

Abstract
Objective:
We sought to define the significance of brachial amyotrophic diplegia (flail arm syndrome [FA]) and the pseudopolyneuritic variant (flail leg syndrome [FL]) of amyotrophic lateral sclerosis (ALS; motor neuron disease).
Methods:
We analyzed survival in clinic cohorts in London, UK (1,188 cases), and Melbourne, Australia (432 cases). Survival from disease onset was analyzed using the Kaplan- Meier method and Cox proportional hazards model.
Results:
In the London cohort, the FA syndrome represented 11% and the FL syndrome 6% of the sample. Median survival was 35 months for limb onset and 27 months for bulbar onset ALS, whereas this was 61 months for FA syndrome (p < 0.001) and 69 months for FL syndrome (p < 0.001). Five-year survival in this cohort was 8.8% for bulbar onset, 20% for limb onset, 52% for FA syndrome, and 64% for FL syndrome. The ratio of men to women was 4:1 in the FA group compared to 2:1 in other limb onset cases. Excluding lower motor neuron FA and FL cases, progressive muscular atrophy comprised 4% of the sample and had a prognosis similar to typical limb onset ALS. In the Melbourne cohort, median survival for limb onset ALS was 31 months, bulbar onset 27 months, FA syndrome 66 months (p < 0.001), and FL syndrome 71 months (p = 0.001).
Conclusions:
The flail arm (FA) and flail leg (FL) syndromes had significantly better survival than typical amyotrophic lateral sclerosis (ALS) or progressive muscular atrophy cases that were not classified as FA or FL. Our findings underline the clinical and prognostic importance of the FA and FL variants of ALS.
GLOSSARY
- ALS
= amyotrophic lateral sclerosis;
- CI
= confidence interval;
- DTR
= deep tendon reflex;
- FA
= flail arm syndrome;
- FL
= flail leg syndrome;
- LL
= lower limbs;
- LMN
= lower motor neuron;
- MND
= motor neuron disease;
- NIV
= noninvasive ventilation;
- PMA
= progressive muscular atrophy;
- UL
= upper limbs;
- UMN
= upper motor neuron.
Amyotrophic lateral sclerosis (ALS) comprises several clinical phenotypes united by a common cellular and molecular pathology.1 The three main clinical categories defined by Aran, Charcot, Duchenne, and others in the 19th century and which were subsequently shown to have both diagnostic and prognostic significance were progressive bulbar palsy (bulbar onset ALS), classic limb onset (Charcot) ALS, and a lower motor neuron form termed progressive muscular atrophy (PMA).2–5 Prognostic factors in these forms of ALS have been delineated through clinic and population-based studies.6,7 Bulbar onset tends to have a worse prognosis than limb onset, and both forms have a worse prognosis than PMA.2,4–7 However, these three phenotypic categories do not fully capture the spectrum of clinical heterogeneity in ALS. This heterogeneity may contribute to diagnostic error and delay, and with the advent of large-scale whole genome studies that have the potential to identify genetic variants influencing both risk and phenotype, the definition of clinically and biologically important phenotypic variations is increasingly important.
Apart from these three main ALS subtypes, two other forms have been recognized since the late 19th and early 20th centuries but relatively inadequately studied, these being the flail arm (FA) and flail leg (FL) syndromes. In order to understand the natural history of these syndromes and to test the hypothesis that these distinctive phenotypes may be associated with significant differences in prognosis or in sex ratio (most likely reflecting important biologic differences from other ALS variants), we studied their natural history in two large clinic-based cohorts.
METHODS
Subjects and definitions.
All patients with a diagnosis of ALS (MND) referred to and assessed at the King’s MND Care and Research Center (London, UK) between 1993 and 2007 and the Calvary Health Care Bethlehem MND service (Melbourne, Australia) between 1995 and 2007 were screened. Clinical notes and databases were analyzed using a standardized form for recording data with agreed phenotyping criteria. The London database included 22 of 39 patients with the FA syndrome evaluated between 1993 and 1996 previously reported.8 The Melbourne database contained 10 FA and 6 FL patients who were included in a prospective study of disease progression and survival.9 The study was approved by the ethical review boards of the participating centers.
