

Abstract
Objective:
To use a combined neurogenetic-neuroimaging approach to examine the functional consequences of preclinical dopaminergic nigrostriatal dysfunction in the human motor system. Specifically, we examined how a single heterozygous mutation in different genes associated with recessively inherited Parkinson disease alters the cortical control of sequential finger movements.
Methods:
Nonmanifesting individuals carrying a single heterozygous Parkin (n = 13) or PINK1 (n = 9) mutation and 23 healthy controls without these mutations were studied with functional MRI (fMRI). During fMRI, participants performed simple sequences of three thumb-to-finger opposition movements with their right dominant hand. Since heterozygous Parkin and PINK1 mutations cause a latent dopaminergic nigrostriatal dysfunction, we predicted a compensatory recruitment of those rostral premotor areas that are normally implicated in the control of complex motor sequences. We expected this overactivity to be independent of the underlying genotype.
Results:
Task performance was comparable for all groups. The performance of a simple motor sequence task consistently activated the rostral supplementary motor area and right rostral dorsal premotor cortex in mutation carriers but not in controls. Task-related activation of these premotor areas was similar in carriers of a Parkin or PINK1 mutation.
Conclusion:
Mutations in different genes linked to recessively inherited Parkinson disease are associated with an additional recruitment of rostral supplementary motor area and rostral dorsal premotor cortex during a simple motor sequence task. These premotor areas were recruited independently of the underlying genotype. The observed activation most likely reflects a “generic” compensatory mechanism to maintain motor function in the context of a mild dopaminergic deficit.
GLOSSARY
- BOLD
= blood oxygen level–dependent;
- CMA
= cingulate motor area;
- FDR
= false discovery rate;
- fMRI
= functional MRI;
- HRF
= hemodynamic response function;
- IPS
= intraparietal sulcus;
- M1HAND
= primary motor hand area;
- PD
= Parkinson disease;
- PMd
= dorsal premotor cortex;
- SMA
= supplementary motor area;
- SPM
= statistical parametric mapping;
- SVC
= small volume correction;
- TE
= echo time;
- TMS
= transcranial magnetic stimulation;
- TR
= repetition time;
- VOI
= volumes of interest.
Several genes have been identified that can lead to Parkinson disease (PD), including four recessively inherited forms caused by mutations in the Parkin (PARK2), DJ-1 (PARK7), PINK1 (PARK6), and ATP13A2 (PARK9) genes.1–3 These familial forms of PD show a substantial clinical overlap with sporadic PD. Nonmanifesting individuals who carry a single heterozygous mutation in the Parkin and PINK1 gene associated with recessively inherited PD have attracted particular interest.4 PET of dopaminergic neurotransmission showed that these individuals have a mild presynaptic dopaminergic dysfunction in the striatum.5–8 Therefore, nonmanifesting carriers of a single mutant allele provide a unique model to study the effect of a subclinical loss of dopamine-producing cells in the substantia nigra on the human motor system.9
In a recent functional MRI (fMRI) study, we provided evidence for a compensatory redistribution of neuronal activity within the motor system in nonmanifesting carriers of a heterozygous mutation in the Parkin gene. With internally cued movements, mutation carriers displayed a stronger activation of the right rostral cingulate motor area and left dorsal premotor cortex (PMd) compared to externally cued movements.10 They also showed stronger functional coupling between the rostral cingulate motor area and posterior putamen in the context of internal movement selection. Because mutation and non–mutation carriers performed the task equally well, these activity changes were interpreted as adaptive redistribution of neuronal activity in rostral motor cortical areas which helps to maintain motor function in the context of a latent nigrostriatal dysfunction.10
The present experiment extended our previous fMRI study in two directions. First, we used a different experimental task which required participants to quickly perform a brief “chunk” of three movements. In our previous fMRI study, the experimental task required the selection of single movements. The onset of each movement was externally paced at a low rate and consecutive movements were separated by periods of rest. By using a “real” motor sequence task, we examined how a heterozygous mutation in a gene linked to recessively inherited PD impacts on functional brain networks subserving sequential movements. We hypothesized that the regional expression of functional changes in motor cortical areas critically depends on the particular function probed by the experimental task. Therefore, the adaptive redistribution of cortical activity within preexisting motor networks was expected to be different for the motor sequence task as opposed to the previously used movement selection task. Specifically, we predicted that mutation carriers would show a compensatory recruitment of rostral premotor areas that are specialized for the control of complex motor sequences.
