

Abstract
Objective:
To evaluate the safety and tolerability of natalizumab when added to glatiramer acetate (GA) in patients with relapsing multiple sclerosis. The primary outcome assessed whether this combination would increase the rate of development of new active lesions on cranial MRI scans vs GA alone.
Methods:
This phase 2, randomized, double-blind, placebo-controlled study included patients aged 19 to 55 years who were treated with GA for at least 1 year before randomization and experienced at least one relapse during the previous year. Patients received IV natalizumab 300 mg (n = 55) or placebo (n = 55) once every 4 weeks plus GA 20 mg subcutaneously once daily for ≤20 weeks.
Results:
The mean rate of development of new active lesions was 0.03 with combination therapy vs 0.11 with GA alone (p = 0.031). Combination therapy resulted in lower mean numbers of new gadolinium-enhancing lesions (0.6 vs 2.3 for GA alone, p = 0.020) and new/newly enlarging T2-hyperintense lesions (0.5 vs 1.3, p = 0.029). The incidence of infection and infusion reactions was similar in both groups; no hypersensitivity reactions were observed. One serious adverse event occurred with combination therapy (elective hip surgery). With the exception of an increase in anti-natalizumab antibodies with combination therapy, laboratory data were consistent with previous clinical studies of natalizumab alone.
Conclusion:
The combination of natalizumab and glatiramer acetate seemed safe and well tolerated during 6 months of therapy.
GLOSSARY
- AE
= adverse event;
- CONSORT
= Consolidated Standards of Reporting Trials;
- EDSS
= Expanded Disability Status Scale;
- GA
= glatiramer acetate;
- Gd+
= gadolinium-enhancing;
- GLANCE
= Glatiramer Acetate and Natalizumab Combination Evaluation;
- IFNβ
= interferon β;
- MS
= multiple sclerosis;
- PML
= progressive multifocal leukoencephalopathy.
Interferon β (IFNβ) and glatiramer acetate (GA) are only partially effective for treatment of relapsing multiple sclerosis (MS); approximately two-thirds of patients continue to experience relapses on these therapies over 2 years.1–4 New focal inflammatory lesions in MS are believed to occur when activated T cells cross the blood–brain barrier and initiate a series of events leading to activation of endothelial cells, recruitment of additional lymphocytes and monocytes, release of proinflammatory cytokines, and subsequent demyelination and formation of MS plaques.5 The interaction of α4β1 integrin on leukocytes with vascular cell adhesion molecule 1 on brain endothelial cells is a critical step in migration of leukocytes across the blood–brain barrier.6–8 Natalizumab binds to the α4 subunit of α4β1 integrin, thereby inhibiting leukocyte trafficking into the CNS (by blocking interactions with molecules including the CS-1 fragment of fibronectin and vascular cell adhesion molecule 1) and potentially altering cell–cell interactions and T-cell activation.9–11
In a phase 3 study in patients with relapsing MS, natalizumab reduced sustained progression of disability by 42% and annualized relapse rate by 68% over 2 years.12 Here we report the results of the phase 2 Glatiramer Acetate and Natalizumab Combination Evaluation (GLANCE) study assessing safety and tolerability of GA in combination with natalizumab in patients with relapsing MS. The primary endpoint was the rate of new active lesion development on cranial MRI scan. It was hypothesized that, because the proposed mechanism of action of GA requires cellular entry into the brain,13–15 α4-integrin blockade by natalizumab might impair rather than enhance the efficacy of GA. Furthermore, because GA may induce a shift toward a Th2-biased immune response,16 it was hypothesized that it might modify the immune response to natalizumab, thereby potentially increasing hypersensitivity reactions or immunogenicity. Hence the secondary endpoint was to determine whether combination therapy would increase incidence or severity of adverse events (AEs), particularly hypersensitivity reactions.
METHODS
Patients.
Eligible patients were aged 18–55 years and had a diagnosis of relapsing MS,17 an Expanded Disability Status Scale (EDSS) score of 0 to 5.0,18 had been treated with GA for ≥12 months before randomization and experienced one or more relapses during that time, and had cranial MRI lesions consistent with MS. The study was performed in accordance with the Declaration of Helsinki and its subsequent amendments, Good Clinical Practice, and applicable regulatory requirements. The protocol was approved by the relevant institutional review boards or ethics committees, and all participants gave written informed consent.
