

Abstract
Immune escape driven by selection pressure from virus-specific CD8 T cells has been demonstrated in both chimpanzees and humans infected with the hepatitis C virus (HCV). Although escape mutations have also been characterized in major histocompatibility complex (MHC) class II–restricted HCV epitopes, it is unknown whether selection-driven immune escape by CD4 T cell epitopes is a significant factor in the failure of these responses or contributes to persistent infection. To address this issue, evolution of MHC class I– and class II–restricted HCV epitopes was compared in four chimpanzees persistently infected with the virus for more than 10 years. We identified an amino acid change in a CD4 epitope of the HCV NS3 protein in one of the chimpanzees 3 years after infection. This mutation resulted in diminished activation, cytokine production (interferon-γ and interleukin-2), and proliferation by an epitope-specific CD4 T cell line. We expanded our analysis to determine if mutations were common in multiple CD4 versus CD8 T cell epitopes in the four chronically infected animals. Whereas we observed mutations in over 75% of CD8 T cell epitopes analyzed in this study, only 18% of CD4 T cell epitopes analyzed showed amino acid changes. The frequency of changes in class II epitopes was not different from flanking regions, so CD4 T cells rarely exert selection pressure against the HCV genome.
Conclusion
Apparent mutational escape can occur in MHC class II–restricted epitopes, but this is uncommon when compared with class I–restricted epitopes in the same individual. This indicates that other mechanisms for silencing CD4 T cells are dominant in persistent HCV infections.
Studies of hepatitis C virus (HCV) infection in humans and chimpanzees, the only animal model of natural HCV infection, have documented a critical role of T cell responses in preventing persistent lifelong viremia. In the minority of individuals developing self-limiting infection, control of acute phase virus replication is critically dependent on the expansion of HCV-specific CD4 and CD8 T cells.1–7 In contrast, the establishment of a persistent infection is associated with an impaired virus-specific CD8 T cell response and failure to sustain virus-specific CD4 T cells past the point of apparent control of virus replication.7–11
The failure of CD8 T cell responses directed against HCV epitopes in persistent hepatitis C is likely the result of multiple factors. Functional anergy, including the loss of proliferative capacity, impaired cytokine production, and diminished cytotoxicity, has been shown to be mediated in part by signaling through the inhibitory coreceptors programmed cell death-1 and cytotoxic T lymphocyte antigen-4.12–16 Additionally, due to the low fidelity of the viral polymerase, HCV genomes display a high rate of mutation, and CD8 T cell–mediated immune selection pressure has been demonstrated in both humans and chimpanzees to drive the accumulation of escape mutations, which can subvert the immune response and contribute to viral persistence.6,17–20 These mechanisms are also potentially linked, as demonstrated in a recent study that showed decreased coinhibitory receptor expression on HCV-specific CD8 T cells that recognize mutated versus intact viral epitopes.16
In contrast, much less is known regarding the mechanisms driving virus-specific CD4 T cell failure in chronic HCV infection. Comparisons of HCV-specific CD4 T cell responses in chronic versus resolved HCV infections have shown that an inability to control viral replication is associated with early failure of CD4 T cell help.7–11 Although HCV-specific CD4 T cells have been detected in both the blood and liver during chronic infection, these responses are generally greatly attenuated in breadth, proliferative capacity, and production of T helper 1 cytokines.7,8,10,20,21 Several reports have documented amino acid changes arising in well-characterized major histocompatibility complex (MHC) class II–restricted epitopes of HCV, with some conferring escape from the CD4 T cell response, or skewing it toward a T helper 2 profile of cytokine secretion.7,22–24 These data would suggest the hypothesis that HCV-specific CD4 T cells exert immune selection pressure similar to CD8 T cell responses to HCV, thus driving HCV escape mutations and promoting viral persistence. Nevertheless, no comprehensive analysis has yet been performed to explore how common such CD4 T cell escape mutations are, or to compare their frequency with escape mutations in CD8 T cell epitopes.
To determine if HCV immune escape from CD4 T cells was a significant factor in driving the failure of these virus-specific cells, we analyzed the sequences of multiple CD4 and CD8 T cell HCV epitopes for mutations in the circulating virus of four persistently infected chimpanzees. The input genotype 1a HCV strain (HCV-1/910) was used for comparison, and mutational frequencies in MHC class I– and II–restricted epitopes and nonepitope flanking regions were assessed. We report here that HCV CD4 T cell epitopes exhibit far fewer amino acid changes than CD8 T cell epitopes. Although we identified a mutation in a single CD4 T cell epitope in the NS3 protein that impaired activation of a specific CD4 T cell clone, the late emergence of this mutation makes it unlikely to have been a factor in initial establishment of viral persistence. Finally, we confirmed that amino acid changes occur at elevated frequencies in restricted CD8 T cell epitopes during persistent HCV infection, whereas amino acid replacements in restricted CD4 epitopes occur no more frequently than in nonrestricted epitopes or flanking regions, consistent with the hypothesis that CD8 T cells exert significant selection pressure on HCV to drive immune escape, whereas CD4 T cells do not.
