0837-4. [DOI] [PubMed] [Google Scholar]
- 61.Wyslouzil BE, Waterbury RG, Weathers PJ. Biotechnol Bioeng. 2000;70:143–150. doi: 10.1002/1097-0290(20001020)70:2<143::aid-bit3>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 62.Ferreira JFS, Janick Jules. Plant Cell Tissue and Organ Cult. 1996;44:211–217. [Google Scholar]
- 63.Weathers PJ, Bunk G, McCoy M. In Vitro Cell Dev Biol-Plant. 2005;41:47–53. [Google Scholar]
- 64.Woerdenbag HJ, Luers J, van Uden W, Pras N, Malingre T, Alfermann A. Plant Cell Tissue Cult. 1993;32:247–257. [Google Scholar]
- 65.Putalun W, Luealon W, De-Eknamkul W, Tanaka H, Shoyama Y. Biotechnol Lett. 2007;29:1143–1146. doi: 10.1007/s10529-007-9368-8. [DOI] [PubMed] [Google Scholar]
- 66.Wang JW, Tan RX. Biotechnol Lett. 2002;124:1153–1156. [Google Scholar]
- 67.Wang Y, Zhang H, Zhao B, Yuan X. Biotechnol Lett. 2001;23:1971–1973. [Google Scholar]
- 68.Souret FF, Kim Y, Wyslouzil BE, Wobbe KK, Weathers PJ. Biotechnol Bioeng. 2003;83:653–667. doi: 10.1002/bit.10711. [DOI] [PubMed] [Google Scholar]
- 69.Ferreira JF, Simon JE, Janick J. Planta Med. 1995;61:351–355. doi: 10.1055/s-2006-958098. [DOI] [PubMed] [Google Scholar]
- 70.Wang Y, Weathers PJ. Plant Cell Reports. 2007;26:1073–1081. doi: 10.1007/s00299-006-0295-2. [DOI] [PubMed] [Google Scholar]
- 71.Asadollahi MA, Maury J, Moller K, Nielsen KF, Schalk M, Clark A, Nielsen J. Biotechnol Bioeng. 2008;99:666–677. doi: 10.1002/bit.21581. [DOI] [PubMed] [Google Scholar]
- 72.Kudakasseril GJ, Lam L, Staba EJ. Planta Med. 1987;53:280–284. doi: 10.1055/s-2006-962706. [DOI] [PubMed] [Google Scholar]
- 73.Wang H, Ge L, Ye HC, Chong K, Lie BY, Li GF. Planta Med. 2004;70:347–352. doi: 10.1055/s-2004-818947. [DOI] [PubMed] [Google Scholar]
- 74.Wang H, Liu Y, Ching K, Liu B, Ye HC, Li Z, Yan F, Li GF. Plant Biol (Stuttg) 2007;9:442–446. doi: 10.1055/s-2006-924634. [DOI] [PubMed] [Google Scholar]
- 75.Sa G, Mi H, He-Chun Y, Ben-Ye L, Guofeng L, Kang C. Plant Sci. 2001;160:691–698. doi: 10.1016/s0168-9452(00)00453-2. [DOI] [PubMed] [Google Scholar]
- 76.Chen D, Ye H, Li G. Plant Sci. 2000;155:179–185. doi: 10.1016/s0168-9452(00)00217-x. [DOI] [PubMed] [Google Scholar]
- 77.Chen D, Liu C, Ye H, Li G, Liu B, Meng Y, Chen X. Plant Cell Tissue Org Cult. 1999;57:157–162. [Google Scholar]
- 78.Chang MC, Eachus RA, Trieu W, Ro DK, Keasling JD. Nat Chem BioI. 2007;3:274–277. doi: 10.1038/nchembio875. [DOI] [PubMed] [Google Scholar]
- 79.Maury J, Asadollahi MA, Moller K, Clark A, Nielsen J. Adv Biochem Eng/Biotechnol. 2005;100:19–51. doi: 10.1007/b136410. [DOI] [PubMed] [Google Scholar]
- 80.Kovacs K, Zhang L, Linforth RS, Whittaker B, Hayes CJ, Fray RG. Transgenic Res. 2007;16:121–126. doi: 10.1007/s11248-006-9039-x. [DOI] [PubMed] [Google Scholar]
- 81.Wu S, Schalk M, Clark A, Miles RB, Coates R, Chappell J. Nat Biotechnol. 2006;24:1441–1447. doi: 10.1038/nbt1251. [DOI] [PubMed] [Google Scholar]
- 82.Ducreux LJ, Morris WL, Hedley PE, Shepherd T, Davies HV, Millam S, Taylor MA. J Exper Botany. 2005;56:81–89. doi: 10.1093/jxb/eri016. [DOI] [PubMed] [Google Scholar]
