

Abstract
In Alzheimer's disease (AD), rod-like cofilin aggregates (cofilin–actin rods) and thread-like inclusions containing phosphorylated microtubule-associated protein (pMAP) tau form in the brain (neuropil threads), and the extent of their presence correlates with cognitive decline and disease progression. The assembly mechanism of these respective pathological lesions and the relationship between them is poorly understood, yet vital to understanding the causes of sporadic AD. We demonstrate that, during mitochondrial inhibition, activated actin-depolymerizing factor (ADF)/cofilin assemble into rods along processes of cultured primary neurons that recruit pMAP/tau and mimic neuropil threads. Fluorescence resonance energy transfer analysis revealed colocalization of cofilin-GFP (green fluorescent protein) and pMAP in rods, suggesting their close proximity within a cytoskeletal inclusion complex. The relationship between pMAP and cofilin–actin rods was further investigated using actin-modifying drugs and small interfering RNA knockdown of ADF/cofilin in primary neurons. The results suggest that activation of ADF/cofilin and generation of cofilin–actin rods is required for the subsequent recruitment of pMAP into the inclusions. Additionally, we were able to induce the formation of pMAP-positive ADF/cofilin rods by exposing cells to exogenous amyloid-β (Aβ) peptides. These results reveal a common pathway for pMAP and cofilin accumulation in neuronal processes. The requirement of activated ADF/cofilin for the sequestration of pMAP suggests that neuropil thread structures in the AD brain may be initiated by elevated cofilin activation and F-actin bundling that can be caused by oxidative stress, mitochondrial dysfunction, or Aβ peptides, all suspected initiators of synaptic loss and neurodegeneration in AD.
Introduction
Alzheimer's disease (AD) is a progressive, degenerative dementia histopathologically characterized by neurofibrillary tangles of tau protein and amyloid-β (Aβ) plaques (Goedert and Spillantini, 2006; Haass and Selkoe, 2007). In early stages of AD, hyperphosphorylated microtubule-associated protein (pMAP) tau forms striated thread-like structures in neurites, so-called “neuropil threads” (Velasco et al., 1998; Augustinack et al., 2002), that correlate with cognitive decline and comprise >85% of end-stage cortical tau pathology (Velasco et al., 1998; Braak et al., 2006; Giannakopoulos et al., 2007).
Tau, like other MAPs, stabilizes neuronal microtubules (MTs) and facilitates MT dynamics through its phosphorylation and dephosphorylation (Timm et al., 2003) (for review, see Garcia and Cleveland, 2001). Although normal adult neurons exhibit low levels of tau phosphorylation, neurons of AD brain and other tau-related neurodegenerative diseases show high levels of tau phosphorylation at both physiological and pathological disease-specific residues. This tau hyperphosphorylation prevents binding and stabilization of MTs and causes abnormal translocation of tau from axonal MT tracks to neuropil thread inclusions, dendritic processes, and cell bodies in which it accumulates and aggregates (Terry, 1998; Garcia and Cleveland, 2001). The phosphorylation of tau at Ser262 in the microtubule-binding domain is one of the earliest markers of AD neuropathology, readily detected in “pretangle” neuropil threads (Augustinack et al., 2002).
Another prominent feature widespread in the AD brain is abnormal aggregates of the actin-associated protein cofilin, which forms punctate and rod-like linear arrays through the neuropil (Minamide et al., 2000). Neuronal cofilin plays important roles in learning and memory pathways by modulating actin-rich dendritic spine architecture (Hotulainen et al., 2009) (for review, see Bamburg and Bloom, 2009). The activity of cofilin and related protein actin-depolymerizing factor (ADF) is negatively regulated by phosphorylation of the conserved Ser3 by LIM (Lin-11, Isl-1, and Mec-3) and other kinases and reactivated on its dephosphorylation by slingshot or chronophin phosphatases (Huang et al., 2008) (for review, see Bamburg and Bloom, 2009), allowing it to actively bind and sever filamentous actin (F-actin), thus regulating actin turnover (Carlier et al., 1997; Bamburg and Bloom, 2009).
ADF/cofilin–actin rods comparable with those observed in the AD brain are inducible in neuronal cell culture through inhibition of mitochondrial ATP generation and other neurodegenerative stimuli such as oxidative stress or exposure to Aβ peptides (Minamide et al., 2000; Maloney et al., 2005; Davis et al., 2009). Since actin dynamics in neurons are purported to use ∼50% of total cellular ATP (Bernstein and Bamburg, 2003), ADF/cofilin–actin rods have been proposed to represent an early neuroprotective mechanism during times of transient stress since virtually all ADF/cofilin is sequestered into nondynamic polymers of ADF/cofilin–actin, inhibiting actin turnover and thereby preserving ATP (Bernstein et al., 2006). Although mitochondrial dysfunction has been linked to AD (Smith et al., 2005; Wang et al., 2009), the relationship between mitochondrial dysfunction, the generation of tau inclusions, and their relationship to cofilin aggregates remains elusive.
In this study, we aimed to determine the effects of mitochondrial dysfunction on cellular pMAP/tau distribution compared with ADF/cofilin–actin rod distribution (Minamide et al., 2000; Huang et al., 2008). Using primary neuronal cell culture models, we demonstrate that cytoskeletal rods containing ADF/cofilin sequester and bind pMAP. The resulting striated pMAP-positive rods bear striking resemblance to neuropil threads observed in postmortem AD brain labeled with the same pMAP antibody. This process may well represent an early pathogenic event in AD leading to synaptic loss and neurodegeneration.
Materials and Methods
Antibodies and reagents.
Mouse monoclonal antibodies are actin (1A4; Dako), β-actin (Abcam), β(III)-tubulin (Abcam), tau phosphorylated at Ser202/Thr205 (AT8; Pierce), and Ser262/356 (12E8; Elan) (Seubert et al., 1995). The monoclonal antibody 12E8, raised against Ser262-phosphorylated tau, is known to cross- react with other phosphorylated MAPs such MAP2c, MAP4 (Timm et al., 2003), and doublecortin (Schaar et al., 2004) since these MAPs also contain the 12E8-specific KXGS motifs in their microtubule-binding domains. However, 12E8 has strong affinity for tau, in which the microtubule-binding domain contains several KXGS motifs, the target sequence for 12E8 (Seubert et al., 1995; Yoshida and Goedert, 2002; Timm et al., 2003; Schaar et al., 2004). Rabbit polyclonal antibodies are ADF (1439), ADF (D8815; Sigma-Aldrich), Ser3-phosphorylated ADF/cofilin (Shaw et al., 2004), cofilin (C8736; Sigma-Aldrich), actin (A2066; Sigma-Aldrich), and total tau (A0024; Dako). Phalloidin 488 (Invitrogen) was used to visualize F-actin (Invitrogen). Anti-mouse and anti-rabbit secondary antibodies included Alexa Fluor (488, 555, and 647)-conjugated (Invitrogen) for immunofluorescence and horseradish peroxidase-conjugated (GE Healthcare) for immunoblotting. Lyophilized amyloid peptides were from Bachem.
Cell culture and treatments.
Primary chick neurons were prepared from freshly dissected chicken embryos [embryonic day 7 (E7)], as previously described, and cultured for 7 d in vitro (div) on polylysine-coated 30 mm culture dishes or glass coverslips (Goldsbury et al., 2008). Primary human neurons were prepared and cultured for 14–20 d as previously described (Guillemin et al., 2007). Primary (E18) rat hippocampal neurons were cultured for 5 d as previously described (Minamide et al., 2000). For ATP depletion studies, cells were treated with 1–2 μm antimycin (AM) (a mitochondrial complex III inhibitor; Sigma-Aldrich), 3 μm carbonyl cyanide 3-chlorophenylhydrazone (CCCP) (a mitochondrial uncoupling agent; Sigma-Aldrich), or 100 μm hydrogen peroxide (Sigma-Aldrich) in PBS containing 0.5 mm CaCl2 and 1 mm MgSO4. For ADF/cofilin phosphorylation studies, cells were treated for 15 min with 1 μm AM. Actin drugs were used at 1 μg/ml jasplakinolide (Jasp) (Calbiochem; 420127) or latrunculin B (Lat B) (Calbiochem; 428020). Cells left without a medium change or treated with PBS or DMSO acted as controls. Stock solutions of lyophilized Aβ peptides were solubilized to 2 mm in DMSO, aliquoted, and stored at −20°C. Immediately before application to cells, the stock solutions were diluted to 100 μm in PBS and agitated at 1200 rpm on a laboratory shaker for 30 min to generate mixtures of polymorphic oligomeric and fibrillar aggregates as previously described (Goldsbury et al., 2000). The peptide assemblages were then applied to cells in 24-well plates at final concentrations of 1 or 2 μm. Control wells were treated with the same volume of DMSO in PBS. After treatments, cells were immediately fixed (cells on coverslips) or prepared for Western blot analysis (plated cells).
Luminescent measurements.
After treatments (in triplicate), primary chick neurons were harvested and assessed for total ATP (ATPLite; PerkinElmer) using a Victor III Multilabel plate reader. Protein measurements were determined using the identical samples and the bicinchoninic acid assay (Sigma-Aldrich). Total ATP was normalized against total protein to account for any difference in cell density. Statistical analysis used Prism software (version 3.0; GraphPad Software).
Immunoblotting.