Materials and Methods
Animals
Chimpanzees (Pan troglodytes) were maintained at the University of Louisiana at Lafayette New Iberia Research Center (New Iberia, LA) under standard conditions for humane care and in compliance with National Institutes of Health guidelines. Chimpanzees CBO603, CBO609, and CH-503 were infected with the HCV-1/910 virus in 1992 and developed persistent viremia. Chimpanzee NM1238 developed a chronic infection after challenge with the HCV-1/910 strain in 1982 and had an average viral load of 2.09 × 106 genome equivalents/mL plasma over the 20-month span of this study. Virologic and clinical data for these animals during the acute phase of infection are unknown.
Isolation of Viral RNA and Sequencing of Targeted Epitopes
Complementary DNA was prepared from HCV viral RNA isolated from blood collected in EDTA then subsequently amplified and sequenced as described.25 Sequence data are available through GenBank (accession nos. GQ848648-GQ848872, GQ870457-GQ870618).
Intracellular Cytokine Staining, Proliferation, and Phosphorylation Assays
Intracellular Cytokine Staining
The chimpanzee 4x0287 CD4 T cell clone 3D,26 specific for the NS31376 epitope YGKAIPLEVI, was analyzed for interferon-γ (IFN-γ) and interleukin-2 (IL-2) production by intracellular cytokine staining as described.26,27 Cells were stimulated with either the NS31376 wild-type (WT) (YGKAIPLEVI) peptide or with one of the following mutated peptides at various concentrations as indicated for each experiment: NS31376 M1 (YGKAIPLAAI), NS31376 M2 (YGKAIPLAVI), or NS31376 M3 (YGKAIPLEAI) (Genemed Synthesis, Inc., San Antonio, TX). Cells were stained with anti–CD4-PerCP, anti–CD3-APC, anti–IFN-γ –PE, and anti–IL-2–FITC antibodies (BD Biosciences).
Extracellular Signal-Regulated Kinase (ERK) 1/2 Phosphorylation Assay
Clone 3D cells were stimulated at 37°C for 15 minutes with autologous Epstein-Barr virus–transformed B-lymphoblastoid cells (BLCL) pulsed for 2 hours with 10-fold dilutions from 1 μg/mL to 0.001 μg/mL of the NS31376 WT peptide or the NS31376 M1 peptide. Cells were then stained with anti–CD4-PerCP and anti–CD19-phycoerythrin (BD Biosciences) antibodies in FACS, followed by intracellular staining with anti–phospho-p44/42 mitogen-activated protein kinase (ERK 1/2)–APC (Cell Signaling Technology, Boston, MA) as described above.
Proliferation Assay
The CD4 T cell clone 3D was labeled with 5-(and 6-)carboxyfluorescein diacetate succinimidyl ester (CFSE) (3 μM), then stimulated with either the NS31376 WT peptide or the NS31376 M1 peptide (0.01 μg/mL). Cells were harvested on days 3, 4, or 5 and stained with anti–CD3-APC and anti–CD4-PE antibodies in FACS wash, then counterstained with pro-pidium iodide to exclude dead populations from analysis.
All flow cytometric analyses were performed on a FACSCalibur instrument using CellQuest (BD Biosciences) software.
Statistical Analyses
The rates of synonymous nucleotide substitutions (dS) and nonsynonymous nucleotide substitutions (dN) were calculated in the four study animals for MHC class I– and class II–restricted epitopes, along with nonepitope flanking regions approximately 200 nucleotides upstream and downstream of the epitopes, essentially as described.17,28
Results
Identification of a Mutation in an MHC Class II–Restricted HCV Epitope
To address the impact of CD4 T cell selection pressure on immune escape in HCV infection, we initially looked for mutations in a dominant well-characterized DRB5*0310-restricted MHC class II epitope26 in viruses from CH-503, a persistently infected animal expressing this allele documented to have escape mutations in multiple MHC class I–restricted epitopes.17 Circulating virus was sequenced from 10 months to 9 years after infection, and the Patr-DRB5*0310-restricted NS31376 epitope was monitored for the emergence of mutations (Table 1). An amino acid substitution at P9 (V to A) in the NS31376 epitope was first detected as a minor variant at 23 months, but became nearly fully fixed in the quasispecies 9 years after infection (15/16). A second substitution at P8 of the epitope (E to A) was not observed until 3 years after infection. This substitution was also initially present as a minor population in the viral quasi-species at year 3, but became fully established at 9 years after infection. Despite the emergence of these mutations in the class II–restricted epitope late in infection, NS31376-specific CD4 T cells were still detectable by way of MHC class II tetramer staining at very low frequencies in the peripheral blood, lymph nodes, and liver more than 10 years after infection in chimpanzee CH-503 (data not shown).
Table 1.