Exclusion criteria included a diagnosis of progressive MS,19 MS relapse within 50 days before randomization, clinically significant infectious illness within 30 days of randomization, abnormal laboratory results (or history thereof) indicative of any major organ system disease precluding administration of natalizumab or GA, history of severe allergic or anaphylactic reactions, known drug hypersensitivity, or history of malignancy (excluding nonmetastatic basal cell carcinoma). Women who were pregnant, at risk of or planning to become pregnant, or breast-feeding were excluded.
Other exclusion criteria included any prior treatment with total lymphoid irradiation, cladribine, T-cell or T-cell receptor vaccination, natalizumab, or any other therapeutic monoclonal antibody; treatment with mitoxantrone, cyclophosphamide, or any interferon product within 1 year before randomization; treatment with cyclosporine, azathioprine, methotrexate, IV immunoglobulin, plasmapheresis, cytapheresis, or mycophenolate mofetil within 6 months before randomization; and treatment with 4-aminopyridine or IV or oral corticosteroids within 50 days before randomization.
Study design and intervention.
This randomized, double-blind, placebo-controlled, parallel-group safety study was conducted at 25 centers in the United States and Canada between June 17, 2003, and March 23, 2004. Patients were randomly assigned 1:1 to receive IV natalizumab 300 mg or placebo every 4 weeks in addition to GA 20 mg subcutaneously once daily for up to 24 weeks (“combination therapy” and “GA alone” groups). All study personnel, patients, and sponsor personnel involved in study conduct were blinded to treatment assignments. Investigators were allowed to discontinue treatment in any patient who developed five or more new enhancing lesions per month for 2 consecutive months relative to baseline or those who experienced two or more clinical relapses within the study period.
Safety assessments.
Cranial MRI assessments were performed at screening and at weeks 4, 8, 12, 16, 20, and 24 (or study withdrawal). Contiguous, 3-mm-thick axial slices through the whole brain were acquired. All scans were evaluated at The Institute of Neurology, University College London, UK. Scans were checked for artifacts, compliance with scan parameters, and repositioning before assessment of lesions.
Newly active lesions were recorded on all scans from weeks 4 to 24, the primary endpoint being the rate of development of new active lesions over this period. Newly active lesions were defined as new gadolinium-enhancing (Gd+) lesions and nonenhancing new or enlarging T2-hyperintense lesions. If a new Gd+ lesion was also new or enlarging on the T2 scan, it was counted only once as a newly active lesion. A new Gd+ lesion was defined as an area of enhancement on postcontrast T1-weighted scans in a place that showed no enhancement on a previous scan and where there was also a lesion visible on the T2-weighted scans. An enlarging T2-hyperintense lesion was defined as a T2-hyperintense lesion that was larger in size on two adjacent slices compared with a previous scan.
The week 24 scan was analyzed for the presence of new and enlarging T2-hyperintense lesions and new T1-hypointense lesions when compared with the baseline scan.
AEs and relapses were monitored throughout the study. Relapses were defined as new or recurrent neurologic symptoms, consistent with MS, not associated with fever or infection, lasting at least 24 hours, and accompanied by new objective neurologic findings. EDSS scores were determined at baseline (week 0) and at weeks 12 and 24 (or study withdrawal). Blood chemistry, hematology, urinalysis, and physical examinations were performed at screening and at weeks 12 and 24 (or study withdrawal). Vital signs were performed at screening, at baseline, and every 4 weeks thereafter.
Immunogenicity.
Serum anti-natalizumab antibodies were determined at baseline and every 4 weeks using an ELISA. Patients were categorized as persistently positive (≥0.5 μg/mL at two or more postbaseline evaluations separated by ≥42 days, or at a single time point with no follow-up samples), transiently positive (≥0.5 μg/mL at a single postbaseline evaluation before the final postbaseline evaluation), or antibody negative (<0.5 μg/mL at all postbaseline evaluations).
Statistical methods.