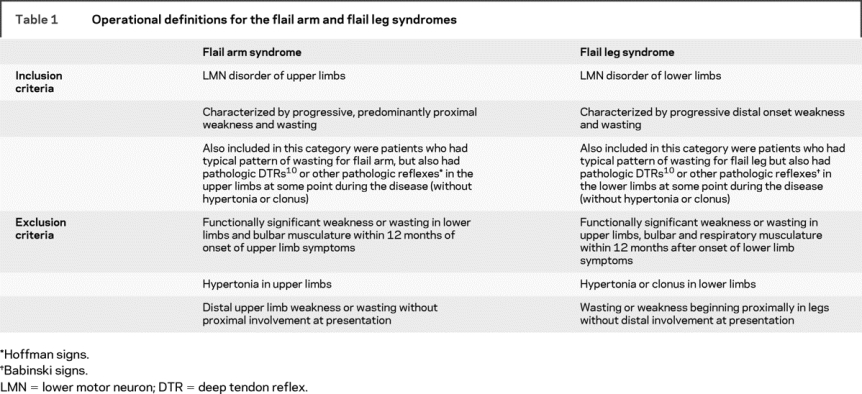
Patients were classified according to the revised El Escorial research diagnostic criteria,10 and categorized according to site of onset (bulbar or limb onset ALS). The operational definitions of the FA and FL syndromes are summarized in table 1. To differentiate these conditions from early limb onset ALS or PMA, we specified that functional involvement must be confined to the flail limb for at least 12 months after onset of symptoms. Patients presenting with a pure lower motor neuron syndrome that was not in keeping with the characteristic pattern of wasting of the FA or FL phenotypes were classified within PMA or limb onset ALS categories, depending on their subsequent clinical picture. Patients diagnosed with conditions such as spinal muscular atrophy, Kennedy syndrome, monomelic amyotrophy, Hirayama syndrome, and multifocal motor neuropathy, which are not considered part of the ALS spectrum, were excluded from the study. Patients with a family history of ALS were excluded from the analysis, as were individuals who developed disease under the age of 18 (juvenile onset).
Table 1 Operational definitions for the flail arm and flail leg syndromes
For the London cases, baseline demographic details and clinical data (date of onset of symptoms, site of onset of symptoms, site and dates of involvement of second and third regions of involvement) were extracted from the patient’s history and correspondence letters. Clinical findings at first examination and last follow-up examinations were noted. As wasting was almost always associated with weakness in the FA and FL patients, and for ALS patients spasticity manifests as weakness, we did not differentiate between those patients whose onset was not weakness, but rather spasticity or wasting without weakness. El Escorial category at presentation was defined according to the revised Airlie House criteria, but patients with pure lower motor syndromes (who could not be classified in the revised criteria) were classified into an additional category of suspected ALS. Use of riluzole was defined as taking riluzole for longer than 2 weeks. Diagnostic delay was the time from symptom onset until a confirmed diagnosis of ALS was made either by the referring neurologist or at the tertiary center, as recorded in the case records. Functional involvement of a CNS region was defined as development of weakness, wasting, or spasticity in a limb or dysarthria and dysphagia in the bulbar region. Timing of spread was based on the date of onset of symptoms and dates of development of functionally significant symptoms in a second and third region, which were gathered from the clinical history. The time (in months) between onset of symptoms and development of functional involvement in a second CNS region was termed spread to second region and time between functional involvement of the second and third CNS regions was termed spread from second to third region. In the Melbourne cases, baseline demographics, clinical features, and information on riluzole use were gathered. Survival times (months) in both populations were considered from onset of symptoms to either death or censoring date of January 1, 2008. Date of death was ascertained by clinic records, death certificates, and contact with the patient’s registered general practitioner.