Evolution of Epitope NS31376 in Chimpanzee CH-503
| Time Point After Infection | NS31376 (DRB5*0310-Restricted) |
|---|---|
| 10 months | YGKAIPLEVI |
| ---------- (16/16) | |
| 23 months | YGKAIPLEVI |
| ---------- (14/15) | |
| ---------- A- (1/15) | |
| 3 years | YGKAIPLEVI |
| ---------- (11/15) | |
| ---------- A- (2/15) | |
| ---------- A-- (2/15) | |
| 9 years | YGKAIPLEVI |
| ---------- AA- (15/16) |
It was unclear whether the amino acid substitutions observed in the NS31376 epitope conferred escape from the CD4 T cell response. To address this, we evaluated the impact of both amino acid changes, individually or combined, for their ability to stimulate various functional activities in an NS31376-specific CD4 T cell clone, 3D, isolated from a Patr-DRB5*0310 –positive animal.26 Three different synthetic peptides were generated expressing each of the mutations alone or in combination with each other. The two peptides that contained the P8 E to A mutation, either alone (peptide NS31376 M2) or in combination with the P9 V to A mutation (peptide NS31376 M1), both diminished IL-2 production compared with cells stimulated with the WT peptide, resulting in an increased IFN-γ+/IL-2lo population as compared with the response generated with the NS31376 WT peptide (Fig. 1A). In contrast, when the CD4 clone 3D was stimulated with a peptide that contained the P9 V to A mutation alone (NS31376 M3), the majority of cytokine-producing CD4 T cells coproduced both IFN-γ and IL-2 at levels similar to those generated by the NS31376 WT peptide, suggesting that the E to A mutation at P8, but not the V to A mutation at P9, is an escape mutation. Responses to both the NS31376 WT and NS31376 M1 peptides over a 5-log range of concentrations revealed a preferential stepwise and progressive loss of cytokine production at decreasing concentrations, beginning first with impaired IL-2 production (Fig. 1B).
Fig. 1.

P8 V to A mutation in the NS31376 epitope impairs IL-2 production in a cognate CD4 T cell clone. (A) Using an overnight intracellular cytokine staining assay, the E to A and V to A amino acid changes at positions P8 and P9, respectively, of the NS31376 (YGKAIPLEVI) epitope were analyzed both separately and in combination for their ability to stimulate IFN-γ and IL-2 production in a CD4 T cell clone specific for the epitope. Representative flow cytometry data are shown, and values in the lower left, upper left, and upper right quadrants represent percentages of gated CD3+CD4+ cells that are IFN-γ−/IL-2−, IFN-γ+/IL-2−, or IFN-γ+/IL-2+, respectively. (B) Titrated concentrations of the NS31376 WT and double mutant NS31376 M1 peptides were analyzed for their ability to stimulate the CD4 T cell clone 3D to produce IFN-γ and IL-2 in an overnight intracellular cytokine staining assay. Representative results are shown. Values in the lower left, upper left, and upper right quadrants represent percentages of gated CD3+CD4+ cells that are IFN-γ−/IL-2−, IFN-γ+/IL-2−, or IFN-γ+/IL-2+, respectively.
Consistent with the observed impairment of cytokine production, levels of phosphorylation of the mitogen-activated protein kinase ERK 1/2, a downstream signaling molecule in the T cell activation pathway, were also reduced following stimulation with the M1 mutant peptide compared with the WT (Fig. 2A). Phosphorylation of ERK 1/2 was observed following stimulation of clone 3D with the NS31376 WT peptide even at concentrations as low as 0.001 μg/mL. In comparison, even at the highest concentration used (1 μg/mL), the NS31376 M1 peptide induced less phosphorylated ERK 1/2 than did the WT peptide and failed to induce phosphorylation at lower concentrations (Fig. 2A). Phosphorylation induced by the mutant peptide was also not increased when the incubation was increased to 30 minutes (data not shown). Impaired capacity of the mutant M1 peptide to stimulate T cell activation was also reflected in reduced proliferation following in vitro stimulation of clone 3D with the NS31376 M1 peptide when compared with the NS31376 WT peptide, as measured by CFSE dilution (Fig. 2B). By day 3, more than 10% of cells stimulated with the WT peptide had undergone one cell division, and at day 4 the majority of these cells (>60%) had undergone at least one cell division, with approximately one-quarter undergoing two or more divisions. In contrast, stimulation with the mutant peptide did not drive significant cell division until day 5, at which time more than half of the cells had still failed to divide, and those that had divided had predominantly undergone only one round of division (Fig. 2B).
Fig. 2.

Mutation in the NS31376 epitope stimulates impaired activation and proliferation in a cognate CD4 T cell clone. (A) Activation of the NS31376-specific CD4 T cell clone 3D was assessed by intracellular staining for the phosphorylated form of the mitogen-activated protein kinase ERK 1/2 following a 15-minute stimulation with various concentrations of the NS31376 WT or NS31376 M1 peptides. Representative histograms are shown. White histograms show phosphorylated ERK 1/2 staining levels in unstimulated control cells; levels in stimulated test cells are depicted by gray histograms. (B) CFSE-labeled CD4 T cell clone 3D cells were stimulated with either the NS31376 WT or NS31376 M1 peptides (0.01 μg/mL) and proliferation, measured as CFSE dilution by way of flow cytometric analysis, was determined on day 3, 4, or 5. Representative plots show gated live CD3+CD4+ cells with frequencies of cells that either remained undivided (CFSEhi) or had undergone up to four rounds of division as indicated above each plot.
Our analysis was next expanded to look for escape of this epitope in other persistently infected animals that did or did not express the restricting MHC class II allele (Fig. 3). Chimpanzee CB0609 was also DRB5*0310-positive, and the viral sequence from this animal displayed a V to A mutation at P9, but not the E to A mutation at P8. Interestingly, chimpanzee CB0603 also possessed the P9 V to A mutation, but did not express the restricting MHC allele, and the other DRB5*0310-negative chimpanzee (NM1238) possessed neither of these mutations. These data further suggest that the E to A mutation at P8 is, in fact, an escape mutation potentially driven by immune selection pressure, whereas the V to A mutation at P9 is not.
