

Abstract
The ability of an organism to adapt during stress has a significant impact on long-term survival and health. Maladaptive responses to stress have been associated with susceptibility to the development of mood disorders, including major depressive disorder (MDD) and generalized anxiety disorder. Importantly, dysfunction of the hypothalamic-pituitary-adrenal (HPA) axis, the endocrine stress response, has been linked to these diseases. Here, we review recent data on the region-specific role of glucocorticoid receptor (GR) signaling in the behavioral, molecular and endocrine response to stress. Using a conditional deletion approach, we have shown that disruption of GR function in the forebrain of mice induces alterations in despair-like behavior and HPA axis function, reminiscent of MDD. Furthermore, in an effort to explore the sub-regional specificity of GR activity, we have developed a model to disrupt GR in the central nucleus of the amygdala. In our initial efforts to characterize these mice, we have demonstrated a critical role for GR in the formation of fear memory.
Keywords: glucocorticoid receptor, conditional knockout, behavior, HPA axis, stress, lentivirus
1. Introduction
An adaptive response is initiated whenever an organism is faced with a situation that introduces a deviation from the physical or psychological basal state. When the deviation involves traumatic circumstances, the stress response system, including the sympathetic nervous system and the hypothalamic-pituitary-adrenal (HPA) axis, is activated. These systems provide the necessary energy, attention and general arousal needed to deal with the stressor. During stress, HPA axis activation through a variety of circuits induces neurons in the paraventricular nucleus of the hypothalamus (PVN) to release vasopressin and corticotropin-releasing hormone (CRH). These neuropeptides bind to receptors in the anterior pituitary gland to cause the secretion adrenocorticotrophic hormone (ACTH), which then causes the release of corticosteroids, cortisol in humans and corticosterone in mice and rats. Corticosteroid levels are modulated through feedback loops when corticosteroids bind to either type I, mineralocorticoid receptors, or type II, glucocorticoid receptors (GR), at the level of the PVN, anterior pituitary gland and other brain regions causing both negative (the classical and predominant influence) and positive modulation of HPA axis activity.
A variety of psychiatric disorders, including major depressive disorder (MDD) and generalized anxiety disorder are associated with stress. In some circumstances, the development of psychiatric illness is precipitated either by an acute trauma (Corcoran et al., 2003) or a stressful experience during development (Nemeroff, 2004). In addition, numerous studies have indicated that hyperactivity of the HPA axis is an important correlate of psychiatric illness (see (Claes, 2004; Stokes, 1995) for review).
Compared with healthy individuals, depressed patients often have enlarged adrenal glands (Nemeroff, 1992), elevated levels of plasma cortisol (Brown, 2004; Carpenter, 1971), increased CSF CRH (Arato et al., 1989), increased PVN CRH (Blanchard et al., 2001; Raadsheer et al., 1994) and impaired inhibition of the HPA axis as measured by the dexamethasone suppression test (DST) (Carroll et al., 1980; Holsboer et al., 1982). In normal adults, dexamethasone, a corticosteroid receptor agonist, will induce a dramatic reduction in plasma cortisol. However, depressed patients often show elevated levels of cortisol in the DST, thus implicating impaired negative feedback in depression.
Furthermore, individuals afflicted with Cushing’s disease are at a much greater risk for developing depression (Sonino and Fava, 2001), demonstrating that a global dysfunction in the HPA axis may be involved in the pathogenesis of the disorder. Finally, lower rates of remission are correlated with a reversal of the HPA axis disruption after antidepressant treatment (Pariante and Miller, 2001).
Overall, studies in humans and other animals have revealed a reproducible connection between HPA axis activity and symptoms reminiscent of depressive or anxious states (see (Claes, 2004) for review). These findings provide critical information for those interested in developing new and more effective pharmacological agents to combat psychiatric illness. Investigators have begun using pharmaceuticals that alter activity of GRs with some success in clinical trials (Murphy et al., 1993; Young et al., 2004). However, observations from animal studies have made it clear that the effectiveness of agonists or antagonists may depend on the area of the brain targeted. For instance, glucocorticoids binding in the PVN largely downregulate the HPA axis (Feldman and Weidenfeld, 2002) while activation of the amygdala during stress has been associated with an increase in HPA axis activity (Beaulieu et al., 1986; Shepard et al., 2003).
To investigate this regional action of GR in the brain for behavioral and endocrine function, our group has recently taken two conditional deletion approaches to disrupt GR expression throughout the forebrain or more specifically in the central nucleus of the amygdala (CeA) (Fig. 1). These approaches have an advantage over traditional pharmacological approaches in that they allow detection of GR disruption in a quantitative fashion and, along with other mutant models of GR (see (Kolber et al., 2008b) for review), provide additional evidence for regional differences in GR function. Here, we will describe these two models of GR deletion highlighting the new and important components of GR activity that have been revealed.
Fig. 1.