After treatments, cells were lysed and prepared for SDS-PAGE. For equal gel loading, protein concentrations were determined by the Lowry assay (Bio-Rad). Proteins transferred to nitrocellulose membranes were detected with ECL Western Blotting Detection System (GE Healthcare) on a ChemiDoc XRS (Bio-Rad). For analysis, band densities were measured using ImageJ (version 1.38x; National Institutes of Health freeware; http://rsb.info.nih.gov/ij), and background intensity was subtracted and normalized to individual β-actin or tubulin loading controls. An average and SEM for each treatment condition was determined, and results are presented as a percentage of the mean control band intensities.
Plasmids and transfection.
Plasmid-mediated expression of wild-type human cofilin has been previously described (Davis et al., 2009). Human cofilin cDNA in a pET vector (a gift from Alan Weeds, Medical Research Council Laboratory of Molecular Biology, Cambridge, UK) was modified by PCR with a 5′-PCR primer containing an EcoRI site and a 3′-primer that removed the stop codon and introduced an XmaI site at the 3′-end. EcoRI and XmaI were then used to cut the PCR product and the cDNA was ligated into pEGFP-N1 (Clontech) in-frame with the green fluorescent protein (GFP) to encode cofilin-GFP. A plasmid vector for expressing small interfering RNAs (siRNAs) for chick ADF was made by inserting DNA oligonucleotides (Macromolecular Resources) into a plasmid expression vector (pSuper) (Brummelkamp et al., 2002) containing the H1 polymerase III promoter. The oligonucleotide product from the pol III promoter is a double-stranded hairpin RNA (antisense-linker-sense) that is processed into a functional siRNA in the cell. The modified inserts including the H1 pol III promoter from the pSuper vector were excised and ligated into the pAdTrack vector (He et al., 1998). The siRNA sequence contained within the hairpin used for chick ADF is 5′-GTGGAAGAAGGCAAAGAGATT-3′. A plasmid made identically but that makes a hairpin RNA to silence human Pak2 (5′-GTCTCTGGGTATCATGGCTAT-3′) was used as a transfection control in the chick cells. The ability of the small hairpin RNA (shRNA)-expressing plasmid to effectively knock down ADF in chick cells was tested in cultures of chick skin fibroblasts (B. M. Marsick, K. C. Flynn, J. R. Bamburg, and P. C. Letourneau, unpublished observations). For plasmid-mediated expression, neuronal cultures at 3 d were transfected using Lipofectamine 2000 (Invitrogen). Four days after transfection, neurons were treated with AM (1 or 2 μm for 10 or 20 min).
Microscopy.
Cells were fixed with 4% paraformaldehyde at 37°C for 30 min, permeabilized with 0.05% Triton X-100 or 100% ice-cold methanol, blocked in 5% goat serum, and stained for immunofluorescence. To optimize visualization of ADF/cofilin–actin rods, 0.1% EM-grade glutaraldehyde was added to the fixative. Epifluorescence images were obtained on a Zeiss Axioplan 2 microscope, captured with a CCD camera driven by AxioVision software. Single-labeled cells/sections were used to check for bleed-through in all double-label immunofluorescence studies. All captured images were converted to tagged image files for subsequent presentation. For fluorescence resonance energy transfer (FRET) analysis, cells (plated on glass-bottomed MatTek dishes) were imaged in PBS using a Zeiss LSM 510 and C-Apochromat 40× water-immersion objective and the argon (488 nm) (for GFP donor) and HeNe1 (543) (for Alexa Fluor 555 acceptor) laser lines. For photobleaching of acceptor, regions of interest in the transfected cells were bleached using the 543 nm laser line at 100% power. FRET efficiency was calculated from the increase in the fluorescence intensity of the donor after the acceptor was selectively photobleached (Gervásio et al., 2008). The donor fluorescence was measured from at least five bleached and five unbleached rods per cell.
Analysis and statistics.
For quantification of cells containing rod structures, cells with rods were counted for each treatment condition from randomly selected fields on each coverslip, and cells containing rod structures were then expressed as a percentage of the total cell population or mean number of rods per cell. Treatments and measurements were repeated in triplicate using independently prepared cell cultures. For determination of relative fluorescence intensities for phalloidin or ADF, immunofluorescent intensity was measured using ImageJ. Cells with abnormal nuclei, as indicated by 4′,6′-diamidino-2-phenylindole (DAPI) staining, were excluded from the data. After adjustment to background, mean and SEM intensities were calculated for each condition. Significance was measured using Student's t test.
Human tissue and rat hippocampal slices.
Free-floating sections (45 μm) of formalin-fixed superior frontal cortex and basal forebrain from normal adult and confirmed Alzheimer's disease patients obtained from the New South Wales Brain Bank were immunostained as previously described (Cullen et al., 2005). Sections were incubated overnight at 4°C in primary 12E8 (see Fig. 1a,b) or AT8 antibody (see supplemental Fig. S1a,b, available at www.jneurosci.org as supplemental material). Bound antibody was visualized using Alexa Fluor-conjugated secondary antibody (Invitrogen) or using ABC-peroxidase (Vector Laboratories) and DAB (Sigma-Aldrich).
Figure 1.
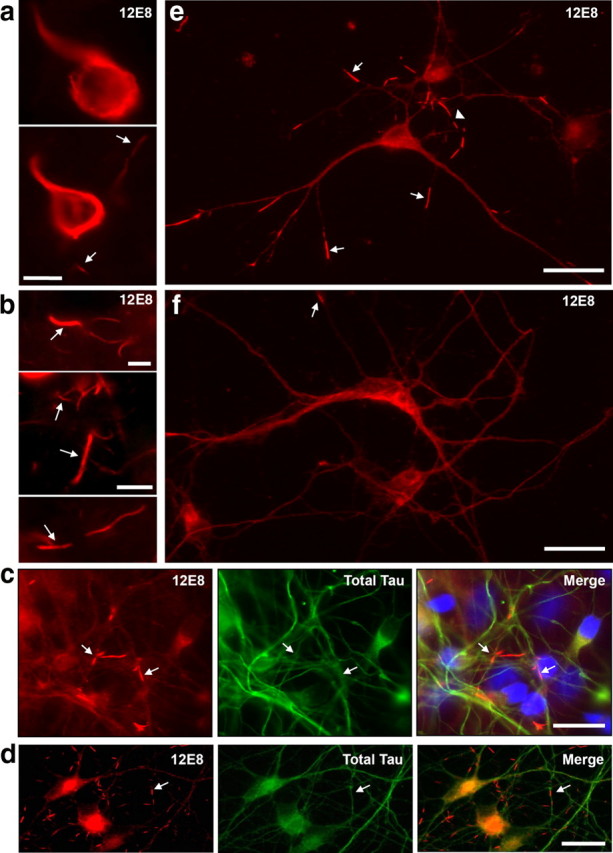
Neuropathological hallmarks of human AD can be recapitulated in ATP-deprived human and chick primary neurons. a, b, Hyperphosphorylation of tau at Ser262/356 (immunolabeled with the 12E8 antibody) and striated neurites are early markers of AD. Immunofluorescent staining of sections of frontal cortex from human AD brain show strong 12E8 reactivity in neurofibrillary tangles in individual neurons (a) and abundant linear arrays of 12E8-labeled inclusions in neurites (a, b, arrows). c, Human primary neuronal cell cultures labeled with the 12E8 antibody (red) and total tau antibody (green), after 30 min AM treatment (plus DAPI in blue in merged image). Tapered pMAP-positive rods were observed throughout the neurites (arrows). d, e, Primary chick neurons (7 div) treated with AM for 15 min also rapidly accumulate rod-like 12E8-positive inclusions (red) that frequently form linear striations within single neurites (e, arrowhead). Rods were not enriched with total tau (c, d, green), indicating that inclusions contain MAP/tau specifically phosphorylated within the microtubule-binding KXGS motifs, as immunostained with 12E8 (red). f, Control chick neurons show evenly distributed 12E8 staining along neurites with only the occasional rod-like accumulation (arrow). Scale bars: a, b, 10 μm; c–f, 20 μm.
Organotypic rat hippocampal slices were grown for 14 d on membranes as described previously (Davis et al., 2009). Slices were treated with 2 μm AM in PBS for 1 h, fixed in 4% formaldehyde for 1 h, permeabilized 90 s in 0.05% Triton X-100 in PBS, and immunostained overnight with 1439 (cofilin) (2 μg/ml) and mouse 12E8 (4 μg/ml) antibodies. Secondary antibody incubations were for 2 h.
Results
Neuritic pMAP accumulation in striated rods, resembling structures in postmortem Alzheimer brain, is induced in primary neurons by energy depletion
We used the monoclonal phosphorylation-dependent 12E8 antibody raised against the pMAP tau, an established early marker for neuropil threads in AD (Augustinack et al., 2002), to determine whether pMAP accumulates in primary neuronal models derived from human, rat, or chick after mitochondrial inhibition. Probing AD brain sections with the 12E8 antibody (Fig. 1a,b) or AT8 antibody (supplemental Fig. S1a,b, available at www.jneurosci.org as supplemental material) showed numerous striated rod-like inclusions within neuropil threads and cytoplasmic accumulations of pMAP, indistinguishable from previous observations in human AD (Augustinack et al., 2002). Additionally, linear striations of rod accumulations labeled with cofilin antibodies were also observed in AD brains (supplemental Fig. S1c, available at www.jneurosci.org as supplemental material).