The primary endpoint was rate of development of new active lesions. The pooled SD was estimated to be 2.53, which was based on a previous phase 2 study20 comparing natalizumab with placebo and the assumptions that 1) the addition of GA to placebo would reduce the rate of development of new active lesions by 30% and 2) natalizumab plus GA would have similar efficacy to natalizumab alone. Using a two-sample t test at the two-sided 5% level of significance, it was estimated that a sample size of 55 patients per treatment group would provide 80% power to detect a mean difference of 1.37 between rates of development of new active lesions.
The rate of development of new active lesions (defined as new Gd+ lesions and nonenhancing new or newly enlarging T2 lesions) was analyzed by calculating the ordinary standard least squares slope of cumulative new active lesions over 4 to 24 weeks for each patient, and was compared between treatment groups using the Wilcoxon rank sum test. The cumulative number of new active lesions at week 24 and number of new or enlarging T2-hyperintense and new T1-hypointense lesions at week 24 compared with baseline were summarized and compared between treatment groups using the Wilcoxon rank sum test. The number of patients remaining relapse free at 6 months was analyzed using the Fisher exact test. Patients who withdrew from the study before week 24 were assumed to have had a relapse. Annualized relapse rate was analyzed using Poisson regression with variance adjustment by correcting for over dispersion, and was adjusted for the number of relapses in the 1 year before study entry, baseline EDSS score (≤3.5 vs >3.5), presence of Gd+ lesions (present vs absent), and age (<40 vs ≥40 years). Between-group EDSS scores were compared at weeks 12 and 24 using analysis of covariance adjusted for baseline score.
All analyses followed the intent-to-treat principle, and all reported p values are two-tailed. Mean values are reported ± SD.
RESULTS
Patients.
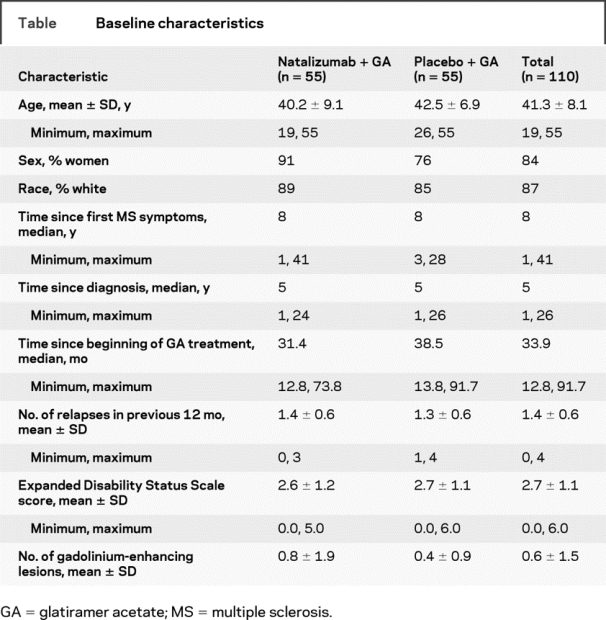
Of the 110 patients enrolled, 55 were randomly assigned to receive natalizumab plus GA (combination therapy), and 55 were randomly assigned to receive placebo plus GA (GA alone) (figure 1). All randomized patients received at least one infusion of study drug. Demographic and baseline clinical characteristics were generally similar between treatment groups (table); however, the proportion of men was slightly higher in the GA alone group. The median time since MS diagnosis was 5 years, and the median time since initiation of current GA treatment was 33.9 months. Patients experienced a mean of 1.4 ± 0.6 relapses during the 12 months before study enrollment. At baseline, the mean EDSS score was 2.7 ± 1.1, and the mean number of Gd+ lesions was 0.6 ± 1.5.

Figure 1 Consolidated Standards of Reporting Trials (CONSORT) flowchart
Table Baseline characteristics
MRI.
Adding natalizumab to GA did not increase the number of new active lesions. The mean rate of development of new active lesions over the 24-week study was lower with combination therapy (0.03) vs GA alone (0.11; p = 0.031). The difference between treatment groups became apparent after approximately 4 to 8 weeks of treatment. The mean cumulative number of new active lesions at 24 weeks was 0.9 ± 2.1 with combination therapy and 2.6 ± 5.4 with GA alone (p = 0.057). With combination therapy, 60% of patients had no new active lesions vs 51% with GA alone.