Statistical analysis.
Clinical and demographic variables were compared between subgroups using one-way analysis of variance for continuous variables (with subsequent post hoc Dunnett tests) and χ2 test for categorical variables. Assumptions were tested by inspection of residual plots and variances, and variables which were non-normally distributed (diagnostic delay, spread to second region, spread from second to third region) were normalized by log transformation. Survival times were analyzed using the Kaplan-Meier method and survival curves between groups were compared using the log-rank test. Censoring date for survival data was January 1, 2008. The Cox proportional hazards model was used to assess the simultaneous effects of several independent variables on survival. Significance was tested at the 5% level and all analyses were done using SPSS 15.0 software package.
RESULTS
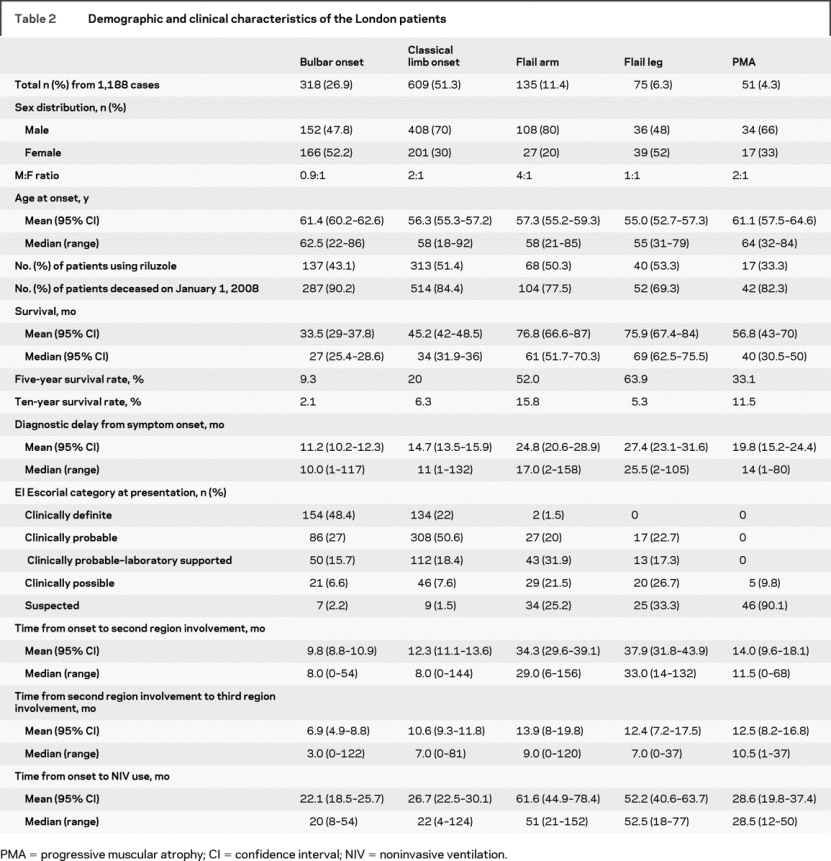
The demographic features of the London cases are shown in table 2. In the London database, a total of 1,311 cases met the inclusion criteria for analysis and survival data were complete for 1,188 (90.6%) cases. Of the 1,188 cases included in the survival analysis, 318 (27%) had the bulbar onset form of ALS, 609 (51%) had the classic limb onset form of ALS, 135 (11%) had the FA syndrome, and 75 (6%) had the FL syndrome. Fifty-one individuals (4%) with a diagnosis of PMA were included in the survival and clinico-demographic analysis for comparison. The M:F ratio for typical ALS was 1.5:1 overall (bulbar = 0.9:1, limb = 2:1) but the FA group had a M:F ratio of 4:1 while the FL group showed an equal M:F ratio. The mean age at symptom onset was different between phenotype groups (F = 16.5; p < 0.001), but post hoc analysis showed there was no difference between limb onset ALS and FA cases (p = 0.89) or FL cases (p = 0.90). In the limb onset ALS group, 73% of cases had either clinically definite or clinically probable disease according to the revised El Escorial criteria at presentation, whereas this was 22% for FA and 23% for FL. Twenty-five percent of FA cases and 33% of FL cases did not fall within the revised criteria at presentation. A similar proportion of patients were using riluzole across the subgroups except for PMA cases.