Treatment of primary human CNS neurons cultured for 7 d in vitro, with the mitochondrial complex III inhibitor AM elicited a rapid accumulation of 12E8-labeled protein into rod inclusions (Fig. 1c, arrows) comparable with those seen in the AD neurons (Fig. 1b, arrows). The rods sequestered the fraction of MAP serine-phosphorylated in the microtubule-binding domain KXGS motifs specific for the 12E8 antibody, rather than total MAP/tau because a polyclonal antibody against total tau yielded a more uniform labeling along neurites of both human and chick neurons (Fig. 1c,d). During AM treatment, primary chick CNS neurons derived from embryonic tectum also generated pMAP-positive rod-like structures (Fig. 1d,e) morphologically identical with those from human neurons. Thus, the readily accessible chick neurons are a useful model system for studying the mechanism by which pMAP accumulates into rods. The conserved 12E8 epitopes in the pMAP sequence reside within the microtubule binding domain in human tau (Ser262/Ser356 residues) and chicken tau (Ser253/Ser378) (supplemental Fig. S2a,b, available at www.jneurosci.org as supplemental material) (Yoshida and Goedert, 2002). Primary chick tectal neurons express five isoforms of tau that are highly homologous to human tau isoforms and exhibit conservation of phosphorylation-specific epitopes recognized by antibodies against human tau (supplemental Fig. S2c, available at www.jneurosci.org as supplemental material) (Yoshida and Goedert, 2002).
pMAP-containing striations induced by ATP depletion colocalize with ADF/cofilin–actin rods
Actin dynamics are highly dependent on ATP availability (Bernstein and Bamburg, 2003), and ATP depletion is associated with an increase in cellular F-actin and an increase in ADF/cofilin activity (Minamide et al., 2000). It follows that acute inhibition of mitochondrial function used here to generate pMAP-immunostained rods, may result in changes to the actin cytoskeleton. Consistent with this, AM-treated chick neurons exhibited a 1.9-fold increase in overall phalloidin staining in cell bodies indicative of increased F-actin content (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). The pMAP-positive rods, however, did not overlap with phalloidin labeling (supplemental Fig. S3b, available at www.jneurosci.org as supplemental material).
In ATP-depleted rat hippocampal neurons, the rapid dephosphorylation of cofilin leads to the development of cofilin–actin rods in neurites (Minamide et al., 2000). These rods contain actin, as evidenced by both antibody immunostaining and ultrastructure, but do not stain with phalloidin, indicating they are probably saturated with ADF/cofilin, which stabilizes the “twisted” form of the filament and eliminates the phalloidin binding site (McGough et al., 1997). We therefore asked whether rods induced by ATP depletion contain both pMAP and ADF/cofilin. Double labeling revealed that ADF/cofilin–actin rods in part colocalize with pMAP in chick (Fig. 2a–c, asterisks), human (Fig. 2d, asterisk), and rat hippocampal neurons (Fig. 2e, asterisk). Colocalization of cofilin and pMAP in rod structures in ATP-depleted organotypic rat hippocampal slice cultures was also revealed (Fig. 3). Moreover, staining for β(III)-tubulin did not accumulate at rods, further suggesting the specificity of pMAP and ADF/cofilin in formation of neuritic rods (supplemental Fig. S4b,c, available at www.jneurosci.org as supplemental material). However, ADF/cofilin-stained rods that do not stain for pMAP (Fig. 2c,e, arrowheads) and pMAP-stained rods that do not stain for ADF (Fig. 2b, arrow) were both observed. Because optimal immunostaining for pMAP requires Triton X-100 permeabilization (avoiding methanol) and optimal immunostaining for ADF/cofilin requires methanol (avoiding Triton), structures that stain for one and not the other may be suboptimally permeabilized or immunostained. However, we cannot exclude that the detected inclusions may represent a heterogeneous population of rod structures. Surprisingly, in hippocampal neurons derived from rat, there was no change in the diffuse and uniform distribution of pMAP staining after ATP depletion, fixation in 4% paraformaldehyde and 0.1% glutaraldehyde, and brief (90 s) 0.05% Triton X-100 permeabilization, which looked identical with the staining of pMAP in untreated human, rat, and chick neurons (Fig. 1f). However, rat neurons subjected to AM-induced ATP depletion, followed by fixation in 4% formaldehyde and permeabilization as above, did exhibit rod-like 12E8 immunostaining (Fig. 2e), suggesting that glutaraldehyde fixation of pMAP when it is in a rod-like structure masks its epitope from 12E8 binding. Together, these results link ADF/cofilin–actin and pMAP inclusions within the same rod structures in neurites, which can be induced in cell culture by the common mechanism of acute ATP depletion through mitochondrial dysfunction.
Figure 2.
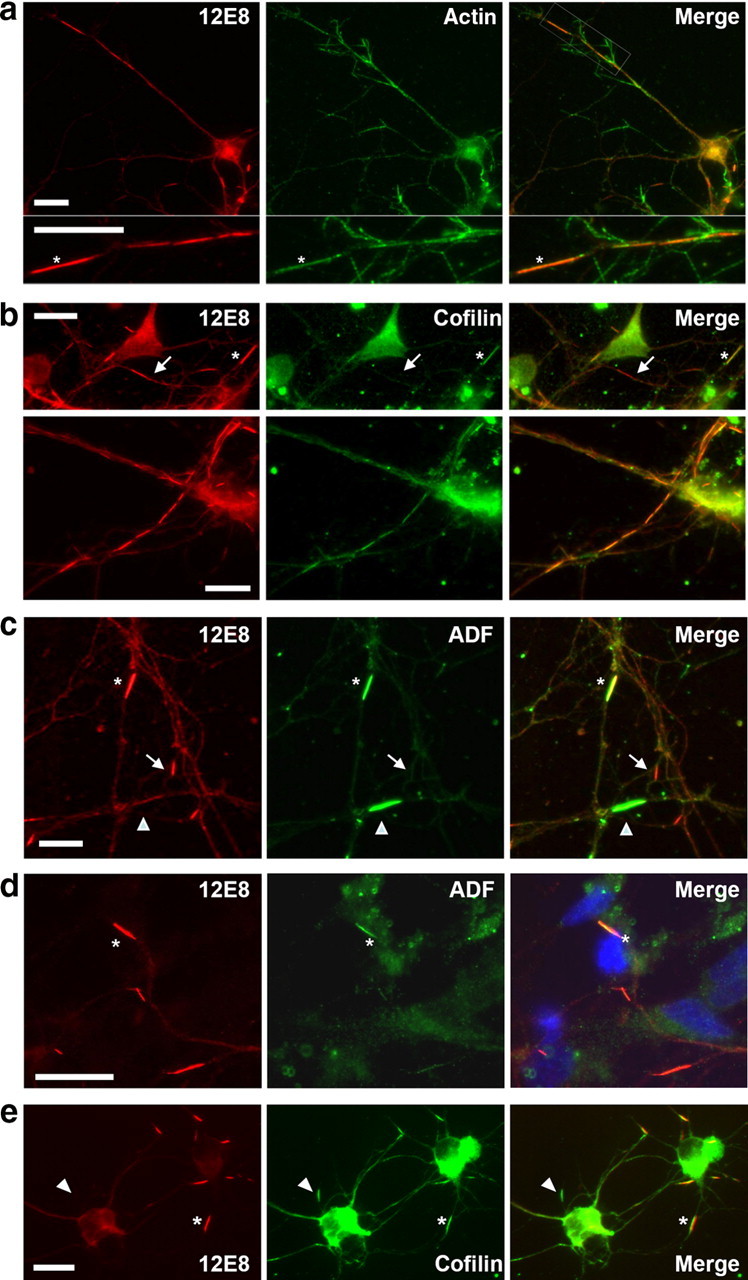
Colocalization of actin, cofilin, and ADF occurs in pMAP inclusions after ATP depletion. Chick neurons treated with AM were double-labeled with 12E8 (red) and actin (a), cofilin (C8736; Sigma-Aldrich) (b), or ADF (D8815; Sigma-Aldrich) (c) antibodies as indicated (green). Merged images are shown on the right. Rods containing pMAP also stained positively for actin (a, asterisk), cofilin (b, asterisk in top panel), and ADF (c, asterisk). Double-labeled strings of rods were often observed within single neurites (b, bottom panel). pMAP inclusions in primary human (d) and rat (e) hippocampal (5 div) neurons also colabeled with ADF (D8815) (d) and cofilin (1439) (e). Some rods contained pMAP but were weakly labeled or negative for cofilin or ADF (b, c, arrows), whereas others stained strongly for ADF or cofilin but only weakly or negative for pMAP (c, e, arrowheads). This is suggestive of a heterogeneous population of rod-like structures and/or suboptimal immunostaining conditions for the respective structures (see text). Scale bars, 10 μm.
Figure 3.

Colocalization of cofilin and pMAP occurs in ATP-depleted rat organotypic hippocampal slices. After ATP depletion, rod-like accumulations are evident in organotypic rat (postnatal day 8) hippocampal slices and are positive for both cofilin (labeled with 1439) and pMAP (labeled with 12E8) (arrows). Scale bar, 10 μm.
To establish the activation state of ADF/cofilin during pMAP accumulation, we used an antibody specific for phosphorylated (inactive) ADF/cofilin (Minamide et al., 2000; Maloney et al., 2005). ADF/cofilin actin-binding activity is negatively regulated by phosphorylation of the N-terminal Ser3 (Huang et al., 2008; Kim et al., 2009). On ATP depletion with AM, ADF/cofilin was rapidly dephosphorylated (activated) in chick tectal neurons, as early as 2 min after treatment (Fig. 4a,b), consistent with studies in primary rat hippocampal neurons (Minamide et al., 2000). Peroxide (H2O2)-treated tectal neurons also showed nearly complete ADF/cofilin activation after 30 min (Fig. 4a,b). The time course for the redistribution of pMAP immunostaining into the rod-like inclusions (Fig. 4c) correlated with the rapid dephosphorylation of ADF/cofilin and the formation of ADF/cofilin–actin rods.