Fig. 3.
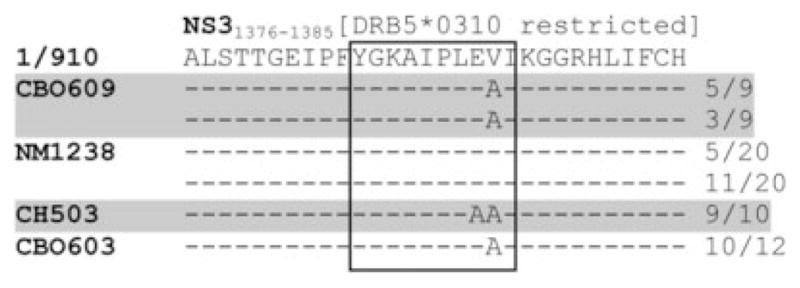
Comparison of the NS31376 epitope in animals with and without the restricting MHC class II allele. Viral RNA was isolated from the serum of four persistently infected chimpanzees (CB0603, CB0609, CH-503, and NM1238) and amplified by PCR. Subsequent molecular cloning and sequencing were then performed to assess the sequence of the NS31376 epitope in two DRB5*0310-positive chimpanzees (CB0609 and CH-503; highlighted) and two chimpanzees (NM1238 and CB0603) that did not express this restricting MHC class II allele. The frequencies of each identified sequence are indicated on the right.
Higher Frequency of Mutations in CD8 Epitopes than in CD4 Epitopes in Persistent HCV Infection of Chimpanzees
Observations in CH-503 and in other studies7,23,29 suggest that escape in MHC class II–restricted epitopes can occur in HCV infection; however, the frequency of such events has not been studied. To address this question, we broadened our analysis to look for escape in multiple CD4 T cell epitopes throughout the HCV polyprotein in four animals persistently infected with HCV.
Ten additional MHC class II–restricted HCV epitopes26,27 (unpublished data) were analyzed in four animals with chronic infection (Fig. 4). In addition to the mutations observed in the NS31376 epitope in animal CH-503, an I to V amino acid change was found in P8 of the NS41842 epitope in chimpanzees CBO603 and CBO609. However, whereas CBO603 expresses the MHC class II allele that presents this epitope, CBO609 does not, suggesting that this mutation was not driven by immune selection pressure. A change from K to R was observed at P13 of the NS31387 epitope in chimpanzee CBO603, and although the epitope would be restricted in this animal, the mutation is not present in the dominant quasispecies after more than 10 years of chronic infection, suggesting that selection pressure is not a dominant factor. Other mutations in the dominant viral quasispecies were observed for the CD4 T cell epitopes NS41825 and NS5a2079, and in the minority quasispecies in the NS41912 epitope, but only in animals that did not express the MHC class II allele that presented the epitopes. We cannot rule out the possibility that these represent mutations in as yet undefined CD8 T cell epitopes in these animals.
Fig. 4.

Infrequent sequence variation in MHC class II–restricted HCV epitopes in persistently infected chimpanzees. HCV RNA isolated from the serum of four persistently infected animals (CBO603, CBO609, CH-503, and NM1238) was amplified by way of PCR, and multiple molecular clones were sequenced to identify amino acid changes in 10 CD4 T cell epitopes compared with a consensus sequence of the infecting genotype 1a 1/910 strain. The frequency of each sequence is indicated to the right in each box. Viral sequences from animals expressing the restricting class II MHC allele are highlighted.
Previously, we described escape mutations in MHC class I–restricted epitopes from three animals included in this study.17 In the present study, we extended that analysis to additional recently defined class I epitopes (unpublished data) and incorporated a fourth animal with chronic HCV infection (Fig. 5). Of the six defined epitopes located in structural proteins, there were five mutations in the dominant viral quasispecies for four epitopes in chimpanzees with the appropriate allele, and one mutation in the dominant viral quasispecies of an epitope in an animal without the appropriate allele (Fig. 5A). For epitopes in nonstructural proteins, 15 mutations were identified in the dominant viral quasispecies in 10 of the 12 epitopes in animals with the appropriate allele, whereas there were three mutations in the dominant viral quasispecies in three of the 12 epitopes in animals without the appropriate restricting allele (Fig. 5B). In total, over 75% of restricted CD8 T cell epitopes analyzed in this study contained amino acid sequences different from the input HCV-1/910 strain, consistent with our previous findings. In contrast, only 18% of restricted CD4 T cell epitopes showed any amino acid variances from the HCV-1/910 strain used to infect the animals. However, of the three amino acid changes we observed in two different CD4 T cell epitopes, only one was exclusive to an animal with the necessary MHC class II allele to present the epitope.
Fig. 5.

Amino acid changes in CD8 T cell–targeted epitopes of HCV proteins in persistently infected chimpanzees. Multiple clonal sequences of class I MHC-restricted epitopes in HCV structural (A) and nonstructural (B) genes were compared from the four persistently infected chimpanzees to the consensus HCV-1/910 sequence. Epitopes in animals that express the appropriate restricting MHC class I allele are highlighted, and frequencies for the different sequences are shown on the right side of each box.