Table 2 Demographic and clinical characteristics of the London patients
FA and FL cases much more commonly remain restricted to one region at 18, 24, and 36 months from symptom onset compared to other phenotypes (see table e-1 on the Neurology® Web site at www.neurology.org). For FA, symptoms were confined to the arms for 18 months in 56% of cases, for 24 months in 46% of cases, and for 36 months in 27% cases. For FL, symptoms were confined to the legs for 18 months in 63% of cases, for 24 months in 48% of cases, and for 36 months in 28% of cases. The median time for symptoms to spread to a second CNS region was similar in bulbar onset and limb onset. There was no difference in time to spread between PMA and limb onset ALS (p = 0.3). Time to spread was longer in FA and FL cases in comparison to both limb onset ALS and PMA (both p < 0.001). The time to spread to a second region correlated with survival for all phenotypes (Pearson r = 0.65; p < 0.001). However, the time to spread from second to third region was only different between bulbar onset cases and other phenotypes (p < 0.001). Diagnostic delay was longer in both FA and FL subgroups compared to limb onset ALS (both p < 0.001). Similarly, the median time from onset to noninvasive ventilation use was longer in the FA and FL patients compared to limb onset cases (both p < 0.001).
The overall median survival from onset of symptoms in the London cohort was 35 months. The bulbar onset survival was 27 months and limb onset 34 months. The longest survival was found in the FL (69 months) and FA (61 months) phenotypes. Consequently, the 5-year survival rates were much higher in FL and FA phenotypes (FL = 63.9%, FA = 52%) in comparison with limb onset ALS (20%). The 10-year survival rates for FA remained higher than other phenotypes, but for FL this reduced to a similar level to limb onset ALS phenotype. The Kaplan-Meier survival curves for the five phenotype subgroups (figure 1A) were different overall (log rank χ2 = 112.1; p < 0.001). Post hoc tests show the curve for FA differs from limb onset (p < 0.001) and PMA (p = 0.008). Similarly, the curve for FL differed from limb onset (p < 0.001) and PMA (p = 0.002). Patients with FA, FL, and PMA fared better than typical ALS at any given time point as indicated by the curves. During the observed time period, 41.8% of FA patients and 58.9% of FL patients had or developed at least one UMN sign in the flail region. There was no difference in survival between patients who had a pure LMN syndrome and those who had at least one focal UMN sign (log rank χ2 = 0.894; p = 0.344).

Figure 1 Survival curves for each phenotype category in the London population
(A) Kaplan-Meier survival curves for each phenotype category in the London population. (B) Survival curves for each phenotype after adjusting for age at onset, gender, riluzole use, El Escorial category at presentation, and diagnostic delay at the covariate means using Cox regression model. PMA = progressive muscular atrophy.
A Cox model (table e-2) was constructed on the London cohort with phenotype, age at onset, gender, bulbar onset, riluzole use, El Escorial category at presentation, and diagnostic delay as independent variables (χ2 = 371.1; df = 12; p < 0.001) and the resulting curves for adjusted survival at the covariate means are shown in figure 1B. Gender was not a significant predictor of survival in either Kaplan-Meier analysis or in the Cox model (p = 0.302). Bulbar onset was significant in a Cox model that included only phenotype, age at onset, and gender as variables, but was not significant in the final model when diagnostic delay, El Escorial category at presentation, and riluzole use were added (p = 0.117). Compared to having limb onset ALS, having FA phenotype reduced the hazard by 37.9% (p < 0.001) and FL reduced hazard by 32% (p = 0.014). Age at onset increased the hazard by 2.7% for each year the patient’s age at onset is above the mean (p < 0.001). Riluzole use reduced the hazard by 25.9% (p < 0.001). Compared to having at presentation clinically definite disease by El Escorial criteria, having clinically suspected disease reduced the hazard by 53.6%; clinically possible disease by 41.8%; clinically probable–laboratory supported disease by 35.5%; and clinically probable disease by 25.9% (all p < 0.001). For diagnostic delay, hazard is reduced by 3.9% for every month longer it took to confirm the diagnosis, above the mean diagnostic delay (p < 0.001).