Figure 4.
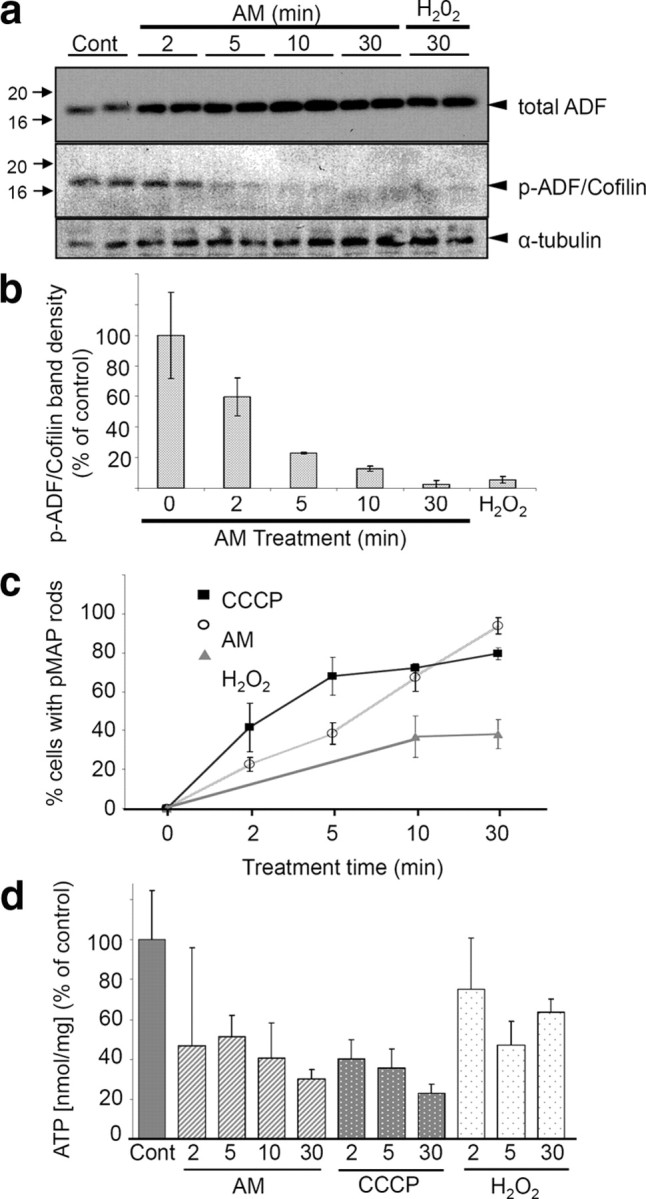
Mitochondrial inhibition induces activation of ADF/cofilin and is correlated to pMAP-positive rod formation. Dephosphorylation at the N-terminal (Ser3) leads to ADF/cofilin activation. a, Chick neurons were treated with AM or H2O2 for the indicated times, lysed, and immunoblotted. Whereas total ADF levels (probed with 1439) remained the same (or slightly increased) compared with control cells, ATP depletion resulted in rapid dephosphorylation of ADF/cofilin as early as 2 min after AM treatment (p-ADF/cofilin antibody is Ser3-specific). Similarly, H2O2 treatment induced almost complete dephosphorylation. b, Graph shows time course of ADF/cofilin dephosphorylation. Band intensities from duplicate samples were normalized to α-tubulin loading controls and calculated as a percentage of control band intensities. c, Coverslips were treated with AM, CCCP, or H2O2 and immunostained in parallel for quantification of the extent of rod generation. AM-treated cells ultimately contained the greatest abundance of pMAP-positive rods, although CCCP-treated cells initially developed rods more rapidly. Approximately 40% of H2O2-treated cells developed rods over 30 min. Data points and error bars represent mean and SEM values from three independent experiments. d, Luminescence measurements were performed to ascertain ATP levels after AM, CCCP, or H2O2 treatment. Compared with control samples, cells treated with AM and CCCP showed a rapid decline in ATP levels, whereas those treated with H2O2 showed a more modest decline. Error bars represent SEM of triplicate samples. Together, these data suggest an inverse correlation between number of pMAP rods formed and level of ATP.
Based on the above results, we expected that the extent of pMAP incorporation into rods would be proportional to the level of intracellular ATP. To explore this relationship, we measured ATP levels in tectal neurons before and after treatment with AM, CCCP (a mitochondrial uncoupling agent), or H2O2 (Fig. 4d). Consistent with the notion that activation of ADF/cofilin induced by intracellular ATP depletion correlates with pMAP accumulation, there was an inverse relationship between the percentage of cells in the culture that contained pMAP-stained rods (Fig. 4c) and the level of ATP detected in the cell treatment groups (Fig. 4d). Cell viability tests revealed 95% cell survival in 10 min AM-treated chick neuronal cultures (compared with control cultures) and 77% survival in 30 min treated cultures (supplemental Fig. S4d, available at www.jneurosci.org as supplemental material). Likewise, abundant pMAP-positive rod formation was recapitulated in primary human neurons, after 30 min AM treatment (mean ± SD, 24 ± 2 rods per field for AM-treated compared with 8 ± 1 rods per field in controls; p < 0.001). Together, these results suggest ATP depletion is an important correlate for redistribution of pMAP into rods.
Activated cofilin sequesters and is closely associated with pMAP in rods
Since ADF/cofilin–actin rods costained for pMAP and rod formation depends on activation of ADF/cofilin, we asked whether transfected cofilin-GFP could sequester pMAP into rods. Previous work has shown that expression of cofilin-GFP leads to increased pools of activated cofilin and cofilin–actin rod formation in neurites of rat hippocampal neurons (Minamide et al., 2000; Bernstein et al., 2006). We investigated the effects of ATP depletion on the reorganization of pMAP in relation to cofilin-GFP rods by inhibiting mitochondria. ATP depletion of neuronal cultures led to rapid cofilin-GFP rod formation in transfected cells and strong pMAP colabeling (Fig. 5a,b; supplemental Fig. S5a, available at www.jneurosci.org as supplemental material). Conversely, cofilin-GFP rods did not sequester β(III)-tubulin label when all other conditions were identical (Fig. 5c). When non-neuronal cells in culture formed cofilin-GFP rods, these were negative for both pMAP and β(III)-tubulin (Fig. 5d), neither of which is expressed in non-neuronal cells. Together, these results suggest that activated cofilin first accumulates into rods and subsequently sequesters pMAP into these cytoskeletal inclusions.
Figure 5.
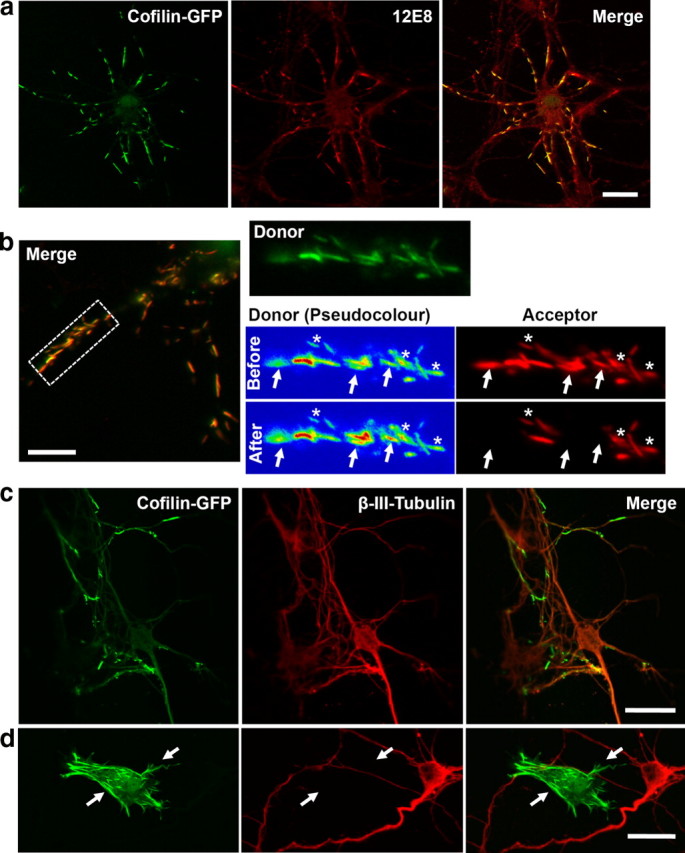
Cofilin-GFP forms rods that sequester pMAP during ATP depletion. Primary chick neurons were transfected with human cofilin-GFP (green) for 24 h, treated with AM to induce rod assembly, and pMAP immunolabeled with 12E8 and Alexa 555 secondary anti-mouse (red). a, Colocalization of cofilin-GFP rods (green) with 12E8 immunolabel (red) is evident in merged images (right panel, yellow). b, FRET measurements revealed a close proximity between pMAP immunolabel and cofilin-GFP rods. Images of donor (cofilin-GFP, green or pseudocolor) before and after photobleaching of acceptor (Alexa 555, red) are shown. Pseudocolor images of the donor signal demonstrate an increase in GFP fluorescence in acceptor-bleached rods (arrows) but not nonbleached rods (asterisks). A FRET efficiency of 41 ± 3% (mean ± SEM; n = 52; p < 0.0001) was quantified by measuring the percentage increase in donor fluorescence after acceptor bleaching. No significant FRET signal was seen in nonbleached rods (b, asterisks) in the same cells (donor fluorescence increase, mean ± SEM, 2.2 ± 1.1%; n = 52). c, β(III)-tubulin remains smooth and evenly distributed throughout cofilin-GFP transfected cells and is not enriched at cofilin-GFP rods. d, The specificity of pMAP sequestration is further illustrated in the occasional non-neuronal cell that was transfected with cofilin-GFP. These cells formed cofilin-GFP rods (arrows) but were negative for both β(III)-tubulin (red) and pMAP (data not shown). This also provides evidence that cofilin rods form in the absence of pMAP. Scale bars: a, c, d, 20 μm; b, 10 μm.