Comparison of Nonsynonymous Mutation Rates Between CD4 and CD8 Epitopes
Previously, we reported that CD8 T cell selection pressure on MHC class I–restricted HCV epitopes facilitated escape and was predictive of a failure to resolve infection.17 Whether a similar mechanism was at play with HCV CD4 T cell responses was next assessed by a similar statistical analysis of mutational frequencies, comparing amino acid changes in CD4 versus CD8 T cell epitopes.
Sequences of both CD4 and CD8 T cell epitopes from the circulating virus of the four chimpanzees were compared with the sequences of multiple molecular clones of the input HCV-1/910 strain of virus to calculate the rates of nonsynonymous nucleotide substitutions (dN) and synonymous nucleotide substitutions (dS). This provides a direct comparison between not only restricted and non-restricted epitopes, but also between CD4 and CD8 T cell epitopes. dS and dN were also calculated for upstream and downstream flanking regions, where positive selection pressure exerted by T cells should be absent. Synonymous nucleotide substitutions do not result in a change in the encoded amino acid, whereas nonsynonymous nucleotide substitutions result in amino acid substitutions and can potentially impact overall viral fitness. In the absence of selection pressure, it is predicted that the dS/dN ratio will be relatively high, reflective of the principle that synonymous mutations are neutral to viral fitness.
Consistent with our previous analysis, we confirmed that MHC class I–restricted CD8 T cell responses exert positive selection pressure on the virus (Fig. 6).17 The median dN for presented CD8 T cell epitopes (0.0431) was significantly higher than for nonpresented epitopes (0.0077; P < 0.001) or flanking regions (0.0089; P < 0.01). In contrast, there was no significant difference between median dN in restricted CD4 T cell epitopes and flanking regions (0.0046 and 0.0089, respectively; P < 0.05). Whereas median dS values for both presented CD4 and CD8 T cell epitopes were similar (0.0610 and 0.0578, respectively), the median dN value for presented CD8 T cell epitopes was nearly 10-fold higher than for presented CD4 T cell epitopes (0.0431 versus 0.0046). These data are consistent with the hypothesis that HCV-specific CD4 T cells do not exert significant selection pressure to drive amino acid changes in the virus.
Fig. 6.
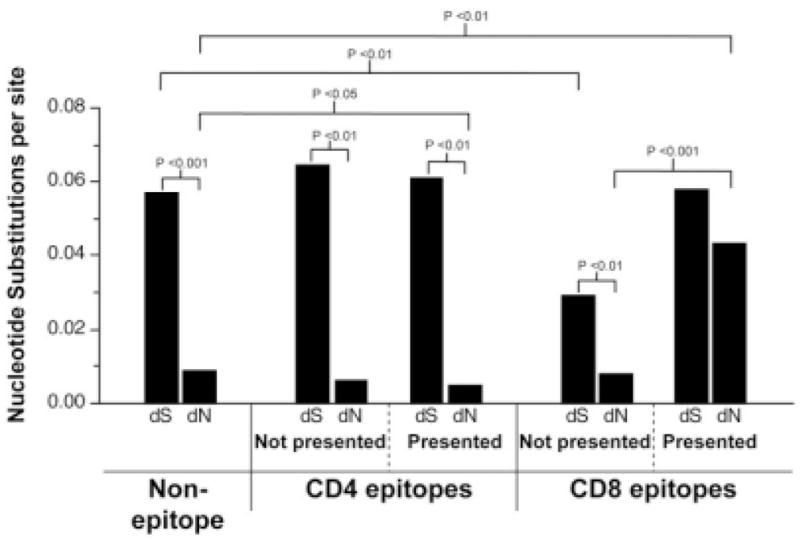
HCV-specific CD8 T cells, but not CD4 T cells, exert significant pressure to drive increased nonsynonymous mutation rates. Median rates of synonymous substitutions per synonymous site (dS) and of nonsynonymous substitutions per nonsynonymous site (dN) were computed separately for nonepitope regions, and for CD4 and CD8 T cell epitopes both in animals that express the specific restricting MHC alleles and in animals that lack the necessary MHC allele to present the epitope to specific CD4 or CD8 T cells (data not shown). Tests of the hypotheses that (1) the median dS is equal to the median dN and (2) an individual dS or dN value in presented epitopes equals that in nonpresented epitopes were performed using the Wilcoxon signed-rank test. Tests of the hypothesis that an individual dS or dN value in epitopes equals that in nonepitope regions were performed using the Mann-Whitney test.
Discussion
Although several studies have explored the phenomenon of immune escape in MHC class II–restricted HCV epitopes during persistent infection, they have often focused on a limited set of CD4 T cell epitopes or viral proteins.7,22–24,29 In particular, where CD4 T cell escape was analyzed in humans with persistent hepatitis C, the input strain that initially infected the subjects was unknown, complicating the analysis of virus evolution.