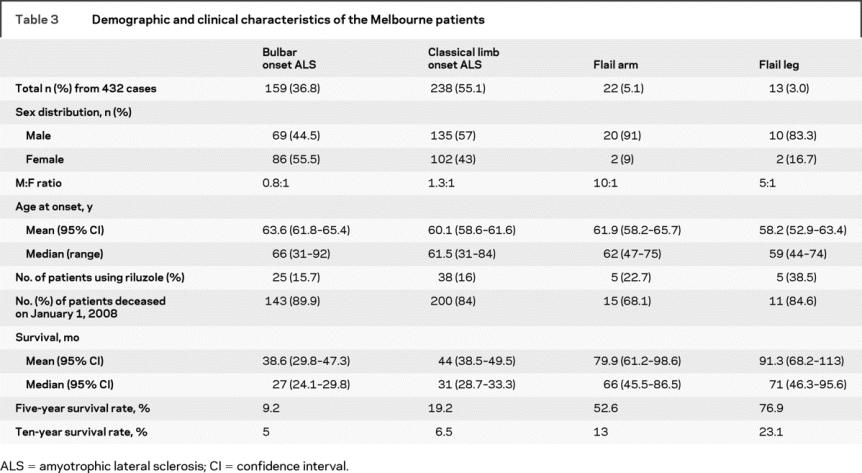
The demographic features of the Melbourne cases are shown in table 3. The database contained 567 cases meeting the inclusion criteria and clinical data were complete for 432 cases (76.1%). Of the 432 cases included in the analysis, 159 (36.8%) had bulbar onset ALS, 238 (55.1%) had classic limb onset, 22 (5.1%) had FA, and 13 (3%) were identified with FL syndrome. The M:F ratio was 1.1:1 for typical ALS (bulbar = 0.8:1, limb = 1.3:1), 10:1 for FA, and 5:1 for FL. Mean age at onset was different between bulbar and limb onset (p = 0.003) but not between limb onset, FA (p = 0.85), and FL (p = 0.9) subgroups. Riluzole use was lower in the Melbourne patients as it was not licensed for use in Australia until August 2003.
Table 3 Demographic and clinical characteristics of the Melbourne patients
The overall median survival from onset in the Melbourne cases was 31 months for all patients. The shortest median survival was in the bulbar onset group (27 months), followed by limb onset (31 months), FA (66 months), with the longest survival being in the FL group (71 months). Five-year survival was 9% for bulbar onset, 19% for limb onset, 53% for FA, and 77% for FL. The survival curves for each phenotype are shown in figure 2. The log-rank test indicated that overall survival patterns were different (χ2 = 43.0, p < 0.001). Post hoc comparisons showed the curve for limb onset differed from that of FA (p < 0.001) and FL (p < 0.001).