Second, we included nonmanifesting individuals carrying a single mutant allele in the Parkin or PINK1 gene. This enabled us to test whether the adaptive redistribution of neuronal activity in motor brain regions is specifically linked to mutations in a specific gene associated with recessively inherited PD. Given the closely related dysfunctional effects of mutations in both proteins in a drosophila model,11,12 our prediction was that the functional phenotype at a brain network level would be similar for both groups.
METHODS
Participants.
We studied 13 subjects (mean age 38.9 ± 5.8 years, seven men) carrying a single heterozygous mutation in the Parkin gene, either a deletion of exon seven (n = 7) or a single base-pair deletion in exon nine (c.del1072T) (n = 6).13 Nine other subjects (mean age 41.9 ± 5.7 years, seven men) carried a heterozygous c.1366C>T nonsense mutation of the PINK1 gene.14 Three of the Parkin 13,15 and five of the PINK1 14,16 mutation carriers had minor motor signs upon careful clinical examination, but were not aware of the motor signs and motor signs did not interfere with their daily activities. None of these subjects had a Unified Parkinson’s Disease Rating Scale score of more than 4 or met the international accepted diagnostic criteria of probable PD. Nine of the heterozygous carriers of a Parkin mutation had previously undergone 18F-DOPA PET showing a presynaptic dopaminergic deficit in the striatum.8
We also studied two groups of healthy age-matched controls: 13 volunteers (mean age 38.7 ± 5.5 years, seven men) who served as controls for the nonmanifesting Parkin mutation carriers and 10 volunteers (mean age 40.0 ± 5.9 years, seven men) formed the control group for the nonmanifesting PINK1 mutation carriers. Controls were recruited from a departmental register of volunteers and did not have mutations in Parkin or PINK1.
Participants had no history of a previous neuropsychiatric disease nor had they previously received dopaminergic or other antiparkinsonian drug treatment. All participants were consistent right-handers according to the Edinburgh handedness inventory.17 Written informed consent was obtained prior to the study. The experimental procedures had the approval of the local ethics committee.
Experimental design.
The fMRI experiment consisted of 10 alternating blocks of REST and TASK. During the TASK periods, participants repeatedly performed sequential finger movements with their right dominant hand. Each sequence consisted of three thumb-to-finger opposition movements instructed by external visual cues. Participants produced three different motor sequences in pseudorandom order. The details of the experimental task are given in figure 1A. Before fMRI, participants were familiarized with the task and practiced the respective finger sequences for approximately 5 minutes.
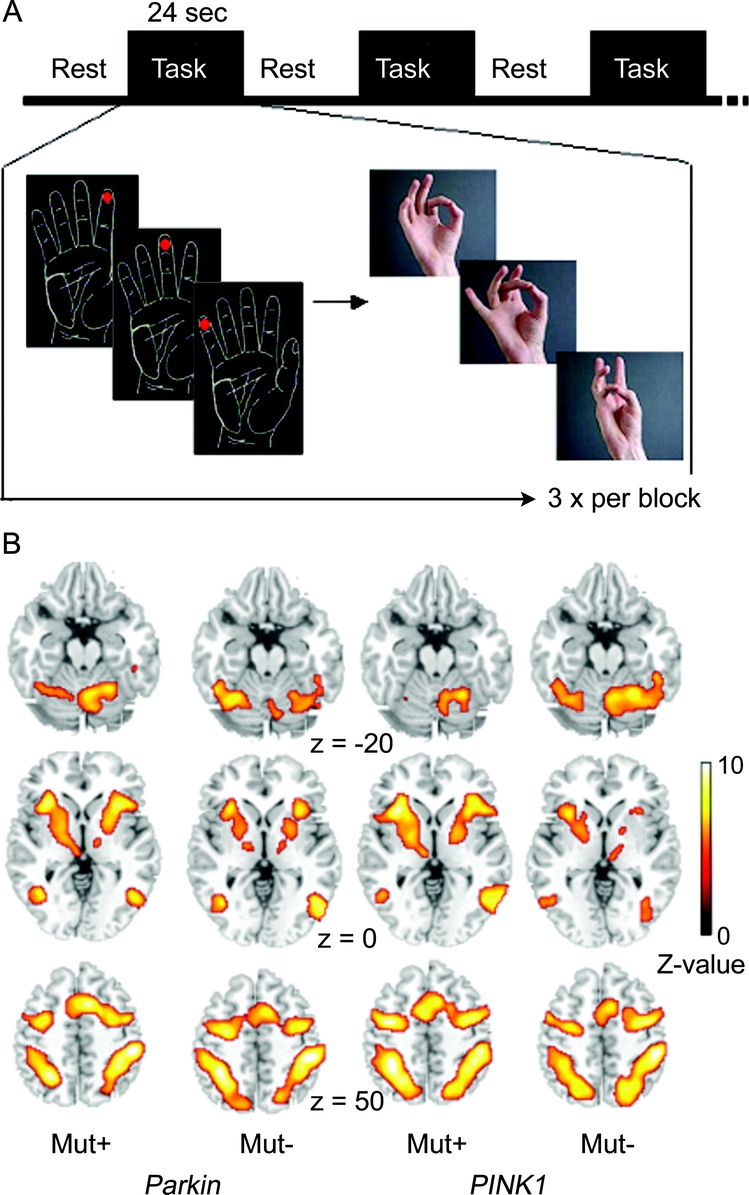
Figure 1 Experimental design (A) and main effect of motor task (B)
(A) Experimental design. The fMRI session consisted of 10 alternating periods without movements (REST) or sequential movements (TASK). Each block lasted for 24 seconds. There were 10 blocks of TASK and 10 blocks of REST. A two-dimensional drawing of the palm of the right hand was continuously presented in the center of the visual field throughout the fMRI session. During the REST periods, the line drawing of the hand was continuously presented but without dots. Participants were instructed to remain still and fixate the hand with their eyes. During each block of TASK, participants performed three motor sequences. Each sequence consisted of three thumb-to-finger opposition movements. At the onset of each movement trial, the