In the present study, our goal was to determine the frequency of mutations in HCV CD4 T cell epitopes by comparing amino acid changes in multiple CD4 and CD8 T cell epitopes within the same subject. In this report, we confirmed and extended our previous findings,17 specifically that mutational escape in HCV CD8 T cell epitopes is common, occurring in over 75% of the epitopes analyzed. Reports of humans persistently infected with HCV have also documented similarly high frequencies of mutations arising in CD8 viral epitopes, with as low as 45% and as high as 75% of the analyzed epitopes accumulating mutations.18,19 In contrast, our comparison of mutations in CD8 and CD4 T cell epitopes in the same animals revealed that nonsynonymous mutations were far less common in the latter (18% of epitopes analyzed), and, importantly, occurred no more frequently than in nonepitope flanking regions. A similar unpublished study has also documented that CD4 T cell epitopes of HCV are highly conserved in persistently infected humans. Additionally, of the four observed amino acid changes in restricted HCV CD4 T cell epitopes that we described, only one is likely to cause escape from immune recognition (NS31376).
Although MHC class II–restricted epitope mutation in HCV has been documented in persistent infection, it has not been universally observed,22,29 and our study demonstrates that it is not common. The low frequency of mutations in CD4 T cell epitopes, especially compared with the high frequency of mutations reported for CD8 T cell epitopes within the same individual, suggests that CD4 T cell HCV epitopes are relatively stable. Although CD4 T cell epitope escape has also been described for HIV,30 another virus subject to high mutation rates, CD4 T cell epitope escape in viral infection has been rarely reported. However, the correlation of broad, functional HCV-specific CD4 T cell responses with infection resolution, and of impaired virus-specific CD4 T cell responses with persistent hepatitis C, suggests that these cells play an essential role in controlling infection and thus may be able to exert selection pressure on the virus. Although CD4 T cells generally lack cytotoxic capabilities, they can produce cytokines such as IFN-γ that contribute to the antiviral state in the host, which may be one potential mechanism for selection pressure. However, this is not likely to be as direct and immediate an influence on viral evolution as cytotoxic activity by CD8 T cells or antibody-mediated neutralization.6 That the CD4 response to HCV wanes very rapidly as acute infection is not controlled also suggests that any pressure to select for mutated CD4 T cell epitopes might be only early and fleeting.7–11,20,31
Although mutational escape in CD4 T cell epitopes of HCV is much less likely than in CD8 T cell epitopes, such mutations do in fact occur, as evidenced by the mutation we observed in the NS31376 epitope and documented mutations reported by others.7,23,24,32 The observed mutation in the NS31376 epitope diminished activation, proliferation, and cytokine production in a cognate CD4 T cell clone. Interestingly, decreased cytokine production was marked by a pattern observed in persistent lymphocytic choriomeningitis virus infection of mice, as well as chronic HCV and HIV infection in humans—namely, a preferential loss of IL-2 production over IFN-γ production in virus-specific CD4 T cells (Fig. 1).33–36 In HIV infection, virus-specific CD4 T cells producing IFN-γ, but not IL-2, had impaired proliferative capacity and were short-lived. The late emergence of the NS31376 CD4 T cell epitope mutation, however, suggests that other mechanisms drive the early loss of CD4 responses in persistent HCV infection.
Boosting HCV-specific CD4 T cell responses by vaccination could improve the ability of the host immune response to control infection while simultaneously exerting greater selection pressure on the virus. In a study by Puig et al.7 in which a chimpanzee was immunized against the HCV non-structural proteins prior to challenge, escape mutations in two separate CD4 T cell epitopes, located in the NS3 and NS5a proteins, emerged as viral control was lost and persistence was established. Interestingly, chimpanzee CH-503, the animal in which the NS31376 mutation was originally identified, had received a therapeutic vaccination against the Core, E1, E2, NS3, and NS4 HCV proteins 4 years after infection (M. Houghton, personal communication). However, the NS31376 mutation was already observed as a minor population in the viral quasispecies at 3 years after infection, 1 year prior to the therapeutic vaccination. However, infrequent sampling before and after the therapeutic vaccination makes it difficult to completely rule out a role for therapeutic vaccination in further driving this escape mutation.
In this study, we demonstrate that, within the same individual, mutations in HCV CD4 T cell epitopes occur much less frequently than in CD8 T cell epitopes. These data suggest that, unlike HCV-specific CD8 T cells, CD4 T cell selection pressure to drive escape mutations is not a significant factor in the failure of these responses and the establishment of viral persistence. Whether such mutations might play a role in maintaining viral persistence, or promoting persistence if transmitted to a new host, remains unclear.
Acknowledgments
Supported by Public Health Service Grant U19A148231 (to C. M. W.). M. J. F. was supported by a postdoctoral fellowship from the American Cancer Society. D. G. B. was supported by a CJ Martin Fellowship from the National Health and Medical Research Foundation, Australia, and a Postdoctoral Research Fellowship from the American Liver Foundation. N. H. S. was supported by postdoctoral fellowships from the Canadian Institute for Health Research and the American Liver Foundation. B. C. is supported in part by an award from the American Liver Foundation. A. L. H. was supported by National Institutes of Health grant GM43940. This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number C06 RR016483-01 from the National Center for Research Resources, National Institutes of Health.
We would like to thank the staff of the University of Louisiana at Lafayette New Iberia Research Center, New Iberia, LA, for their expert veterinary and technical support. Excellent technical assistance in viral sequencing and sequence data analysis were provided by Jennifer Rutkiewicz and Genevieve Mulroy.