Figure 2 Kaplan-Meier survival curves for each phenotype category in the Melbourne population
DISCUSSION
The FA phenotype was described by Vulpian11 in 1886 as a syndrome of proximal weakness and wasting of the upper limbs (scapulohumeral variant of progressive muscular atrophy or forme scapulo-humérale). The condition has been variously termed the Vulpian-Bernhardt syndrome,12,13 hanging-arm syndrome,14 neurogenic man-in-a-barrel syndrome,15 brachial amyotrophic diplegia,15 or the FA syndrome.8 The syndrome typically presents with progressive upper limb weakness and wasting that is often symmetric and proximal, without significant functional involvement of lower limbs or bulbar muscles. Most patients with FA have or later develop EMG evidence of lower limb involvement15,16 and bulbar involvement develops in 27–77% of patients.8,17,18 Electrophysiologic studies indicate that cortical and peripheral hyperexcitability are present in FA syndrome as in typical ALS.18 Pathologic studies on two cases have shown anterior horn cell loss with Bunina bodies, ubiquitin-positive skein-like inclusions, and Lewy body-like inclusions in the remaining motor neurons, typical of ALS.16,19
The FL syndrome was first recognized by Pierre Marie and first described by his student Patrikios20 and was known as the pseudopolyneuritic variant of ALS (forme pseudopolynévritique de la sclérose latérale amyotrophique),2,21 the Marie-Patrikios form, or the peroneal form of ALS.22 Marie and Patrikios described a syndrome of distal onset weakness and wasting of the lower limbs which was asymmetric in onset, with absent lower limb tendon reflexes, slow progression, and subtle or late UMN signs. Population and clinic-based studies have shown that FL had the longest median and 5-year survival rates.23–25 Central motor conduction times are markedly prolonged despite the absence of pyramidal signs.26 The pathology of this condition is that of ALS, with extensive myelinated fiber loss in the lateral corticospinal tracts of the thoracic and lumbar spinal cord segments.27,28
We have shown in this study that the natural history of the FA and FL syndromes differs from more typical forms of ALS. The FA and FL syndromes have a significantly better prognosis in terms of median and 5-year survival rates compared to bulbar and limb onset ALS. The longer FA is confined to the arms, and FL is confined to the legs, the longer is survival. However, for all phenotypes, a longer duration to involvement of a second region was associated with better prognosis. In addition, our data suggest that the FA and FL cases that may have been classified as PMA in previous studies probably account for the supposed better prognosis of PMA,6,7,29,30 since in the London cohort PMA cases falling outside the definitions of FA and FL syndromes had a survival probability identical to that of typical ALS. We acknowledge that specification of case definitions is likely to account to some degree for the differences. The distinction between FA without UMN signs and PMA cases that remain confined to arms for >12 months was made on the pattern of muscle wasting at presentation, and it is not surprising that more generalized disease at onset is associated with worse prognosis. We also confirmed that the FA syndrome is more common in men, with a male to female ratio of 4:1. While this is less striking than the ratios observed in earlier studies of 9:18 or 10:1,18 our observations based on a much larger sample are more likely to be robust. For FL, contrary to the original description where this syndrome was considered to be more common in women, the M:F ratio was 1:1 in the London cohort, although in the smaller Melbourne cohort the excess of men is most likely an artifact of sample size.
The validity of our observations based on the London database is strengthened by the findings from the Melbourne cohort. The natural history data are similar, suggesting that our observations are valid for ALS populations drawn from a similar (predominantly Caucasian) genetic background, despite the biases inherent in any clinic cohort. Nevertheless, we consider it unlikely that selection bias seriously undermines the general validity of our observations. A population-based cohort study is less likely to be biased, but it is difficult to achieve the same level of clinical detail and sample size required for analysis of the natural history of relatively uncommon subgroups such as the FA and FL syndromes.23,24 Nonetheless, median survival from symptom onset in the bulbar and limb onset ALS groups in our study is comparable to that found in previous large clinic-based cohort studies6,31,32 and in large population registry studies,4,5 making it unlikely that we have overestimated survival. In the future, large population-based samples comprising detailed and standardized phenotypic information will be required to validate or modify our conclusions.
AUTHOR CONTRIBUTIONS
L. Wijesekera and A. Al-Chalabi performed the statistical analysis.
ACKNOWLEDGMENT
The authors thank the Motor Neuron Disease Association of England, Wales, and Northern Ireland for support of the King’s MND Care and Research Center since 1994; Dr. Daniel Stahl for advice on statistical analysis of the data; Dr. Tibor Hortobagyi for help with translation; and the staff and patients at both participating centers.