index, middle, ring, or little finger was labeled with a red dot on a two-dimensional drawing of the palm of the right hand. The position and order of the red dot specified the motor sequence that had to be performed within a given trial. When the instruction cue disappeared from the screen, participants sequentially tapped with the tip of their right thumb onto the tip of the indicated fingers. They were asked to move at a convenient speed and to perform the task as accurately as possible. (B) Main effect of the motor task. The axial slices show the motor regions that showed a task-related increase in BOLD signal during the sequential finger movement task. The statistical parametric maps are superimposed onto a T2-weighted structural MRI template provided by MRIcro (http://www.sph.sc.edu/comd/rorden/mricro.html). The voxels of the activation maps are color-coded according to their Z values and thresholded at p < 0.05 using the FWE method as implemented in SPM2.
By choosing a short sequence, we kept the task simple, favoring automatic performance without a high level of monitoring. The use of longer sequences would have increased the load on working memory, possibly forcing subjects to divide the sequence into separate chunks.18 We randomly presented three sequences rather than repeating the same sequence during a given block. This forced the participants to continuously switch between different motor representations of simple overlearned sequences.
Our decision to select sequential finger movements as experimental task was based on two considerations. First, sequential finger movements have been extensively studied in PD, providing evidence for compensatory overactivity in the PMd and intraparietal sulcus in PD during sequential movements.19–21 Second, healthy controls show a linear increase in activity with sequence complexity in the rostral part of the supplementary motor area (referred to as pre-SMA) and the rostrodorsal portion of the right PMd.22,23 Therefore, we hypothesized that the latent dopaminergic dysfunction in presymptomatic carriers of a Parkin or PINK1 mutation results in a compensatory recruitment of the pre-SMA and right PMd to maintain motor performance within a normal range.
Participants performed 30 consecutive sequences per fMRI session. To assess performance during fMRI, we taped aluminum foil to the tips of the thumb and the fingers of the right hand. When the thumb and finger tips contacted each other, an electrical circuit was closed which was specific to a given finger. For each trial, we recorded the time during which the tip of the thumb had contact with the index, middle, ring, or little finger. This enabled us to calculate the time that elapsed between the first and last finger-to-thumb contact of the motor sequence, referred to as Tap1-Tap3 interval. To assess the stability of motor performance, we calculated the mean Tap1-Tap3 interval for 10 consecutive trials during the fMRI session.
MRI data acquisition.
Whole-brain MRI was performed on a 1.5 T Magnetom Symphony scanner (Siemens, Erlangen, Germany) equipped with a standard head coil. We used a T2*-weighted gradient-echo echoplanar sequence (repetition time [TR] = 3,000 msec, echo time [TE] = 40 msec, flip angle = 90°, matrix 64 × 64 voxels, field of view = 256 × 256 mm2, 30 axial slices, slice thickness: 4 mm) to map task-related changes in the blood oxygen level–dependent (BOLD) signal. A total of 160 brain volumes were acquired per session. We also obtained a whole-brain structural MRI dataset using a three-dimensional T1-weighted FLASH sequence (TR = 15 msec, TE = 5 msec, 192 axial slices, voxel size = 1 × 1 × 1 mm3, axial field of view = 256 × 256 mm2).