Abbreviations
- CFSE
5-(and 6-)carboxyfluorescein diacetate succinimidyl ester
- dN
rate of nonsynonymous nucleotide substitution
- dS
rate of synonymous nucleotide substitution
- ERK
extracellular signal-regulated kinase
- HCV
hepatitis C virus
- IFN-γ
interferon-γ
- IL-2
interleukin-2
- MHC
major histocompatibility complex
- WT
wild-type
Footnotes
Potential conflict of interest: Nothing to report.
References
- 1.Cooper S, Erickson AL, Adams EJ, Kansopon J, Weiner AJ, Chien DY, et al. Analysis of a successful immune response against hepatitis C virus. Immunity. 1999;10:439–449. doi: 10.1016/s1074-7613(00)80044-8. [DOI] [PubMed] [Google Scholar]
- 2.Missale G, Bertoni R, Lamonaca V, Valli A, Massari M, Mori C, et al. Different clinical behaviors of acute hepatitis C virus infection are associated with different vigor of the anti-viral cell-mediated immune response. J Clin Invest. 1996;98:706–714. doi: 10.1172/JCI118842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thimme R, Oldach D, Chang KM, Steiger C, Ray SC, Chisari FV. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med. 2001;194:1395–1406. doi: 10.1084/jem.194.10.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shoukry NH, Grakoui A, Houghton M, Chien DY, Ghrayeb J, Reimann KA, et al. Memory CD8+ T cells are required for protection from persistent hepatitis C virus infection. J Exp Med. 2003;197:1645–1655. doi: 10.1084/jem.20030239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Major ME, Mihalik K, Puig M, Rehermann B, Nascimbeni M, Rice CM, et al. Previously infected and recovered chimpanzees exhibit rapid responses that control hepatitis C virus replication upon rechallenge. J Virol. 2002;76:6586–6595. doi: 10.1128/JVI.76.13.6586-6595.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grakoui A, Shoukry NH, Woollard DJ, Han JH, Hanson HL, Ghrayeb J, et al. HCV persistence and immune evasion in the absence of memory T cell help. Science. 2003;302:659–662. doi: 10.1126/science.1088774. [DOI] [PubMed] [Google Scholar]
- 7.Puig M, Mihalik K, Tilton JC, Williams O, Merchlinsky M, Connors M, et al. CD4+ immune escape and subsequent T-cell failure following chimpanzee immunization against hepatitis C virus. Hepatology. 2006;44:736–745. doi: 10.1002/hep.21319. [DOI] [PubMed] [Google Scholar]
- 8.Day CL, Lauer GM, Robbins GK, McGovern B, Wurcel AG, Gandhi RT, et al. Broad specificity of virus-specific CD4+ T-helper-cell responses in resolved hepatitis C virus infection. J Virol. 2002;76:12584–12595. doi: 10.1128/JVI.76.24.12584-12595.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urbani S, Amadei B, Fisicaro P, Tola D, Orlandini A, Sacchelli L, et al. Outcome of acute hepatitis C is related to virus-specific CD4 function and maturation of antiviral memory CD8 responses. Hepatology. 2006;44:126–139. doi: 10.1002/hep.21242. [DOI] [PubMed] [Google Scholar]
- 10.Thimme R, Bukh J, Spangenberg HC, Wieland S, Pemberton J, Steiger C, et al. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc Natl Acad Sci U S A. 2002;99:15661–15668. doi: 10.1073/pnas.202608299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smyk-Pearson S, Tester IA, Klarquist J, Palmer BE, Pawlotsky JM, Golden-Mason L, et al. Spontaneous recovery in acute human hepatitis C virus infection: functional T-cell thresholds and relative importance of CD4 help. J Virol. 2008;82:1827–1837. doi: 10.1128/JVI.01581-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, et al. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol. 2006;80:11398–11403. doi: 10.1128/JVI.01177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yee LJ, Perez KA, Tang J, van Leeuwen DJ, Kaslow RA. Association of CTLA4 polymorphisms with sustained response to interferon and ribavirin therapy for chronic hepatitis C virus infection. J Infect Dis. 2003;187:1264–1271. doi: 10.1086/374561. [DOI] [PubMed] [Google Scholar]
- 14.Radziewicz H, Ibegbu CC, Fernandez ML, Workowski KA, Obideen K, Wehbi M, et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol. 2007;81:2545–2553. doi: 10.1128/JVI.02021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamoto N, Cho H, Shaked A, Olthoff K, Valiga ME, Kaminski M, et al. Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/CTLA-4 blockade. PLoS Pathog. 2009;5:e1000313. doi: 10.1371/journal.ppat.1000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rutebemberwa A, Ray SC, Astemborski J, Levine J, Liu L, Dowd KA, et al. High-programmed death-1 levels on hepatitis C virus-specific T cells during acute infection are associated with viral persistence and require preservation of cognate antigen during chronic infection. J Immunol. 2008;181:8215–8225. doi: 10.4049/jimmunol.181.12.8215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erickson AL, Kimura Y, Igarashi S, Eichelberger J, Houghton M, Sidney J, et al. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity. 2001;15:883–895. doi: 10.1016/s1074-7613(01)00245-x. [DOI] [PubMed] [Google Scholar]