The mean cumulative number of new Gd+ lesions at 24 weeks was lower with combination therapy vs GA alone (0.6 ± 1.8 vs 2.3 ± 5.3; p = 0.020; figure 2). Furthermore, over the 24-week study period, more patients treated with combination therapy remained free of new Gd+ lesions compared with patients receiving GA alone (69% vs 55%). A similar pattern was observed for new or enlarging T2-hyperintense lesions (figure 2). At week 24, the mean number of all new or enlarging T2-hyperintense lesions compared with baseline was 0.5 ± 1.1 with combination therapy and 1.3 ± 2.1 with GA alone (p = 0.029), representing a 62% reduction with combination therapy. With combination therapy, 67% of patients had no new or enlarging T2-hyperintense lesions vs 51% with GA alone. At week 24, slightly more natalizumab-treated patients were free of new T1-hypointense lesions compared with patients receiving placebo (73% vs 67%); the mean numbers of T1-hypointense lesions were 0.2 ± 0.5 and 0.4 ± 0.8 (p = 0.359).
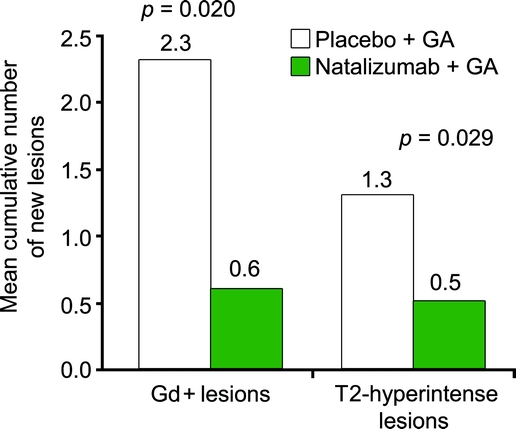
Figure 2 Cumulative number of new Gd+ lesions and T2-hyperintense lesions at week 24 in the placebo + GA group and the natalizumab + GA group
Gd+ = gadolinium-enhancing; GA = glatiramer acetate.
Adverse events.
At least one AE was reported by 91% of patients receiving combination therapy and 93% receiving GA alone. Overall, the most common AEs (combination therapy vs GA alone) were headache (31% vs 27%), MS relapse (16% vs 25%), nasopharyngitis (13% vs 20%), and nausea (16% vs 15%). AEs that occurred at an incidence >5% higher with combination therapy vs GA alone included upper respiratory infection (16% vs 9%), sinusitis (16% vs 7%), back pain (16% vs 7%), arthralgia (9% vs 2%), and flushing (11% vs 2%). No deaths occurred during the study. Serious AEs were reported in one patient receiving combination therapy (elective hip surgery) and two patients receiving GA alone (hospitalization for MS relapse, anaphylactic reaction to GA). One patient in each group discontinued treatment because of an AE (combination therapy: rigors during infusion of natalizumab; GA alone: shoulder pain). Two patients receiving GA alone withdrew from the study because of previously mentioned AEs (shoulder pain, MS relapse).
The overall incidence of infection was 60% with combination therapy and 65% with GA alone. The most common infections (combination therapy vs GA alone) were nasopharyngitis (13% vs 20%), upper respiratory infection (16% vs 9%), sinusitis (16% vs 7%), and urinary tract infection (5% vs 11%). No serious or opportunistic infections were observed in either treatment group.
Infusion reactions (any event occurring within 2 hours after the start of infusion) occurred in 11% of patients receiving combination therapy and 13% receiving GA alone. Most infusion reactions were classified as nervous system disorders (headache [7% placebo plus GA, 7% natalizumab plus GA] and dizziness [2%, 0%]). Hypersensitivity reactions were defined as “hypersensitivity,” “allergic reaction,” or “anaphylactic/anaphylactoid” and were categorized by the investigator based on clinical judgment and severity, as well as any report of “urticaria,” “allergic dermatitis,” or “hives.” No hypersensitivity reactions occurred during natalizumab infusions. Two patients receiving GA alone experienced postinjection systemic reactions, one of which was reported as a serious AE.
GA-related AEs, such as injection-site erythema, injection-site pain, and flushing, occurred more often with combination therapy than with GA alone (16% vs 5%). One patient receiving GA alone experienced depressed mood, and another experienced increased depression; worsening depression was observed in three natalizumab-treated patients.
Other safety assessments.