Data analysis.
Using the mean Tap1-Tap3 interval as dependent variable, we performed a two-factorial repeated-measures analysis of variance with the within-subject factor TIME (three levels: trial 1 to 10, trials 11–20, and trials 21–30) and between-groups factor GROUP (four levels: nonmanifesting PINK1 or Parkin mutation carriers and their respective control groups without mutation). The Greenhouse-Geisser method was used to correct for nonsphericity if appropriate. Depending on a significant F value, post hoc t tests were performed. Data are given as mean and onefold SD. A p value of <0.05 was considered significant.
The fMRI data were processed and analyzed using statistical parametric mapping (SPM) software (SPM2; Wellcome Trust Centre for Neuroimaging, London, UK; http://www.fil.ion.ucl.ac.uk/spm). The first two scans of each session were discarded to allow for steady-state magnetization. The remaining images were realigned to the first image and spatially normalized to MNI stereotactic space using a standard EPI template as implemented in SPM2. The normalized images were spatially smoothed with a Gaussian kernel of 9 mm at full-width half-maximum.
At the individual level, task-related changes in BOLD signal were estimated at each voxel by modeling the time course of alternating blocks as delta functions convolved with a hemodynamic response function (HRF). Based on this model we computed a t statistic for each voxel that tested for regional increases in BOLD signal during the finger sequence task. The result of the t statistics was used to generate a SPM of task-related increases in BOLD signal.
The contrast images obtained in each subject were entered into a two-sample t test for between-groups comparisons to test for between-group differences in brain activations between mutation carriers and their respective control groups without mutation. The individual motor Unified Parkinson’s Disease Rating Scale scores were included in the analysis as covariate of no interest. The resulting t values were corrected for multiple comparisons at voxel level, using the false discovery rate (FDR) correction method. Significance level was set at a corrected p value of p < 0.05. Any task-related BOLD signal change that reached an uncorrected p value of 0.001 but failed to survive FDR correction is descriptively reported as statistical trend.
We defined the pre-SMA, PMd, and intraparietal sulcus of both hemispheres as volumes of interest (VOI). The selection of cortical VOIs was based on previous neuroimaging studies. On the one hand, the pre-SMA and right PMd are increasingly active with the complexity of sequential finger movements in healthy individuals.22,23 On the other hand, patients with PD show bilateral increases in activity in PMd and intraparietal sulcus during sequential movements.19–21 Therefore, nonmanifesting mutation carriers should show an additional recruitment of the pre-SMA, PMd, and intraparietal sulcus during the finger sequence task. At the subcortical level, ROIs were placed in the posterior part of the putamen bilaterally. The putaminal ROIs were motivated by our recent morphometric study in which in a nearly identical group asymptomatic mutation carriers showed a putaminal increase in gray matter on T1-weighted structural MRIs.24
Each VOI was covered by a sphere. The diameter of the sphere was 27 mm which was threefold the FWHM used for Gaussian filtering. The spheres were centered on the peak increase in BOLD signal for the main effect of task. For these predefined VOIs, correction for multiple comparisons only considered voxels within the sphere. Outside the VOIs, all results were corrected across the whole brain.
RESULTS
Behavior.
All participants found the thumb-to-finger opposition tasks easy to perform. The maximum error rate was two sequential errors per session. Analysis of variance revealed no difference in mean Tap1-Tap3 interval among groups (p > 0.5). The mean Tap1-Tap3 interval was 1.44 ± 0.18 s among individuals carrying a Parkin mutation and 1.45 ± 0.08 s among controls without mutation. The mean Tap1-Tap3 interval was 1.40 ± 0.11 s in individuals with a PINK1 mutation and 1.36 ± 0.08s in the corresponding controls.
Functional MRI.
Epoch related analysis identified a bilateral set of sensorimotor areas where the BOLD signal increased when participants performed the finger sequence task (figure 1B and tables e-1 through e-3 on the Neurology® Web site at www.neurology.org). Mutation carriers showed increased activation in right rostral PMd and the pre-SMA compared to controls (figure 2, table 1). The overactivity in these rostral premotor areas was independent of the genotype (figure 2, table 1). The pre-SMA and rostral PMd showed the most prominent increase in task related activation as compared to any other area in the brain. No additional activations emerged in any other brain area, even when we lowered the statistical threshold to an uncorrected p value of 0.01 (extent threshold: 20 voxels). Mutation carriers showed no differences in task related deactivations relative to healthy controls.