Supplementary Material
Address correspondence and reprint requests to Prof. P.N. Leigh, MRC Center for Neurodegeneration Research, Kings College London, Institute of Psychiatry, PO 41, Department of Clinical Neuroscience, London SE5 8AF, UK pnigel.leigh@iop.kcl.ac.uk
Supplemental data at www.neurology.org
*A.A.-C. and P.N.L. are joint senior authors who contributed equally.
Disclosure: The authors report no disclosures.
Received May 20, 2008. Accepted in final form January 8, 2009.
REFERENCES
- 1.Kato S, Shaw P, Wood-Allum C, Leigh PN, Shaw CE. Amyotrophic lateral sclerosis. In: Dickson DW, ed. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. ISN Neuropath Press; 2003: 350–371. [Google Scholar]
- 2.Bonduelle M. Amyotrophic lateral sclerosis. In: Vinken PJ, Bruyn GW, Klawans HL, de Jong JU, eds. Handbook of Clinical Neurology. North-Holland Publishing Company; 1975: 281–338. [Google Scholar]
- 3.Visser J, de Jong JMBV, Visser MD. The history of progressive muscular atrophy: syndrome or disease? Neurology 2008;70:723–727. [DOI] [PubMed] [Google Scholar]
- 4.Logroscino G, Traynor BJ, Hardiman O, et al. Descriptive epidemiology of amyotrophic lateral sclerosis: new evidence and unsolved issues. J Neurol Neurosurg Psychiatry 2008;79:6–11. [DOI] [PubMed] [Google Scholar]
- 5.del Aguila MA, Longstreth WT, Jr, McGuire V, Koepsell TD, van Belle G. Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology 2003;60:813–819. [DOI] [PubMed] [Google Scholar]
- 6.Norris F, Shepherd R, Denys E, et al. Onset, natural history and outcome in idiopathic adult motor neuron disease. J Neurol Sci 1993;118:48–55. [DOI] [PubMed] [Google Scholar]
- 7.Chancellor AM, Slattery JM, Fraser H, Swingler RJ, Holloway SM, Warlow CP. The prognosis of adult-onset motor neuron disease: a prospective study based on the Scottish Motor Neuron Disease Register. J Neurol 1993;240:339–346. [DOI] [PubMed] [Google Scholar]
- 8.Hu MT, Ellis CM, Al-Chalabi A, Leigh PN, Shaw CE. Flail arm syndrome: a distinctive variant of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 1998;65:950–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Talman P, Forbes A, Mathers S. Clinical phenotypes and natural progression for motor neuron disease: analysis from an Australian database. Amyotroph Lateral Scler Epub 2008 Jun 18:1–6. [DOI] [PubMed]
- 10.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 11.Vulpian A. Maladies du Système Nerveux (Moelle Épinière). Paris: Octave Dion; 1886:346. [Google Scholar]
- 12.Gamez J, Cervera C, Codina A. Flail arm syndrome of Vulpian-Bernhart’s form of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 1999;67:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gamez J, Cervera C. Brachial amyotrophic diplegia: a slowly progressive motor neuron disorder. Neurology 2000;54:2355. [PubMed] [Google Scholar]
- 14.Mulder DW. The clinical syndrome of amyotrophic lateral sclerosis. Proc Staff Meet Mayo Clin 1957;32:427–436. [PubMed] [Google Scholar]
- 15.Katz JS, Wolfe GI, Andersson PB, et al. Brachial amyotrophic diplegia: a slowly progressive motor neuron disorder. Neurology 1999;53:1071–1076. [DOI] [PubMed] [Google Scholar]
- 16.Sasaki S, Iwata M. Atypical form of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 1999;66:581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Couratier P, Truong C, Khalil M, Deviere F, Vallat JM. Clinical features of flail arm syndrome. Muscle Nerve 2000;23:646–648. [DOI] [PubMed] [Google Scholar]