Expected changes in white blood cell counts, including lymphocytes, occurred with natalizumab treatment. Mean values remained within the normal ranges. No other clinically significant changes were observed for laboratory parameters. In addition, no clinically significant differences were noted between treatment groups in changes from baseline for physical examination findings or vital signs.
Relapse rate/disability.
Adding natalizumab to GA did not increase clinical disease activity. The adjusted annualized relapse rate throughout 24 weeks was 0.40 with combination therapy and 0.67 with GA alone (p = 0.237). Over the 24-week study, 78% of patients receiving combination therapy remained relapse free, compared with 73% receiving GA alone (p = 0.658). EDSS scores remained stable throughout the study, with no major differences observed between treatment groups. At week 24, the median EDSS score was 2.5 in both groups.
Immunogenicity.
Data on the presence of anti-natalizumab antibodies were available for 54 patients receiving combination therapy. Forty patients (74%) were antibody negative, 7 (13%) were transiently antibody positive, and 7 (13%) were persistently antibody positive. Two of the patients who were persistently positive were positive at a single time point and had no follow-up sample drawn. Persistently positive patients showed a reduction in serum natalizumab levels before the week 12 and 20 infusions, as well as decreased α4-integrin saturation, compared with transiently positive or antibody negative patients.
Antibody-negative patients had an overall AE incidence of 95%. In patients who tested positive for antibodies at any time (transient and persistent), the incidence was 86%. Patients who were persistently positive for antibodies had a higher incidence of MS relapse (two cases [29%] vs one transiently positive case [14%] and six antibody-negative cases [15%]) and certain infusion-related AEs, such as flushing (three cases [43%] vs one case [14%] and two cases [5%]) and rigors (two cases [29%] vs no cases and one case [3%]).
DISCUSSION
The GLANCE study assessed the safety of natalizumab add-on therapy in patients who had experienced at least one episode of breakthrough disease while receiving GA during the preceding year. When the study was planned, it was assumed that some clinicians might administer natalizumab in combination with GA for relapsing forms of MS, particularly in patients with breakthrough disease activity during GA treatment. However, this previously untested combination of therapies led to safety concerns, including the possibility that natalizumab might adversely affect the efficacy and safety of GA and that GA exposure might increase the immunogenicity of natalizumab, and hence adversely affect natalizumab efficacy or increase the risk of infusion reactions.
Safety was primarily assessed from the perspective of a potential efficacy loss with combination therapy. The primary endpoint assessed whether the rate of new active lesion development increased after natalizumab was added in patients already receiving GA. However, the rate of new active MRI lesion development (figure 2) was lower when natalizumab was added to GA compared with GA alone. In addition, fewer new Gd+ T1 lesions and fewer new or enlarging T2-hyperintense lesions were observed with the combination therapy. Although not directly comparable, the pattern of efficacy seems qualitatively similar to that seen in studies of natalizumab monotherapy12,20 or in combination with IFNβ.21 Without a natalizumab monotherapy cohort, we could not discern whether the effects on MRI and clinical outcomes resulted from natalizumab alone or from a synergistic effect of the combination with GA.
The novel primary outcome in this study was the rate of new MRI lesion development rather than lesion count or volume change. This method was sufficiently sensitive in a population of patients with relapsing–remitting MS so that a major MRI benefit was detected within a relatively short study duration involving a small number of patients and where an active treatment (GA) probably reduced disease activity in the control arm. Although it was used as a safety measure here, it could also be used to assess efficacy in future phase 2 studies.