Figure 2 Regional increases in task-related blood oxygen level–dependent (BOLD) signal changes in nonmanifesting carriers of a Parkin or PINK1 mutation
(A) Statistical parametric maps. Sagittal, coronal, and axial slices highlighting those voxels in the pre–supplementary motor area (SMA) and adjacent dorsal premotor cortex (PMd) that showed a relative increase in BOLD signal during the sequential finger movement task in mutation carriers relative to controls without a mutation. The statistical parametric maps are superimposed onto a T2-weighted structural MRI template provided by MRIcro (http://www.sph.sc.edu/comd/rorden/mricro.html). The voxels of the activation maps are color-coded according to their Z values. For illustrative purposes, the maps are thresholded at an uncorrected p value of p < 0.01. (B) Parameter estimates of task-related BOLD signal changes in the right and left pre-SMA and right dorsomedial PMd. The column plots give the mean β values (as estimated by the general linear model) for the task-related change in BOLD signal during the sequential finger movement task for each of the four groups (red columns = mutation carriers; yellow columns = non–mutation carriers). The β values are given in arbitrary units (AU) and refer to the voxel showing a peak difference between mutation carriers and noncarriers. Error bars equal the 95% confidence interval of the mean.
Table 1 Differences in task-related blood oxygen level–dependent (BOLD) signal changes between mutation carriers of a Parkin or PINK1 mutation and healthy controls without mutation
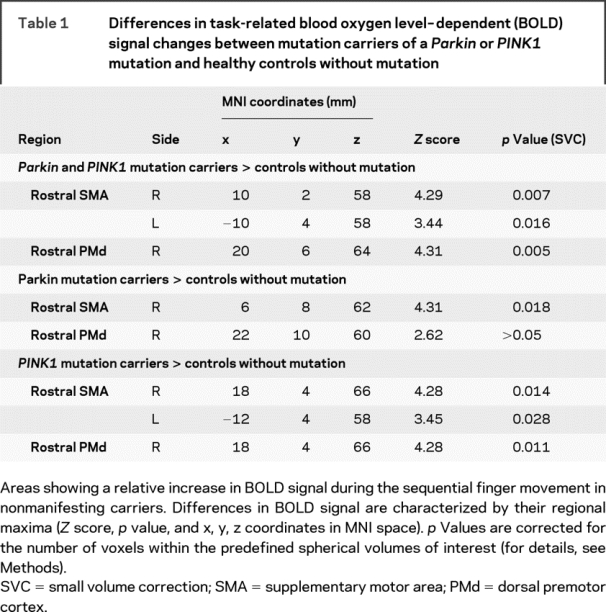
Relative to the corresponding control group, Parkin mutation carriers displayed an increased activation in the pre-SMA as well as a trend toward a stronger activation in the right rostral PMd. Likewise, PINK1 mutation carriers showed a bilateral overactivity in the pre-SMA which extended to the adjacent PMd. Controls without mutation showed no task-related regional increases in BOLD signal relative to the mutation carriers. The putamen showed a consistent task-related activation in all four groups. No between-group differences in task-related activity were detected in the VOIs covering the right and left putamen.
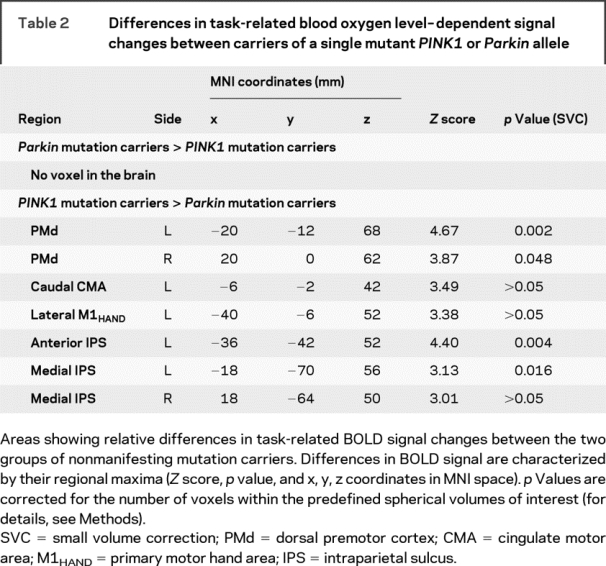
There were also differences in task-related BOLD signal changes between the two groups of mutation carriers. The left and right PMd as well as the anterior and medial portion of the left intraparietal sulcus displayed a stronger activation in PINK1 mutation carriers relative to Parkin mutation carriers (figure 3, table 2). Additional trends toward an increased activation were observed in the right medial intraparietal sulcus, the anterior cingulate cortex, and left primary motor cortex. There was no relative increase in BOLD signal with sequential finger movements in Parkin as opposed to PINK1 mutation carriers.