To further evaluate the interaction between cofilin and pMAP in rods, we exploited FRET between the GFP (donor) and Alexa 555 (acceptor) fluorophors. FRET between colocalized cofilin-GFP and pMAP/Alexa 555 was measured using acceptor photobleaching (Fig. 5b). The cofilin-GFP fluorescence signal in rods increased significantly after bleaching the Alexa 555 fluorophor (Fig. 5b, right, arrows), with a measured FRET efficiency of 41 ± 3% (mean ± SEM; n = 52; p < 0.0001) as measured by the percentage increase in donor fluorescence after acceptor bleaching. In contrast, cofilin-GFP fluorescence did not significantly increase in adjacent rods in the same cells in which the Alexa 555 fluorophor was not bleached (Fig. 5b, right, asterisks). Furthermore, a much reduced FRET signal was detected in neurons between cofilin-GFP and Alexa 555 immunostained pMAP in adjacent non-rod-forming regions of neurites and cell bodies (supplemental Fig. S5b, available at www.jneurosci.org as supplemental material). Control experiments demonstrated no FRET between cofilin-GFP and control antibody-labeled epitopes—β(III)-tubulin/Alexa 555 or Src/Alexa 555—even in regions in which the fluorescence appeared colocalized (supplemental Fig. S5c,d, available at www.jneurosci.org as supplemental material). Cells expressing free GFP and treated with AM to generated pMAP-positive rods labeled with 12E8/Alexa 555 also never produced FRET (supplemental Fig. S5d, available at www.jneurosci.org as supplemental material). These results demonstrate that cofilin-GFP and the secondary antibody labeling pMAP are colocalized <10 nm apart within the cytoskeletal rod complex, indicating a close proximity of pMAP and cofilin in these inclusions.
Silencing ADF prevents pMAP accumulation in rods
Since induction of ADF/cofilin rods leads to the reorganization and sequestering of pMAP, we asked whether silencing ADF or cofilin would prevent sequestering of pMAP under the same conditions. During chick brain development, ADF comprises ∼75% of the total ADF/cofilin from embryonic day 14 onward (Devineni et al., 1999). We therefore chose to knock down the total cellular pool of ADF specifically, using a plasmid for expression of a shRNA that yields an siRNA when expressed and processed. The shRNA was transfected into chick neurons and coexpressed from the same plasmid as GFP for visualization of transfected cells. Four days posttransfection, cells were immunolabeled for ADF (Fig. 6a) and quantified, revealing 75% knockdown of ADF (Fig. 6e). Treating transfected cultures with AM (1 μm for 10 min) and staining for pMAP revealed that significant silencing of ADF inhibited accumulation of pMAP into rod structures, whereas surrounding untransfected cells contained an abundance of rods (Fig. 6d, asterisks). In control chick neuronal cultures transfected for 4 d with shRNA specific for human PAK2, immunolabeling revealed no decrease in total cellular ADF (Fig. 6b,e). pMAP accumulated in rod-like inclusions both in surrounding nontransfected neurons and in shRNA PAK2 transfected neurons (Fig. 6c, asterisks and arrowhead). Together, these results suggest that the presence of ADF/cofilin is necessary for sequestration of pMAP into rod-like structures.
Figure 6.
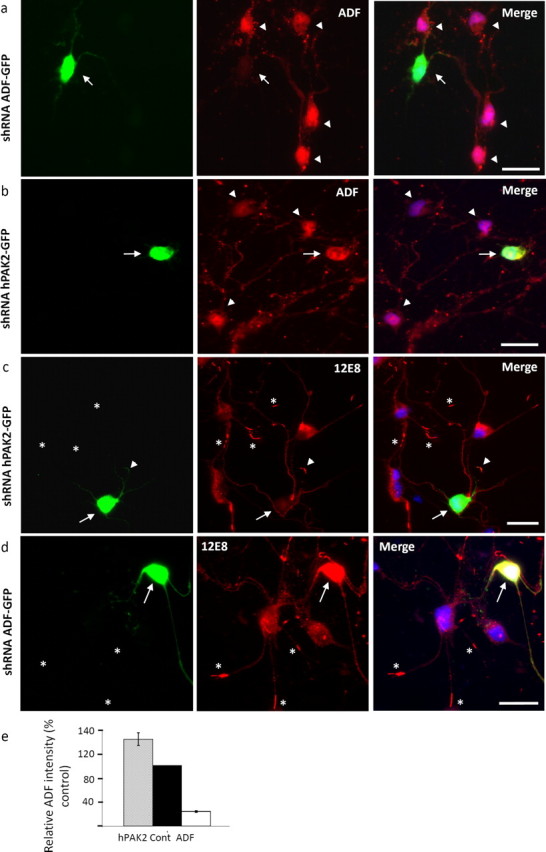
Reduction of the cellular ADF/cofilin pool inhibits the formation of rods and sequestration of pMAP. a, ADF was knocked down in primary chick tectal neurons by expression of shRNA from a plasmid also encoding GFP for visualization of transfected cells. In cultures transfected for 4 d, ADF immunolabeling using the 1439 antibody demonstrated significant ADF knockdown in transfected cells (arrow) compared with surrounding nontransfected cells (arrowheads). b, Cells transfected with an shRNA specific for human PAK2 as a control (arrow) showed no reduction of ADF compared with surrounding nontransfected cells (arrowheads). c, d, Transfected cultures were treated with 1 μm AM and immunolabeled with 12E8 to determine whether pMAP accumulates into rod-like structures after significant reduction of ADF. Whereas hPAK2 shRNA-transfected cells (c, arrow) formed pMAP-positive rods (arrowhead) comparable with surrounding nontransfected cells (asterisks), ADF shRNA-transfected cells (d, arrow) never accumulated pMAP-positive rods, although surrounding cells frequently did (asterisks). Images represent single examples from >50 ADF shRNA transfected cells in three independent experiments. e, Quantification of ADF (1439) staining intensity revealed a 75% knockdown in ADF shRNA-transfected cells compared with nearby nontransfected (control) cells (transfected cell staining intensity, mean ± SEM, 24.1 ± 1.5, expressed as percentage of control cells; n = 25). In contrast, ADF staining intensity of hPAK2 shRNA-transfected cells compared with surrounding nontransfected cells was not reduced (intensity, mean ± SEM, 144.2 ± 10.8%; n = 25). Scale bars, 20 μm.
Latrunculin B enhances and jasplakinolide represses pMAP staining in rods
Since activation of ADF/cofilin coincided with pMAP recruitment into ADF/cofilin–actin rods, we asked whether other manipulations of F-actin assembly could influence pMAP sequestration to rods. To address this, we used pharmacological manipulation (enhancing or suppressing) of F-actin pools and stained for pMAP. Latrunculins sequester monomeric G-actin to form a nonpolymerizable 1:1 complex, thereby inhibiting F-actin reassembly and thus promoting overall F-actin depolymerization (Coué et al., 1987). Latrunculins, however, compete weakly with ADF/cofilin for actin binding (Bernstein et al., 2006) and can actually induce ADF/cofilin–actin rod formation (Pendleton et al., 2003). The decline in the phalloidin-stainable F-actin pool after Lat B treatment is indeed evident (Fig. 7a), although it should be noted that phalloidin cannot stain ADF/cofilin saturated F-actin. In contrast, Jasp binds and stabilizes F-actin resulting in a net decrease in the G-actin pool (Bubb et al., 1994). Since Jasp competes for the phalloidin-binding site on F-actin, stabilized F-actin cannot be visualized with fluorescent phalloidin in the presence of Jasp (Fig. 7a). However, ADF/cofilin cannot bind to phalloidin-stabilized F-actin (Minamide et al., 2000) and thus also would not likely bind to the Jasp-stabilized actin filaments.
Figure 7.