- 18.Cox AL, Mosbruger T, Mao Q, Liu Z, Wang XH, Yang HC, et al. Cellular immune selection with hepatitis C virus persistence in humans. J Exp Med. 2005;201:1741–1752. doi: 10.1084/jem.20050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ray SC, Fanning L, Wang XH, Netski DM, Kenny-Walsh E, Thomas DL. Divergent and convergent evolution after a common-source outbreak of hepatitis C virus. J Exp Med. 2005;201:1753–1759. doi: 10.1084/jem.20050122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tester I, Smyk-Pearson S, Wang P, Wertheimer A, Yao E, Lewinsohn DM, et al. Immune evasion versus recovery after acute hepatitis C virus infection from a shared source. J Exp Med. 2005;201:1725–1731. doi: 10.1084/jem.20042284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerlach JT, Diepolder HM, Jung MC, Gruener NH, Schraut WW, Zachoval R, et al. Recurrence of hepatitis C virus after loss of virus-specific CD4(+) T-cell response in acute hepatitis C. Gastroenterology. 1999;117:933–941. doi: 10.1016/s0016-5085(99)70353-7. [DOI] [PubMed] [Google Scholar]
- 22.Penna A, Missale G, Lamonaca V, Pilli M, Mori C, Zanelli P, et al. Intrahepatic and circulating HLA class II-restricted, hepatitis C virus-specific T cells: functional characterization in patients with chronic hepatitis C. Hepatology. 2002;35:1225–1236. doi: 10.1053/jhep.2002.33153. [DOI] [PubMed] [Google Scholar]
- 23.Wang JH, Layden TJ, Eckels DD. Modulation of the peripheral T-Cell response by CD4 mutants of hepatitis C virus: transition from a Th1 to a Th2 response. Hum Immunol. 2003;64:662–673. doi: 10.1016/s0198-8859(03)00070-3. [DOI] [PubMed] [Google Scholar]
- 24.von Hahn T, Yoon JC, Alter H, Rice CM, Rehermann B, Balfe P, et al. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology. 2007;132:667–678. doi: 10.1053/j.gastro.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Kimura Y, Gushima T, Rawale S, Kaumaya P, Walker CM. Escape mutations alter proteasome processing of major histocompatibility complex class I-restricted epitopes in persistent hepatitis C virus infection. J Virol. 2005;79:4870–4876. doi: 10.1128/JVI.79.8.4870-4876.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woollard DJ, Grakoui A, Shoukry NH, Murthy KK, Campbell KJ, Walker CM. Characterization of HCV-specific Patr class II restricted CD4+ T cell responses in an acutely infected chimpanzee. Hepatology. 2003;38:1297–1306. doi: 10.1053/jhep.2003.50478. [DOI] [PubMed] [Google Scholar]
- 27.Shoukry NH, Sidney J, Sette A, Walker CM. Conserved hierarchy of helper T cell responses in a chimpanzee during primary and secondary hepatitis C virus infections. J Immunol. 2004;172:483–492. doi: 10.4049/jimmunol.172.1.483. [DOI] [PubMed] [Google Scholar]
- 28.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 29.Scotta C, Garbuglia AR, Ruggeri L, Spada E, Laurenti L, Perrone MP, et al. Influence of specific CD4+ T cells and antibodies on evolution of hypervariable region 1 during acute HCV infection. J Hepatol. 2008;48:216–228. doi: 10.1016/j.jhep.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 30.Harcourt GC, Garrard S, Davenport MP, Edwards A, Phillips RE. HIV-1 variation diminishes CD4 T lymphocyte recognition. J Exp Med. 1998;188:1785–1793. doi: 10.1084/jem.188.10.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Day CL, Seth NP, Lucas M, Appel H, Gauthier L, Lauer GM, et al. Ex vivo analysis of human memory CD4 T cells specific for hepatitis C virus using MHC class II tetramers. J Clin Invest. 2003;112:831–842. doi: 10.1172/JCI18509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frasca L, Del Porto P, Tuosto L, Marinari B, Scotta C, Carbonari M, et al. Hypervariable region 1 variants act as TCR antagonists for hepatitis C virus-specific CD4+ T cells. J Immunol. 1999;163:650–658. [PubMed] [Google Scholar]
- 33.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iyasere C, Tilton JC, Johnson AJ, Younes S, Yassine-Diab B, Sekaly RP, et al. Diminished proliferation of human immunodeficiency virus-specific CD4+ T cells is associated with diminished interleukin-2 (IL-2) production and is recovered by exogenous IL-2. J Virol. 2003;77:10900–10909. doi: 10.1128/JVI.77.20.10900-10909.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Younes SA, Yassine-Diab B, Dumont AR, Boulassel MR, Grossman Z, Routy JP, et al. HIV-1 viremia prevents the establishment of interleukin 2-producing HIV-specific memory CD4+ T cells endowed with proliferative capacity. J Exp Med. 2003;198:1909–1922. doi: 10.1084/jem.20031598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Semmo N, Day CL, Ward SM, Lucas M, Harcourt G, Loughry A, et al. Preferential loss of IL-2-secreting CD4+ T helper cells in chronic HCV infection. Hepatology. 2005;41:1019–1028. doi: 10.1002/hep.20669. [DOI] [PubMed] [Google Scholar]