A suggestion of increased immunogenicity was observed with respect to persistence of natalizumab neutralizing antibodies: this occurred in 13% of patients receiving natalizumab plus GA combination therapy compared with 6% of patients in phase 3 studies of natalizumab. It has been shown that GA treatment in humans induces a broad repertoire of T-cell response with a tendency for Th2 deviation as well as a broad repertoire of B-cell activation and concomitant antibody production.22,23 Some of the antibodies induced by GA may also recognize natalizumab; this may explain the increase in natalizumab neutralizing antibodies seen with the natalizumab plus GA combination. Consistent with the phase 3 natalizumab studies, patients who developed persistent neutralizing antibodies seemed to be at greater risk for MS relapse and for certain infusion-related AEs.12,21
Patients who completed this study were eligible to enroll in a safety-extension study during which they continued to receive combination therapy. No evidence of progressive multifocal leukoencephalopathy (PML) was observed during the GLANCE study or subsequent extension phase. However, the emergence of PML as a complication of natalizumab in combination with IFNβ in two extension study patients who had originally participated in another study resulted in the discontinuation of further investigation of the natalizumab plus GA combination as well.24 Because of ongoing concern about the risk of PML, combination of natalizumab with any other immunomodulatory drug is currently not recommended.25 Although the GLANCE study did not directly compare monotherapy with natalizumab vs GA, our data suggest that natalizumab has a strong effect on disease activity in relapsing–remitting MS, underpinning the use of natalizumab monotherapy in patients with breakthrough MS.
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by M.Y.
ACKNOWLEDGMENT
The authors acknowledge the contributions of Jillian Licata, Paul Benfield, and Matthew Hasson for administrative and medical editing assistance in the preparation of the manuscript. They are employees of Scientific Connexions, Newtown, PA, which was retained by Biogen Idec for this assistance.
DISCLOSURE
This study was supported by Biogen Idec, Inc. and Elan Pharmaceuticals, Inc. A.D.G. has received has received research support or honoraria as a speaker or consultant from Acorda, Anacor, Bayer, Biogen Idec, Elan Pharmaceuticals, EMD-Serono, Genentech, Genzyme, Novartis, Pfizer, and Teva Neuroscience. H.R. has received research support and honoraria as a speaker, consultant, and member of advisory boards for Biogen Idec and Elan Pharmaceuticals, as well as research support from Genentech, Roche, Merck, Teva Neuroscience, Centocor, UCB, and EMD Serono. A.B.-O. has received grant support, consulting fees, and honoraria from Aventis, Bayhill Therapeutics, Berlex, Biogen Idec, BMD, Genentech, Merck-Serono, and Teva Neuroscience. A.M. has received research support from Biogen Idec, Genentech, Genzyme, Immune Tolerance Network, and Teva Neuroscience; has served as a consultant for Acorda, Avigen, Barofold, Biogen Idec, Daiichi-Sankyo, Genentech, GlaxoSmithKline, Medicinova, Merck Serono, and Sanofi-Aventis; and has received honoraria for serving on speakers bureaus for Biogen Idec, EMD Serono, Pfizer, and Teva Neuroscience. D.H.M. has received research grants (held by University College London) from Biogen Idec, GlaxoSmithKline, Schering AG, and Novartis, and has received honoraria and travel expenses for advisory committee work or as an invited speaker from Bayer-Schering, Biogen Idec, GlaxoSmithKline, and the US National Institutes of Health. K.S. has received research support and honoraria from Biogen Idec and has received research support from Schering and EMD Serono. F.L. has received research support or honoraria as a speaker or consultant from Acorda, Actelion, AmGen, Avigen, Bayer, Biogen Idec, BioMS, Genentech, GenMab, Genzyme, Medicinova, Novartis, Pfizer, Serono, and Teva Neuroscience and has an ownership interest in Cognition Pharma. O.K. has received research support and honoraria from Biogen Idec, Inc., as well as research support from Acorda, Bayer Healthcare, BioMS/Eli Lilly, EMD Serono, Genentech, GlaxoSmithKline, Novartis, Sanofi-Aventis, and Teva Neuroscience. N.M.B., M.Y., M.A.P., and A.W.S. are employees of and have equity interest in Biogen Idec, Inc.
APPENDIX
GLANCE Investigators.