Figure 3 Relative increases in task-related blood oxygen level–dependent (BOLD) signal changes in nonmanifesting carriers of a PINK1 mutation compared with nonmanifesting carriers of a Parkin mutation
Axial slices showing voxels in dorsal frontoparietal cortex with significant increase in BOLD signal during the sequential finger movement task in PINK1 mutation carriers relative to Parkin mutation carriers. The statistical parametric maps are superimposed onto T2-weighted structural MR images provided by MRIcro (http://www.sph.sc.edu/comd/rorden/mricro.html). The voxels of the activation maps are color-coded according to their Z values. For illustrative purposes, the maps are thresholded at an uncorrected p value of p < 0.01.
Table 2 Differences in task-related blood oxygen level–dependent signal changes between carriers of a single mutant PINK1 or Parkin allele
DISCUSSION
When nonmanifesting heterozygous carriers of a Parkin or PINK1 mutation perform a simple motor sequence task, they recruit the pre-SMA and right rostral PMd which are not utilized by healthy controls without mutation. This finding extends our recent morphometric MRI study showing an increase in gray matter volume in the basal ganglia in a comparable group of nonmanifesting carriers of a Parkin or PINK1 mutation.25 Together, the functional and structural MRI data suggest that mutations in the Parkin and PINK1 gene produce a very similar functional and structural endophenotype. This implies that single heterozygous mutations in these two genes have a similar impact on the human motor system.
Converging evidence from neuroimaging and transcranial magnetic stimulation (TMS) show that in healthy individuals, the pre-SMA mainly contributes to motor sequence control in nonroutine situations. Accordingly, functional neuroimaging demonstrated an activation of the pre-SMA and rostral right PMd with new or complex motor sequences but not with sequences that were highly overlearned or easy to perform.22,23,26 The activation of the pre-SMA during sequential movements was attributed to the formation of and switch between visuomotor associations rather than the control of the movements per se.26,27 In the presence of a mutant Parkin or PINK1 allele, the “extra-recruitment” of the pre-SMA and adjacent PMd most likely reflects an adaptive mechanism by which the motor system counteracts the preexisting latent nigrostriatal dysfunction.5,6,8 Mutation carriers and controls showed equal performance when performing the simple motor sequence task. We propose that the mechanism by which mutation carriers maintain a normal level of performance is to recruit additional premotor regions that are specialized for handling complex sequential movements. We argue that the subclinical nigrostriatal neurotransmission in nonmanifesting mutation carriers produced a dysfunction of the corticobasal ganglia-thalamocortical motor loops involved in the control of overlearned motor sequences. This latent dysfunction rendered the simple sequence task more demanding in terms of neuronal motor control and specifically called on the “support” of rostromedial premotor cortex to maintain task performance.28 Because the pre-SMA and rostromedial PMd are reciprocally connected with prefrontal areas29,30 and receive inputs from the “nonmotor” (associative) territories of the cerebellum and basal ganglia,31 the increased activation of the pre-SMA and rostral PMd might be driven by a compensatory increase in neuronal input from connected prefrontal or subcortical areas during the task.
The adaptive recruitment of cortical premotor areas was restricted to the pre-SMA and the dorsomedial part of right rostral PMd. In a previous fMRI study, we also found an increase in rostral motor areas in nonmanifesting carriers of a Parkin mutation, but the spatial pattern of increased activity was different from the one found in the present study. When participants selected a finger movement with their right hand based on internal cues, individuals with a mutant Parkin allele showed a stronger activation of the rostral cingulate motor area and the ventrolateral part of left rostral PMd but not in rostral SMA and right rostromedial PMd.10 These findings lend support to the notion that a latent nigrostriatal dopaminergic dysfunction gives rise to variable patterns of activity changes in rostral premotor regions which critically depend on the specific motor functions probed by the experimental task. The observation that the compensatory recruitment of cortical motor areas is task-specific underscores the capacity of human sensorimotor networks to flexibly adapt to a regional dysfunction.32
We also identified some differences between carriers of mutations in the Parkin or PINK1 gene. In PINK1 mutation carriers, we observed an additional mainly left-hemispheric recruitment of frontoparietal areas, including distinct areas in left caudal PMd and intraparietal sulcus. It is unclear whether this reflects a true genotype-specific pattern of functional adaptation in the frontoparietal cortex. One possibility is that nonmanifesting PINK1 mutation carriers have a stronger functional impairment of the motor system, requiring recruitment of additional frontoparietal loops. Future studies could resolve this issue by correlating adaptive redistribution of cortical activity to the depth of the nigrostriatal dopaminergic deficit, for example using nuclear imaging.