- 83.Beyer P, Al-Babili S, Ye X, Lucca P, Schaub P, Welsch R, Potrykus I. J Nutr. 2002;132:506S–510S. doi: 10.1093/jn/132.3.506S. [DOI] [PubMed] [Google Scholar]
- 84.Newman JD, Marshall J, Chang M, Nowroozi F, Paradise E, Pitera D, Newman KL, Keasling JD. Biotechnol Bioeng. 2006;95:684–691. doi: 10.1002/bit.21017. [DOI] [PubMed] [Google Scholar]
- 85.Picaud S, Olsson ME, Brodelius PE. Protein Expression and Purification. 2007;51:71–79. doi: 10.1016/j.pep.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 86.Lee PC, Schmidt-Dannert C. Appl Microbiol Biotechnol. 2002;60:1–11. doi: 10.1007/s00253-002-1101-x. [DOI] [PubMed] [Google Scholar]
- 87.Martin VJ, Pitera DJ, Withers ST, Newman JD, Keasling JD. Nat Biotechnol. 2003;21:796–802. doi: 10.1038/nbt833. [DOI] [PubMed] [Google Scholar]
- 88.Lindahl AL, Olsson ME, Mercke P, Tollbom O, Schelin J, Brodelius M, Brodelius PE. Biotechnol Lett. 2006;28:571–580. doi: 10.1007/s10529-006-0015-6. [DOI] [PubMed] [Google Scholar]
- 89.Suthers PF, Burgard AP, Dasika MS, Nowroozi F, Van Dien S, Keasling JD, Maranas CD. Metab Eng. 2007;9:387–405. doi: 10.1016/j.ymben.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Julsing MK, Koulman A, Woerdenbag HJ, Quax WJ, Kayser O. Biomol Eng. 2006;23:265–279. doi: 10.1016/j.bioeng.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 91.Roberts SC. Nat Chem BioI. 2007;3:387–395. doi: 10.1038/nchembio.2007.8. [DOI] [PubMed] [Google Scholar]
- 92.Aharoni A, Jongsma MA, Bouwmeester HJ. Trends Plant Sci. 2005;10:594–602. doi: 10.1016/j.tplants.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 93.Eckstein-Ludwig U, Webb RJ, Van Goethem ID, East JM, Lee AG, Kimura M, O’Neill PM, Bray PG, Ward SA, Krishna S. Nature. 2003;424:957–961. doi: 10.1038/nature01813. [DOI] [PubMed] [Google Scholar]
- 94.Kannan R, Kumar K, Sahal D, Kukreti S, Chauhan VS. Biochem J. 2005;385(Pt 2):409–418. doi: 10.1042/BJ20041170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jung M, Kim H, Nam KY, No KT. Bioorg Med Chem Lett. 2005;15:2994–2997. doi: 10.1016/j.bmcl.2005.04.041. [DOI] [PubMed] [Google Scholar]
- 96.Uhlemann AC, Cameron A, Eckstein-Ludwig U, Fischbarg J, Iserovich P, Zuniga FA, East M, Lee A, Brady L, Haynes RK, Krishna S. Nat Struct Mol Biol. 2005;12:628–629. doi: 10.1038/nsmb947. [DOI] [PubMed] [Google Scholar]
- 97.Benoit-Vical F, Lelievre J, Berry A, Deymier C, Dechy-Cabaret O, Cazelles J, Loup C, Robert A, Magnaval JF, Meunier B. Antimicrobial Agents Chemother. 2007;51:1463–1472. doi: 10.1128/AAC.00967-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ploypradith P. Acta Tropica. 2004;89:329–342. doi: 10.1016/j.actatropica.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 99.Posner GH, McRiner AJ, Paik IH, Sur S, Borstnik K, Xie S, Shapiro TA, Alagbala A, Foster B. J Med Chem. 2004;47:1299–1301. doi: 10.1021/jm0303711. [DOI] [PubMed] [Google Scholar]
- 100.Galal AM, Ross SA, Jacob M, ElSohly MA. J Nat Prod. 2005;68:1274–1276. doi: 10.1021/np050074u. [DOI] [PubMed] [Google Scholar]