Alpha Neurology, PC, Staten Island, NY— A.B. Perel, A.N. Babu; Center for Neurological Disorders, Milwaukee, WI—B.O. Khatri, V.K. Saxena; CHVO Hospital de Hull, Quebec, Canada—F. Jacques, D. Halle; The Cleveland Clinic, OH—L. Stone, R.M. Marrie; Colorado Springs Neurological Associates, CO—P.A. Fodor, L.J. Adams; Division of Neurology, Maimonides Medical Center, Brooklyn, NY—A.E. Miller, M.J. Keilson; Elisabeth Bruyere Health Centre, Ottawa, Ontario, Canada—S.N. Christie, T. Mendis; Maryland Center for MS, Baltimore, MD—R. Shin, C. Bever; MeritCare Neuroscience Clinic, Fargo, ND—S.L. Scarberry, R.C. Bailly; Michigan Institute for Neurological Disorders, Farmington Hills, MI—H.S. Rossman, M. Belkin; Montreal Neurological Institute, Quebec, Canada—A. Bar-Or, D. Arnold; The MS Center of Atlanta, GA—J. English, W. Stuart; Raleigh Neurology Associates, NC—S.M. Freedman, W.G. Ferrell; Swedish Medical Center, Seattle, WA—C.H. Smith, S. Hamilton; Texas Neurology, Dallas, TX—J.T. Phillips, D. Heitzman; University of Calgary, MS Clinic, Alberta, Canada—L. Metz, D. Patry; University Campus LHSC, London, Ontario, Canada—G.P.A. Rice, D. Mason; University Hospital Stony Brook, NY—P. Coyle, L. Krupp; University of New Mexico Health Sciences Center, Albuquerque, NM—C. Ford, J. Katsman; University of Ottawa, Ottawa, Ontario, Canada—M. Freedman, H. Rabinovitch; University of Rochester School of Medicine and Dentistry, NY—A.D. Goodman, S.R. Schwid; University of Southern California MS Comprehensive Care Center, Los Angeles, CA—N.J. Kachuck, Q. Zhang; University of Texas Houston, TX—S. Brod, J.S. Wolinsky; Wake Forest University School of Medicine, Winston-Salem, NC—D.R. Jeffery, S. Kumar; Wayne State University, Detroit, MI—O. Khan, R. Lisak.
MRI Analysis Center.
MS NMR Research Unit, Institute of Neurology, University College London, UK—D. Miller, K. Schmierer, K. Miszkiel, D. MacManus, S. Zalita.
Address correspondence and reprint requests to Dr. Andrew D. Goodman, Department of Neurology, University of Rochester Medical Center, 601 Elmwood Ave., Rochester, NY 14642 andrew_goodman@urmc.rochester.edu
*GLANCE study investigators are listed in the appendix.
The MS NMR Research Unit in London is supported by the MS Society of Great Britain and Northern Ireland. K.S. is supported by the WellcomeTrust.
Disclosure: Author disclosures are provided at the end of the article.
Received June 4, 2008. Accepted in final form December 9, 2008.
REFERENCES
- 1. The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis, I: clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology. 1993;43:655–661. doi: 10.1212/wnl.43.4.655. [DOI] [PubMed] [Google Scholar]
- 2. Johnson KP Brooks BR Cohen JA et al. for the Copolymer I Multiple Sclerosis Study Group. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind, placebo-controlled trial. Neurology. 1995;45:1268–1276. doi: 10.1212/wnl.45.7.1268. [DOI] [PubMed] [Google Scholar]
- 3. Jacobs LD Cookfair DL Rudick RA et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol. 1996;39:285–294. doi: 10.1002/ana.410390304. [DOI] [PubMed] [Google Scholar]
- 4. The Prevention of Relapses and Disability by Interferon β-1a Subcutaneously in Multiple Sclerosis (PRISMS) Study Group. Randomised double-blind placebo-controlled study of interferon β-1a in relapsing/remitting multiple sclerosis. Lancet. 1998;352:1498–1504. [PubMed] [Google Scholar]
- 5. Ffrench-Constant C Pathogenesis of multiple sclerosis. Lancet. 1994;343:271–275. doi: 10.1016/s0140-6736(94)91118-5. [DOI] [PubMed] [Google Scholar]
- 6. Yednock TA Cannon C Fritz LC Sanchez-Madrid F Steinman L Karin N Prevention of experimental autoimmune encephalomyelitis by antibodies against α4β1 integrin. Nature. 1992;356:63–66. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- 7. Baron JL Madri JA Ruddle NH Hashim G Janeway CAJr Surface expression of α4 integrin by CD4 T cells is required for their entry into brain parenchyma. J Exp Med. 1993;177:57–68. doi: 10.1084/jem.177.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lobb RR Hemler ME The pathophysiologic role of α4 integrins in vivo. J Clin Invest. 1994;94:1722–1728. doi: 10.1172/JCI117519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rudick RA Sandrock A Natalizumab: α4-integrin antagonist selective adhesion molecule inhibitors for MS. Expert Rev Neurother. 2004;4:571–580. doi: 10.1586/14737175.4.4.571. [DOI] [PubMed] [Google Scholar]