An intriguing question is whether these increases in task-related activity persist, increase, or attenuate in mutation carriers who ultimately develop PD. A recent H2 15O PET study provided some evidence that the extra-recruitment of frontal motor areas still represents an effective mechanism of compensation in the early stage of sporadic PD.33 In that study, patients with PD achieved equal performance with healthy controls when learning short motor sequences. In patients, equal performance was associated with the additional recruitment of cortical areas that are normally specialized for learning more difficult sequences. Additional fMRI studies on high-risk populations as well as cross-sectional studies on drug-naïve patients with newly diagnosed sporadic or monogenic PD are needed to further address this important question.
Supplementary Material
Address correspondence and reprint requests to Dr. Hartwig Siebner, Danish Research Centre for Magnetic Resonance, Hvidovre University Hospital, Kettegaard Allé 30, DK-2650 Hvidovre, Denmark hartwig.siebner@drcmr.dk
Supplemental data at www.neurology.org
Editorial, page 1036
e-Pub ahead of print on November 26, 2008, at www.neurology.org.
Supported by a BMBF grant to H.R.S. (01 GO 0511) and F.B. (01 GO 0512) (NeuroImage-Nord) and by the 6th European Framework (EU-LSHB-CT-2006-037544-GENEPARK). C.K., F.B., and H.S. have been supported by the Volkswagenstiftung. B.F.L.v.N. and B.R.B. were supported by a NWO VIDI research grant (number: 917.76.352).
Disclosure: The authors report no disclosures.
Received May 2, 2008. Accepted in final form September 26, 2008.
REFERENCES
- 1.Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392:605–608. [DOI] [PubMed] [Google Scholar]
- 2.Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003;299:256–259. [DOI] [PubMed] [Google Scholar]
- 3.Valente EM, Salvi S, Ialongo T, et al. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol 2004;56:336–341. [DOI] [PubMed] [Google Scholar]
- 4.Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol 2007;6:652–662. [DOI] [PubMed] [Google Scholar]
- 5.Khan NL, Scherfler C, Graham E, et al. Dopaminergic dysfunction in unrelated, asymptomatic carriers of a single parkin mutation. Neurology 2005;64:134–136. [DOI] [PubMed] [Google Scholar]
- 6.Khan NL, Brooks DJ, Pavese N, et al. Progression of nigrostriatal dysfunction in a parkin kindred: an [18F]dopa PET and clinical study. Brain 2002;125:2248–2256. [DOI] [PubMed] [Google Scholar]
- 7.Khan NL, Valente EM, Bentivoglio AR, et al. Clinical and subclinical dopaminergic dysfunction in PARK6-linked parkinsonism: an 18F-dopa PET study. Ann Neurol 2002;52:849–853. [DOI] [PubMed] [Google Scholar]
- 8.Hilker R, Klein C, Ghaemi M, et al. Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene. Ann Neurol 2001;49:367–376. [PubMed] [Google Scholar]
- 9.vanEimeren T, Siebner HR. An update on functional neuroimaging of parkinsonism and dystonia. Curr Opin Neurol 2006;19:412–419. [DOI] [PubMed] [Google Scholar]
- 10.Buhmann C, Binkofski F, Klein C, et al. Motor reorganization in asymptomatic carriers of a single mutant Parkin allele: a human model for presymptomatic parkinsonism. Brain 2005;128:2281–2290. [DOI] [PubMed] [Google Scholar]
- 11.Park J, Lee SB, Lee S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006;441:1157–1161. [DOI] [PubMed] [Google Scholar]
- 12.Clark IE, Dodson MW, Jiang C, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006;441:1162–1166. [DOI] [PubMed] [Google Scholar]
- 13.Pramstaller PP, Kis B, Eskelson C, et al. Phenotypic variability in a large kindred (Family LA) with deletions in the parkin gene. Mov Disord 2002;17:424–426. [DOI] [PubMed] [Google Scholar]
- 14.Hedrich K, Hagenah J, Djarmati A, et al. Clinical spectrum of homozygous and heterozygous PINK1 mutations in a large German family with Parkinson disease: role of a single hit? Arch Neurol 2006;63:833–838. [DOI] [PubMed] [Google Scholar]