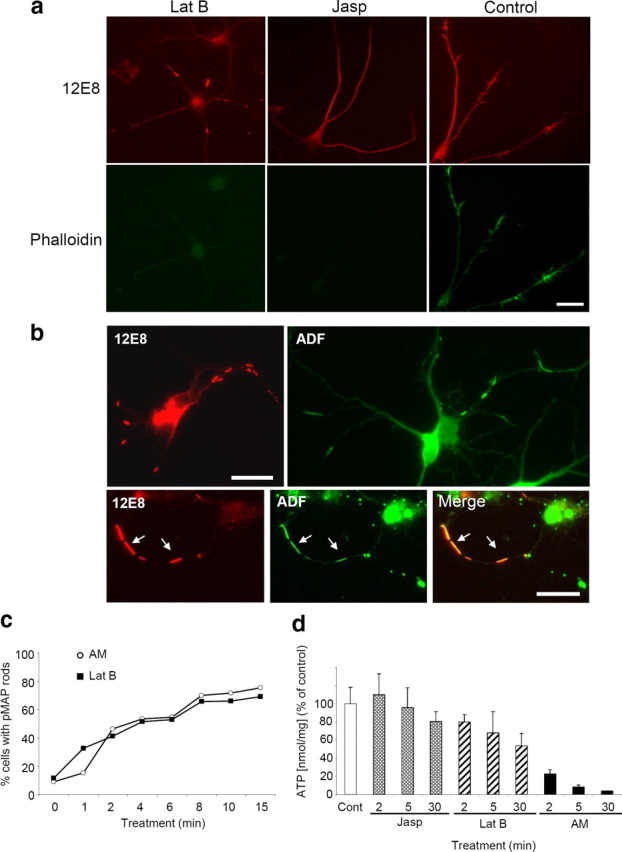
Dynamic F-actin enhances pMAP and actin rod formation. a, After treatment with Lat B but not Jasp, the assembly of pMAP into rod-like structures occurred. The chick neurons were stained for pMAP (12E8) and F-actin (phalloidin-488). Costaining with phalloidin showed only very weak labeling of F-actin in both Lat B- and Jasp-treated cells. b, Treating cells with Lat B induced formation of pMAP (red; 12E8) and ADF (green; 1439) rods, as seen in single stainings. Colabeling revealed frequent colocalization of both proteins (arrows). c, To compare the rate and abundance of rod formation under either mitochondrial inhibition or F-actin manipulation, rods were counted in cells treated with AM or Lat B for increasing times, as indicated. The pattern of rod formation in each condition was comparable. d, To ascertain the relationship between rod formation and ATP levels under these conditions, luminescence assays were conducted. Whereas Jasp-treated cells had levels of ATP comparable with controls and AM-treated cells had a dramatic decrease, only moderate decline in ATP levels in Lat B-treated cells was evident. Error bars represent the SEM from triplicate samples. Scale bars, 20 μm.
Antibody labeling of Lat B-treated neurons revealed formation of both pMAP- and ADF-positive rods, which frequently colocalized in double-labeling experiments (Fig. 7b). In contrast, neurons treated with Jasp (or cotreated with Jasp and AM) exhibited a total absence of pMAP and ADF rods (Fig. 7a). We then compared the rate and abundance of pMAP-stained rods after Lat B compared with AM treatments and found the chronology of their formation indistinguishable between conditions (Fig. 7c). That Lat B treatment only moderately reduced ATP in neurons (Fig. 7d) yet still induced ADF/cofilin–actin and pMAP rod formation comparable with mitochondrial inhibitors suggests that ATP depletion is not a direct cause of rod formation, but an upstream event. These data collectively suggest that subunit release from F-actin is required for the generation of pMAP-positive rods and that in neurons this can be induced by ADF/cofilin activation. It also appears that ADF/cofilin binding to F-actin is essential for the formation of cytoskeletal rods. Dissociated actin subunits saturated with activated ADF/cofilin thereby form cytoskeletal rod inclusions that appear to sequester and accumulate pMAP.
Aβ peptides enhance pMAP accumulation at ADF/cofilin rods
In AD, the relationships between pMAP/tau pathologies, cofilin–actin pathologies, and amyloid pathologies arising from Aβ peptide oligomerization and/or aggregation are poorly understood. Synthetic Aβ1-42 has been shown to induce cofilin–actin rod pathology in up to 20% of neurons in dissociated rodent hippocampal cultures (Maloney et al., 2005), with the majority of these neurons located in the dentate gyrus (Davis et al., 2009). We therefore asked whether Aβ could influence the sequestering of pMAP to ADF/cofilin–actin rods. Primary chick neurons were exposed to 1 or 2 μm Aβ1-40 or Aβ1-42. Before applying to cells, peptide solutions were agitated on a shaker for 30 min to generate mixtures of oligomeric and fibrillar structures (Goldsbury et al., 2000). The solutions were then diluted into cell culture medium and added to cells at a final concentration of 1 or 2 μm. After exposure to the peptides for 20 h, the cells were fixed and immunostained for pMAP. Less rods were generated in cultures treated with Aβ compared with those treated with the mitochondrial inhibitors, consistent with previous studies of ADF/cofilin–actin rod generation in hippocampal neurons treated with Aβ peptides (Maloney et al., 2005). However, when mean numbers of pMAP-positive rods per cell were quantified on vehicle control and Aβ-treated coverslips, a significant increase in the number of rods was revealed in Aβ-treated cells (Fig. 8a). No difference was observed between cells treated with 1 and 2 μm peptide or between cells treated with Aβ1-40 and Aβ1-42. The presence of F-actin-rich active growth cones in Aβ-treated neurons containing pMAP rods in the neurite shafts indicates the continuing viability of the neurons, despite the formation of pMAP-positive rods (Fig. 8b, white arrows). We confirmed that rods in Aβ-treated neurons contained both pMAP and ADF (Fig. 8c, arrows). In conclusion, these results show that synthetic Aβ aggregates can induce the recruitment of pMAP to ADF/cofilin–actin rods in a subset of neurons in these primary cultures.
Figure 8.
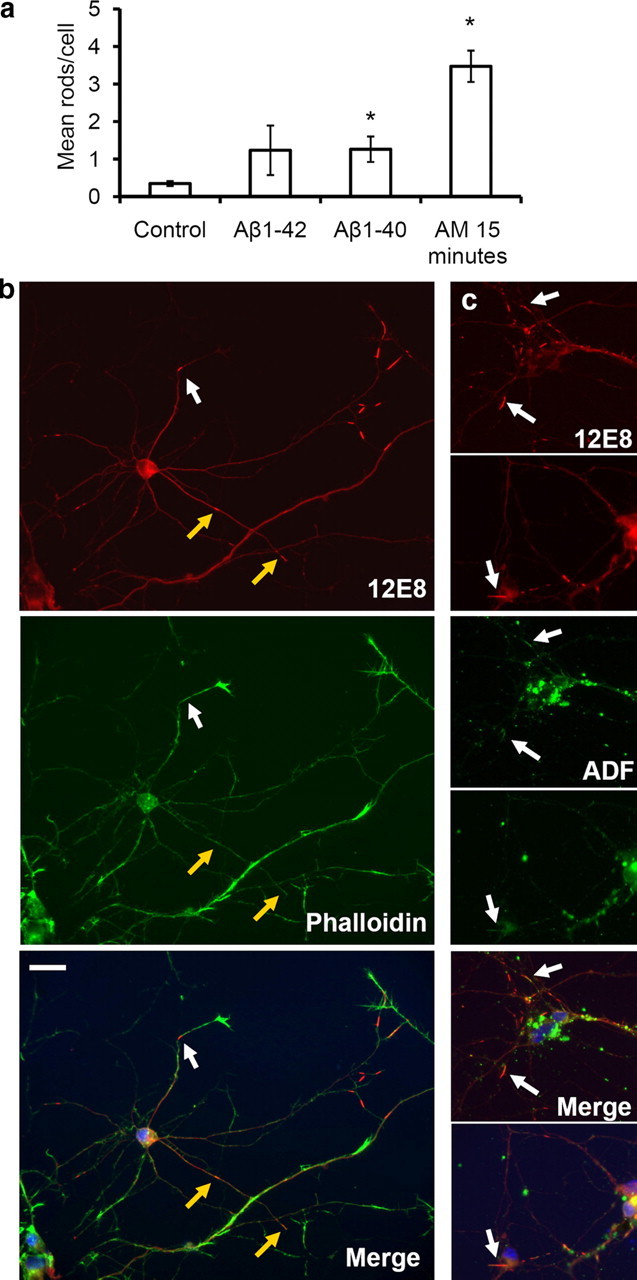
Amyloid peptides enhance pMAP sequestration to rods in primary chick neurons. Primary neurons were treated for 20 h with solutions of Aβ peptides (1 or 2 μm) and double-labeled with the 12E8 antibody (red) and either phalloidin-488 or ADF (both green) as indicated. a, The mean number of pMAP-positive rods per cell was quantified in >10 fields on control coverslips and coverslips treated with Aβ peptides for 20 h or AM for 15 min. pMAP-positive rods per cell increased significantly after incubation with AM or Aβ solutions compared with vehicle (DMSO) control (mean ± SEM, 1.3 ± 0.3 rods/nuclei for both aged Aβ1-40 and Aβ1-42 and 3.5 ± 0.4 rods/nuclei for AM-treated compared with 0.4 ± 0.1 rods/nuclei for control cells; *p < 0.02; n > 20; results are from duplicate experiments). Since no difference was seen between 1 versus 2 μm peptide treatments, results have been pooled. b, pMAP-positive rods in Aβ-treated neurons (shown here for Aβ1-40) often formed at distal regions near the base of phalloidin-positive (F-actin-rich) active growth cones (white arrows). They also formed in neurite shafts associated with collapsed growth cones (b, yellow arrows). c, Rods assembling in Aβ-treated neurons (shown here for Aβ1-40) accumulated both pMAP (red) and ADF (green) (arrows). Merged images (plus DAPI in blue) are shown in bottom panels. Scale bar, 20 μm.
Discussion
Neuropil threads of the AD brain are composed of linear arrays of cytoskeletal inclusions containing the pMAP tau (Velasco et al., 1998; Augustinack et al., 2002). These structures are positive for Ser262/356 tau labeled with the 12E8 antibody before extensive staining by antibodies marking hyperphosphorylation of tau that is observed in late-stage disease (Augustinack et al., 2002). Corresponding to this, formation of neuropil threads is associated with the early clinical stages of dementia (Giannakopoulos et al., 2007). The structure of neuropil threads implicates a disrupted cytoskeletal network that spans the width of the neurite likely contributing to dementia by blocking cargo trafficking to synapses that underlie memory formation and cognition, causing subsequent retraction of distal neurites (Velasco et al., 1998) (for review, see Terry, 1998; Bamburg and Bloom, 2009). Our data open the possibility that abnormal activation of cofilin, an actin-binding protein, in the AD brain and generation of cofilin–actin cytoskeletal rods is a triggering factor for the sequestration and accumulation of Ser262/356 phosphorylated tau in some neuropil threads. This process may well represent an early pathogenic event in AD neurodegeneration.