- 10. Rice GPA Hartung H-P Calabresi PA Anti-α4 integrin therapy for multiple sclerosis: mechanisms and rationale. Neurology. 2005;64:1336–1342. doi: 10.1212/01.WNL.0000158329.30470.D0. [DOI] [PubMed] [Google Scholar]
- 11. Niino M Bodner C Simard M-L et al. Natalizumab effects on immune cell responses in multiple sclerosis. Ann Neurol. 2006;59:748–754. doi: 10.1002/ana.20859. [DOI] [PubMed] [Google Scholar]
- 12. Polman CH O’Connor PW Havrdova E et al. for the Natalizumab Safety and Efficacy Relapsing Remitting Multiple Sclerosis (AFFIRM) Investigators. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 13. Aharoni R Teitelbaum D Sela M Arnon R Copolymer 1 induces T cells of the T helper type 2 that crossreact with myelin basic protein and suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 1997;94:10821–10826. doi: 10.1073/pnas.94.20.10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aharoni R Teitelbaum D Sela M Arnon R Bystander suppression of experimental autoimmune encephalomyelitis by T cell lines and clones of the Th2 type induced by copolymer 1. J Neuroimmunol. 1998;91:135–146. doi: 10.1016/s0165-5728(98)00166-0. [DOI] [PubMed] [Google Scholar]
- 15. Duda PW Schmied MC Cook SL Krieger JI Hafler DA Glatiramer acetate (Copaxone®) induces degenerate, Th2-polarized immune responses in patients with multiple sclerosis. J Clin Invest. 2000;105:967–976. doi: 10.1172/JCI8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang J Hutton G Zang Y A comparison of the mechanisms of action of interferon beta and glatiramer acetate in the treatment of multiple sclerosis. Clin Ther. 2002;24:1998–2012. doi: 10.1016/s0149-2918(02)80094-7. [DOI] [PubMed] [Google Scholar]
- 17. McDonald WI Compston A Edan G et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the Diagnosis of Multiple Sclerosis. Ann Neurol. 2001;50:121–127. doi: 10.1002/ana.1032. [DOI] [PubMed] [Google Scholar]
- 18. Kurtzke JF Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33:1444–1452. doi: 10.1212/wnl.33.11.1444. [DOI] [PubMed] [Google Scholar]
- 19. Lublin FD Reingold SC Defining the clinical course of multiple sclerosis: results of an international survey. Neurology. 1996;46:907–911. doi: 10.1212/wnl.46.4.907. [DOI] [PubMed] [Google Scholar]
- 20. Miller DH Khan OA Sheremata WA et al. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2003;348:15–23. doi: 10.1056/NEJMoa020696. [DOI] [PubMed] [Google Scholar]
- 21. Rudick RA Stuart WH Calabresi PA et al. for the Safety and Efficacy of Natalizumab in Combination with Interferon Beta-1a in Patients with Relapsing Remitting Multiple Sclerosis (SENTINEL) Investigators. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354:911–923. doi: 10.1056/NEJMoa044396. [DOI] [PubMed] [Google Scholar]
- 22. Basile E Gibbs E Aziz T et al. During 3 years treatment of primary progressive multiple sclerosis with glatiramer acetate, specific antibodies switch from IgG1 to IgG4. J Neuroimmunol. 2006;177:161–166. doi: 10.1016/j.jneuroim.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 23. Mosley RL Gordon PH Hasiak CM et al. Glatiramer acetate immunization induces specific antibody and cytokine responses in ALS patients. Amyotroph Lateral Scler. 2007;8:235–242. doi: 10.1080/17482960701374601. [DOI] [PubMed] [Google Scholar]
- 24. Yousry TA Major EO Ryschkewitcsh C et al. Evaluation of patients treated with natalizumab for progressive multifocal leukoencephalopathy. N Engl J Med. 2006;354:924–933. doi: 10.1056/NEJMoa054693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.TYSABRI® (natalizumab) [prescribing information]. Cambridge, MA: Biogen Idec; 2008. [Google Scholar]