- 15.Pramstaller PP, Schlossmacher MG, Jacques TS, et al. Lewy body Parkinson’s disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol 2005;58:411–422. [DOI] [PubMed] [Google Scholar]
- 16.Hiller A, Hagenah JM, Djarmati A, et al. Phenotypic spectrum of PINK1-associated parkinsonism in 15 mutation carriers from 1 family. Mov Disord 2007;22:145–147. [DOI] [PubMed] [Google Scholar]
- 17.Oldfield RC. The assessment and analysis of handedness: the Edinburgh inventory. Neuropsychologia 1971;9:97–113. [DOI] [PubMed] [Google Scholar]
- 18.Kennerley SW, Sakai K, Rushworth MF. Organization of action sequences and the role of the pre-SMA. J Neurophysiol 2004;91:978–993. [DOI] [PubMed] [Google Scholar]
- 19.Carbon M, Eidelberg D. Functional imaging of sequence learning in Parkinson’s disease. J Neurol Sci 2006;248:72–77. [DOI] [PubMed] [Google Scholar]
- 20.Samuel M, Ceballos-Baumann AO, Blin J, et al. Evidence for lateral premotor and parietal overactivity in Parkinson’s disease during sequential and bimanual movements. A PET study. Brain 1997;120(Pt 6):963–976. [DOI] [PubMed] [Google Scholar]
- 21.Catalan MJ, Ishii K, Honda M, Samii A, Hallett M. A PET study of sequential finger movements of varying length in patients with Parkinson’s disease. Brain 1999;122(Pt 3):483–495. [DOI] [PubMed] [Google Scholar]
- 22.Boecker H, Dagher A, Ceballos-Baumann AO, et al. Role of the human rostral supplementary motor area and the basal ganglia in motor sequence control: investigations with H2 15O PET. J Neurophysiol 1998;79:1070–1080. [DOI] [PubMed] [Google Scholar]
- 23.Sadato N, Yonekura Y, Waki A, Yamada H, Ishii Y. Role of the supplementary motor area and the right premotor cortex in the coordination of bimanual finger movements. J Neurosci 1997;17:9667–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weihofen A, Ostaszewski B, Minami Y, Selkoe DJ. Pink1 Parkinson mutations, the Cdc37/Hsp90 chaperones and Parkin all influence the maturation or subcellular distribution of Pink1. Hum Mol Genet 2008;17:602–616. [DOI] [PubMed] [Google Scholar]
- 25.Binkofski F, Reetz K, Gaser C, et al. Morphometric fingerprint of asymptomatic Parkin and PINK1 mutation carriers in the basal ganglia. Neurology 2007;69:842–850. [DOI] [PubMed] [Google Scholar]
- 26.Sakai K, Hikosaka O, Miyauchi S, Sasaki Y, Fujimaki N, Putz B. Presupplementary motor area activation during sequence learning reflects visuo-motor association. J Neurosci 1999;19:RC1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rushworth MF, Hadland KA, Paus T, Sipila PK. Role of the human medial frontal cortex in task switching: a combined fMRI and TMS study. J Neurophysiol 2002;87:2577–2592. [DOI] [PubMed] [Google Scholar]
- 28.Isoda M, Hikosaka O. Switching from automatic to controlled action by monkey medial frontal cortex. Nat Neurosci 2007;10:240–248. [DOI] [PubMed] [Google Scholar]
- 29.Luppino G, Matelli M, Camarda R, Rizzolatti G. Corticocortical connections of area F3 (SMA-proper) and area F6 (pre-SMA) in the macaque monkey. J Comp Neurol 1993;338:114–140. [DOI] [PubMed] [Google Scholar]
- 30.Lu MT, Preston JB, Strick PL. Interconnections between the prefrontal cortex and the premotor areas in the frontal lobe. J Comp Neurol 1994;341:375–392. [DOI] [PubMed] [Google Scholar]
- 31.Akkal D, Dum RP, Strick PL. Supplementary motor area and presupplementary motor area: targets of basal ganglia and cerebellar output. J Neurosci 2007;27:10659–10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee L, Siebner HR, Rowe JB, et al. Acute remapping within the motor system induced by low-frequency repetitive transcranial magnetic stimulation. J Neurosci 2003;23:5308–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mentis MJ, Dhawan V, Feigin A, et al. Early stage Parkinson’s disease patients and normal volunteers: comparative mechanisms of sequence learning. Hum Brain Mapp 2003;20:246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.