Neurodegenerative stimuli including oxidative stress, mitochondrial dysfunction, excitotoxic glutamate, ischemia, and soluble forms of Aβ lead to activation of cofilin and the related protein ADF and the generation of ADF/cofilin–actin rods in primary neuronal cell culture (for review, see Bamburg and Bloom, 2009). These rods resemble the cofilin aggregates that are widely distributed in the AD brain (Minamide et al., 2000). At a low ratio to actin and in an active (dephosphorylated) state, ADF/cofilin maximally enhance subunit turnover of F-actin, dynamically remodeling the actin cytoskeleton and thus playing an integral role in regulating actin-dependent synaptic stabilization (for review, see Bamburg and Bloom, 2009). ADF/cofilin bind cooperatively to F-actin, and, at higher ratios to actin, they can saturate regions of filaments and stabilize the pieces of filaments that remain after severing (Andrianantoandro and Pollard, 2006; Chan et al., 2009). Because ADF/cofilin bind to a minor, slightly twisted conformation of F-actin, they stabilize this “twisted” form, which prevents binding of the commonly used F-actin stain, phalloidin (McGough et al., 1997). In the brain, mitochondrial dysfunction and energy depletion associated with AD (Smith et al., 2005; Wang et al., 2009) could feasibly serve as a pathway for F-actin remodeling in neurons, since up to 50% of neuronal energy is dedicated to actin dynamics (Bernstein and Bamburg, 2003). We propose mitochondrial dysfunction is one potential pathway upstream of the assembly of cytoskeletal rods because direct electron transport chain inhibitors elicit cofilin activation, concomitant with rod assembly in primary neurons and organotypic rat hippocampal brain slices. This pathway results in a precipitous drop in ATP, and the release of the cofilin phosphatase chronophin from an inhibitory complex with Hsp90, resulting in cofilin–actin rod formation (Huang et al., 2008). However, cofilin–actin rods can also be induced while maintaining higher levels of cellular ATP, such as with peroxide (Fig. 4), which activates the cofilin phosphatase slingshot by oxidizing and removing inhibition by 14-3-3 and leads to almost complete cofilin dephosphorylation and cofilin–actin rod formation (Kim et al., 2009). The common thread is an increased pool of active ADF/cofilin with respect to the amount of F-actin such that ADF/cofilin-saturated pieces of F-actin are available and coalesce into rods.
What is the role of pMAP at the cofilin–actin rods? Rods isolated from neurons and non-neuronal cell lines contain ADF/cofilin:actin in a 1:1 complex and indeed can be formed in vitro from these purified proteins (L. S. Minamide, S. Maiti, J. A. Boyle, R. C. Davis, J. A. Coppinger, Y. Bao, T. Y. Huang, J. Yates, G. M. Bokoch, and J. R. Bamburg, unpublished results). Thus, as is also shown here, pMAP is not essential for cofilin–actin rod formation. Nevertheless, pMAP association with most cofilin–actin rods occurs very early in the rod formation process. Rods isolated from cortical neurons and those formed from endogenous proteins from a non-neuronal cell line both have similar stabilities to alterations in pH, ionic strength, reducing agents, Ca2+/EGTA, ATP, and detergents. Therefore the presence of pMAP on the neuronal rods does not appear to confer in them any special property related to their stability. The question then arises—do the rods confer any special property on the associated pMAP? The tandem arrays of the rapidly formed cofilin–actin rods are similar in size and distribution to the neuritic striations of the neuropil threads in AD. The ultrastructure of most striated neuropil threads from AD brain clearly shows these to consist of the ∼20-nm-diameter paired helical filaments (PHFs) (Velasco et al., 1998), not of the cofilin–actin bundles containing ∼10-nm-diameter filaments that we observe in organotypic hippocampal slices treated with Aβ1-42 peptide oligomers (Davis et al., 2009). Thus, we suggest that the cofilin–actin rods serve as a template for recruitment and binding of pMAP, and that this association facilitates additional phosphorylation of pMAP and its self-assembly leading to the eventual replacement of the cofilin–actin rods with PHF bundles. Additional long-term studies are needed to test this hypothesis.
Supporting the concept that pMAP/tau accumulation and toxicity is linked to interactions with the actin cytoskeleton is work in Drosophila models that showed that toxicity of overexpressed hyperphosphorylated tau could be modified by genetic ablation or coexpression of actin-associated proteins (Fulga et al., 2007). Interestingly, tau phosphorylation at the 12E8 antibody epitope was shown to be essential for the initiation of tau toxicity and its downstream hyperphosphorylation in Drosophila (Nishimura et al., 2004). Mutated tau nonphosphorylatable at the 12E8 epitope did not confer toxicity in Drosophila and exhibited reduced downstream phosphorylation at other AD-relevant phospho-tau epitopes (Nishimura et al., 2004). These findings are consistent with the premise that the sequestration of 12E8-specific pMAP to cofilin–actin rods demonstrated here could represent an essential early event in a neurodegenerative pathway. The recent finding that tau reduction can ameliorate toxicity induced by APP (amyloid precursor protein) overexpression or exposure of cells to Aβ oligomers is also consistent with the key involvement of pMAP/tau in response to toxic events (King et al., 2006; Roberson et al., 2007). Potential deleterious effects of cytoskeletal rods on neurons are multifold. First, rods that assemble in axons and dendrites of cultured neurons form transport blockades inhibiting the free movement of vesicles and organelles leading to synaptic dysfunction (Jang et al., 2005; Maloney et al., 2005, 2008). Second, the accumulation of actin and cofilin in rod structures could directly impinge on the availability of cofilin for its function in modulating actin dynamics at the synapse (Hotulainen et al., 2009). Likewise, the sequestration of pMAP/tau to the rod matrix could adversely affect the role of tau in stabilizing and regulating the axonal MT network of tracks for fast axonal transport.
The generation of cytoskeletal rods in response to stressors may be an initial compensatory response to slow down energy-consuming actin dynamics and thereby protect neurons under conditions in which ATP supply is compromised (Bernstein et al., 2006). However, repeated or prolonged stress would likely be detrimental. Increasing evidence suggests declining mitochondrial function and reduced energy metabolism to be early events in the AD brain, preceding severe pathological changes (Smith et al., 2005; Wang et al., 2009). Aging is the major risk factor for sporadic AD, and on the one hand, electron transport chain activity declines with age, whereas on the other, oxidative stress increases, potentially impacting on the availability of ATP in cells (Lin and Beal, 2006). Moreover, the reduction of glucose uptake in AD brain cells and concomitant decline in glycolysis would also negatively impact ATP levels. Precedents of local mitochondrial dysfunction could be multifold and varied in the aging brain, for example, low brain perfusion attributable to vascular insufficiency, stroke, and Aβ peptide-mediated perturbation of mitochondria (Cullen et al., 2005; Du et al., 2008; Cho et al., 2009). Mechanisms linking this metabolic dysfunction with the formation of neuropathological lesions in the AD brain need to be established. Interestingly, it was demonstrated that environmental toxin inhibitors of mitochondrial respiratory chain complexes induce somatic inclusions of phosphorylated tau in neuronal cell culture and neurodegeneration in tau-related disease (Escobar-Khondiker et al., 2007).
Advanced neuropathology involving pMAP and cofilin accumulation and aggregation may be a result of increased vulnerability to normal environmental stress in brains expressing mutant tau (i.e., in hereditary frontotemporal dementias) or from elevated oxidative/mitochondrial stress in brains expressing wild-type endogenous tau (in sporadic AD). In a healthy brain, a fine balance between these stressors and compensatory defense mechanisms must normally occur, given that development of tau pathology and AD are not an inevitable part of aging. Recent evidence showing that neuronal cell bodies attached to dystrophic neurites in AD-related transgenic mice are still viable and otherwise healthy (Adalbert et al., 2009) holds promise for future work to develop methods to disrupt the formation of the cofilin–actin rods and/or the interaction between cofilin and pMAP for establishing new therapeutic strategies to combat this disease early in its development.
Footnotes
This work was supported by The Sir Zelman Cowen Universities Fund, The Judith Jane Mason and Harold Stannett Williams Memorial Foundation, The Rebecca Cooper Foundation, Sydney Medical School (C.G.), Alzheimer Drug Delivery Foundation Grant 281201 (J.R.B.), and National Institutes of Health–National Institute of Neurological Diseases and Stroke Grants NS43115 and NS40371 (J.R.B.). We thank Prof. Peter Seubert (Elan Pharmaceuticals, South San Francisco, CA) for providing the 12E8 antibody.