- 18.Vucic S, Kiernan MC. Abnormalities in cortical and peripheral excitability in flail arm variant amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2007;78:849–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sasaki S, Iwata M. Motor neuron disease with predominantly upper extremity involvement: a clinicopathological study. Acta Neuropathol 1999;98:645–650. [DOI] [PubMed] [Google Scholar]
- 20.Patrikios JS. Contribution à l’Étude des Formes Cliniques et de l’Anatomiepathologique de la Sclérose Latérale Amyotrophique. Paris: Paris University; 1918. [Google Scholar]
- 21.Alema G, Brusa A, Pastorino P, Sacco G. [On 3 cases of the pseudopolyneuritic form of amyotrophic lateral sclerosis: anatomic and electromyographic study.] J Neurol Sci 1967;4:241–257. [DOI] [PubMed] [Google Scholar]
- 22.Hemmer R. [On the peroneal form of amyotrophic lateral sclerosis.] Nervenarzt 1955;26:400–401. [PubMed] [Google Scholar]
- 23.Salemi G, Fierro B, Arcara A, Cassata M, Castiglione MG, Savettieri G. Amyotrophic lateral sclerosis in Palermo, Italy: an epidemiological study. Ital J Neurol Sci 1989;10:505–509. [DOI] [PubMed] [Google Scholar]
- 24.Guidetti D, Bondavalli M, Sabadini R, et al. Epidemiological survey of amyotrophic lateral sclerosis in the province of Reggio Emilia, Italy: influence of environmental exposure to lead. Neuroepidemiology 1996;15:301–312. [DOI] [PubMed] [Google Scholar]
- 25.Mortara P, Bardelli D, Leone M, Schiffer D. Prognosis and clinical varieties of ALS disease. Ital J Neurol Sci 1981;2:237–242. [DOI] [PubMed] [Google Scholar]
- 26.Kachi T, Sobue G, Yamada T, Tamura T, Ando K. [Central motor conduction time in the pseudopolyneuritic form of amyotrophic lateral sclerosis.] Rinsho Shinkeigaku 1991;31:1029–1031. [PubMed] [Google Scholar]
- 27.Terao S, Sobue G, Hashizume Y, Mukai E, Mitsuma T. [A clinicopathological study of the somatic motor efferents in the pseudopolyneuritic form of amyotrophic lateral sclerosis]. Rinsho Shinkeigaku 1991;31:163–169. [PubMed] [Google Scholar]
- 28.Terao S, Sobue G, Hashizume Y, Mitsuma T, Takahashi A. Disease-specific patterns of neuronal loss in the spinal ventral horn in amyotrophic lateral sclerosis, multiple system atrophy and X-linked recessive bulbospinal neuronopathy, with special reference to the loss of small neurons in the intermediate zone. J Neurol 1994;241:196–203. [DOI] [PubMed] [Google Scholar]
- 29.Mortara P, Chio A, Rosso MG, Leone M, Schiffer D. Motor neuron disease in the province of Turin, Italy, 1966–1980: survival analysis in an unselected population. J Neurol Sci 1984;66:165–173. [DOI] [PubMed] [Google Scholar]
- 30.Chio A, Brignolio F, Leone M, et al. A survival analysis of 155 cases of progressive muscular atrophy. Acta Neurol Scand 1985;72:407–413. [DOI] [PubMed] [Google Scholar]
- 31.Testa D, Lovati R, Ferrarini M, Salmoiraghi F, Filippini G. Survival of 793 patients with amyotrophic lateral sclerosis diagnosed over a 28-year period. Amyotroph Lateral Scler Other Motor Neuron Disord 2004;5:208–212. [PubMed] [Google Scholar]
- 32.Haverkamp LJ, Appel V, Appel SH. Natural history of amyotrophic lateral sclerosis in a database population: validation of a scoring system and a model for survival prediction. Brain 1995;118:707–719. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.