References
- Adalbert R, Nogradi A, Babetto E, Janeckova L, Walker SA, Kerschensteiner M, Misgeld T, Coleman MP. Severely dystrophic axons at amyloid plaques remain continuous and connected to viable cell bodies. Brain. 2009;132:402–416. doi: 10.1093/brain/awn312. [DOI] [PubMed] [Google Scholar]
- Andrianantoandro E, Pollard TD. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol Cell. 2006;24:13–23. doi: 10.1016/j.molcel.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Augustinack JC, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- Bamburg JR, Bloom GS. Cytoskeletal pathologies of Alzheimer disease. Cell Motil Cytoskel. 2009;66:635–649. doi: 10.1002/cm.20388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BW, Bamburg JR. Actin-ATP hydrolysis is a major energy drain for neurons. J Neurosci. 2003;23:1–6. doi: 10.1523/JNEUROSCI.23-01-00002.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BW, Chen H, Boyle JA, Bamburg JR. Formation of actin-ADF/cofilin rods transiently retards decline of mitochondrial potential and ATP in stressed neurons. Am J Physiol Cell Physiol. 2006;291:C828–C839. doi: 10.1152/ajpcell.00066.2006. [DOI] [PubMed] [Google Scholar]
- Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Bubb MR, Senderowicz AM, Sausville EA, Duncan KL, Korn ED. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J Biol Chem. 1994;269:14869–14871. [PubMed] [Google Scholar]
- Carlier M-F, Laurent V, Santolini J, Melki R, Didry D, Xia G-X, Hong Y, Chua N-H, Pantaloni D. Actin depolymerizing factor (ADF/Cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C, Beltzner CC, Pollard TD. Cofilin dissociates Arp2/3 complex and branches from actin filaments. Curr Biol. 2009;19:537–545. doi: 10.1016/j.cub.2009.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coué M, Brenner SL, Spector I, Korn ED. Inhibition of actin polymerization by latrunculin A. FEBS Lett. 1987;213:316–318. doi: 10.1016/0014-5793(87)81513-2. [DOI] [PubMed] [Google Scholar]
- Cullen KM, Kócsi Z, Stone J. Vascular relationships of haem-rich deposits in the aging cerebral cortex. J Cereb Blood Flow Metab. 2005;25:1656–1667. doi: 10.1038/sj.jcbfm.9600155. [DOI] [PubMed] [Google Scholar]
- Davis RC, Maloney MT, Minamide LS, Flynn KC, Stonebraker MA, Bamburg JR. Mapping cofilin-actin rods in stressed hippocampal slices and the role of cdc42 in amyloid β-induced rods. J Alzheimers Dis. 2009;18:35–50. doi: 10.3233/JAD-2009-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devineni N, Minamide LS, Niu M, Safer D, Verma R, Bamburg JR, Nachmias VT. A quantitative analysis of G-actin binding proteins and the G-actin pool in developing chick brain. Brain Res. 1999;823:129–140. doi: 10.1016/s0006-8993(99)01147-6. [DOI] [PubMed] [Google Scholar]
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar-Khondiker M, Höllerhage M, Muriel MP, Champy P, Bach A, Depienne C, Respondek G, Yamada ES, Lannuzel A, Yagi T, Hirsch EC, Oertel WH, Jacob R, Michel PP, Ruberg M, Höglinger GU. Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons. J Neurosci. 2007;27:7827–7837. doi: 10.1523/JNEUROSCI.1644-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol. 2007;9:139–148. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- Garcia ML, Cleveland DW. Going new places using an old MAP: tau, microtubules and human neurodegenerative disease. Curr Opin Cell Biol. 2001;13:41–48. doi: 10.1016/s0955-0674(00)00172-1. [DOI] [PubMed] [Google Scholar]
- Gervásio OL, Whitehead NP, Yeung EW, Phillips WD, Allen DG. TRPC1 binds to caveolin-3 and is regulated by Src kinase—role in Duchenne muscular dystrophy. J Cell Sci. 2008;121:2246–2255. doi: 10.1242/jcs.032003. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P, von Gunten A, Kovari E, Gold G, Herrman FR, Hof PR, Bouras C. Stereological analysis of neuropil threads in the hippocampal formation: relationships with Alzheimer's disease neuronal pathology and cognition. Neuropathol Appl Neurobiol. 2007;33:334–343. doi: 10.1111/j.1365-2990.2007.00827.x. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG. A century of Alzheimer's disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- Goldsbury C, Whiteman IT, Jeong EV, Lim Y-A. Oxidative stress increases levels of endogenous amyloid-beta peptides secreted from primary chick brain neurons. Aging Cell. 2008;7:771–775. doi: 10.1111/j.1474-9726.2008.00423.x. [DOI] [PubMed] [Google Scholar]
- Goldsbury CS, Wirtz S, Müller SA, Sunderji S, Wicki P, Aebi U, Frey P. Studies on the in vitro assembly of a beta 1-40: implications for the search for a beta fibril formation inhibitors. J Struct Biol. 2000;130:217–231. doi: 10.1006/jsbi.2000.4259. [DOI] [PubMed] [Google Scholar]
- Guillemin GJ, Cullen KM, Lim CK, Smythe GA, Garner B, Kapoor V, Takikawa O, Brew BJ. Characterization of the kynurenine pathway in human neurons. J Neurosci. 2007;27:12884–12892. doi: 10.1523/JNEUROSCI.4101-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen P, Llano O, Smirnov S, Tanhuanpää K, Faix J, Rivera C, Lappalainen P. Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J Cell Biol. 2009;185:323–339. doi: 10.1083/jcb.200809046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TY, Minamide LS, Bamburg JR, Bokoch GM. Chronophin mediates an ATP-sensing mechanism for cofilin dephosphorylation and neuronal cofilin-actin rod formation. Dev Cell. 2008;15:691–703. doi: 10.1016/j.devcel.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang DH, Han JH, Lee SH, Lee YS, Park H, Lee SH, Kim H, Kaang BK. Cofilin expression induces cofilin-actin rod formation and disrupts synaptic structure and function in Aplysia synapses. Proc Natl Acad Sci U S A. 2005;102:16072–16077. doi: 10.1073/pnas.0507675102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Huang TY, Bokoch GM. Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol Biol Cell. 2009;20:2650–2660. doi: 10.1091/mbc.E09-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King ME, Kan HM, Baas PW, Erisir A, Glabe CG, Bloom GS. Tau-dependent microtubule disassembly initiated by prefibrillar beta-amyloid. J Cell Biol. 2006;175:541–546. doi: 10.1083/jcb.200605187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Maloney MT, Minamide LS, Kinley AW, Boyle JA, Bamburg JR. β-secretase-cleaved amyloid precursor protein accumulates at actin inclusions induced in neurons by stress or amyloid-β: a feedforward mechanism for Alzheimer's disease. J Neurosci. 2005;25:11313–11321. doi: 10.1523/JNEUROSCI.3711-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney MT, Kinley A, Pak C, Bamburg JR. ADF/cofilin, actin dynamics and disease. Protein Rev. 2008;8:83–187. [Google Scholar]
- McGough A, Pope B, Chiu W, Weeds A. Cofilin changes the twist of F-actin: implications for actin filament dynamics and cellular function. J Cell Biol. 1997;138:771–781. doi: 10.1083/jcb.138.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamide LS, Striegl AM, Boyle JA, Meberg PJ, Bamburg JR. Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat Cell Biol. 2000;2:628–636. doi: 10.1038/35023579. [DOI] [PubMed] [Google Scholar]
- Nishimura I, Yang Y, Lu B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell. 2004;116:671–682. doi: 10.1016/s0092-8674(04)00170-9. [DOI] [PubMed] [Google Scholar]
- Pendleton A, Pope B, Weeds A, Koffer A. Latrunculin B or ATP depletion induces cofilin-dependent translocation of actin into nuclei of mast cells. J Biol Chem. 2003;278:14394–14400. doi: 10.1074/jbc.M206393200. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Schaar BT, Kinoshita K, McConnell SK. Doublecortin microtubule affinity is regulated by a balance of kinase and phosphatase activity at the leading edge of migrating neurons. Neuron. 2004;41:203–213. doi: 10.1016/s0896-6273(03)00843-2. [DOI] [PubMed] [Google Scholar]
- Seubert P, Mawal-Dewan M, Barbour R, Jakes R, Goedert M, Johnson GV, Litersky JM, Schenk D, Lieberburg I, Trojanowski JQ. Detection of phosphorylated Ser262 in fetal tau, adult tau, and paired helical filament tau. J Biol Chem. 1995;270:18917–18922. doi: 10.1074/jbc.270.32.18917. [DOI] [PubMed] [Google Scholar]
- Shaw AE, Minamide LS, Bill CL, Funk JD, Maiti S, Bamburg JR. Cross-reactivity of antibodies to actin-depolymerizing factor/cofilin family proteins and identification of the major epitope recognized by a mammalian actin-depolymerizing factor/cofilin antibody. Electrophoresis. 2004;25:2611–2620. doi: 10.1002/elps.200406017. [DOI] [PubMed] [Google Scholar]
- Smith MA, Nunomura A, Lee HG, Zhu X, Moreira PI, Avila J, Perry G. Chronological primacy of oxidative stress in Alzheimer disease. Neurobiol Aging. 2005;26:579–580. doi: 10.1016/j.neurobiolaging.2004.09.021. [DOI] [PubMed] [Google Scholar]
- Terry R. The cytoskeleton in Alzheimer disease. J Neural Transm Suppl. 1998;53:141–145. doi: 10.1007/978-3-7091-6467-9_12. [DOI] [PubMed] [Google Scholar]
- Timm T, Li XY, Biernat J, Jiao J, Mandelkow E, Vandekerckhove J, Mandelkow EM. MARKK, a Ste20-like kinase, activates the polarity-inducing kinase MARK/PAR-1. EMBO J. 2003;22:5090–5101. doi: 10.1093/emboj/cdg447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco ME, Smith MA, Siedlak SL, Nunomura A, Perry G. Striation is the characteristic neuritic abnormality in Alzheimer disease. Brain Res. 1998;813:329–333. doi: 10.1016/s0006-8993(98)01034-8. [DOI] [PubMed] [Google Scholar]
- Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Goedert M. Molecular cloning and functional characterization of chicken brain tau: isoforms with up to five tandem repeats. Biochemistry. 2002;41:15203–15211. doi: 10.1021/bi026464m. [DOI] [PubMed] [Google Scholar]