Expression of neuronal GR expression in wildtype, FBGRKO and CeAGRKO mice. GR is ubiquitously expressed throughout the brain, showing higher expression in a number of important limbic areas (e.g. CeA, PVN, hippocampus). Circles (●) represent neuronal glucocorticoid receptors (GR) in wildtype mice (top panel), FBGRKO mice (middle panel) and CeAGRKO mice (lower panel). Abundance of receptors is given by the relative density of circles in an area. Acc – nucleus accumbens; APit – anterior pituitary gland; BLA – basolateral nucleus of the amygdala; BnST – bed nucleus of the stria terminalis; CA1, CA2, CA3 – hippocampal areas CA1 to CA3; CeA – central nucleus of the amygdala; Cereb – cerebellum; Cing Ctx – cingulate cortex; DG – dentate gyrus; Fr Ctx – frontal cortex; InfC – inferior colliculus; LC – locus coeruleus; MeA – medial nucleus of the amygdala; Occ Ctx – occipital cortex; PAG – periaqueductal gray; Par Ctx – parietal cortex; PVN – paraventricular hypothalamic nucleus; Red – red nucleus; RN – raphe nuclei; Sep –septum; SupC – superior colliculus; SN – substantia nigra; Stri – striatum; Thal – thalamus.
Adapted from (Boyle et al., 2006; Kolber et al., 2008a; Kretz et al., 2001; Morimoto et al., 1996; Steckler and Holsboer, 1999).
2. Conditional GR Disruption Model Systems
Forebrain GR Knockout Mice (FBGRKO)
As an initial investigation into the region-specific role of GR in modulating HPA axis function and behavior, we used the CaMKII promoter to drive expression of Cre-recombinase in the forebrain of mice containing a loxP flanked GR allele (Boyle et al., 2005). Delayed activity of Cre-recombinase in these mice (FBGRKO) induces near complete neuronal disruption of GR throughout the adult hippocampus, cortex, striatum and basolateral nucleus of the amygdala (BLA) while sparing GR populations in the CeA, thalamus, PVN and cerebellum (Boyle et al., 2006) (Fig. 1). Maintenance of PVN GR allowed us to evaluate the role of extrahypothalamic GR in mediating both circadian and stress-activated HPA axis activity. Interestingly, we found increased glucocorticoids in FBGRKO mice at circadian nadir and peak, implying that forebrain GR might promote an overall negative drive to the PVN under basal conditions (Fig. 2A).
Fig. 2.

Endocrine and behavioral function in FBGRKO and CeAGRKO mice. (A) At 6 months of age, FBGRKO mice show a significant increase in basal corticosterone and in peak corticosterone relative to control mice of the same age. (B) FBGRKO mice treated with saline showed decreased activity in the tail suspension test (TST) compared with controls (n = 4–6). However, FBGRKO mice treated chronically with imipramine showed no significant difference compared with controls (n = 3–6). (C) FBGRKO mice show no changes in baseline, post-shock or contextual freezing compared to littermate controls (n=6). In auditory testing, FBGRKO mice show no changes compared to littermate control mice during baseline (pre-cue) or post-cue testing. (D) CeAGRKO show equivalent levels of plasma corticosterone at circadian nadir and circadian peak compared to GFP control mice (n=9). (E) CeAGRKO mice show a deficit in contextual freezing but no change in baseline or post-shock freezing compared to GFP control mice (n=9). In auditory testing, CeAGRKO mice show an attenuation of auditory cued freezing but no change in baseline (pre-cure) freezing compared to GFP control mice. (*p<0.05 versus control group)
Adapted from (Boyle et al., 2005; Kolber et al., 2008a).
Under stressful circumstances, deletion of forebrain GR induced an exaggerated increase in corticosterone (Boyle et al., 2006). Perhaps most interesting, in the DST, FBGRKO mice show no inhibition of corticosterone release compared to control mice (Boyle et al., 2005). Under normal circumstances, endogenous negative feedback by corticosterone is thought to occur in the pituitary gland and extrahypothalamic sites. However, due to the lower brain penetration of dexamethasone compared to corticosterone (De Kloet et al., 1975)), it has been assumed that GRs in the pituitary gland and perhaps the PVN were responsible for HPA axis negative feedback after a dexamethasone challenge. In contrast, our results suggest that GR populations in the hippocampus (or elsewhere in the forebrain) may also contribute to this negative feedback loop. It should be noted that this result may involve changes that occur with long-term loss of GR in the forebrain and may not be entirely representative of the basal HPA axis negative feedback system. For example, persistent changes in circadian corticosterone in FBGRKO mice may lead to altered feedback mechanisms in the anterior pituitary gland. Although we found no changes in GR expression at this site (Boyle et al., 2005), it is nonetheless possible that there are alterations in the pituitary gland that might cause the loss of dexamethasone suppression in the knockout mice.
Behaviorally, FBGRKO mice have been particularly useful in describing the unique role that forebrain GRs may play in depressive-like versus anxiety-like symptoms. FBGRKO mice show an increase in depression-like behavior in the forced swim test, tail suspension test (TST; Fig. 2B) and 2-bottle sucrose preference test (Boyle et al., 2005). Importantly, despair-like behavior was normalized after chronic but not acute treatment with the antidepressant imipramine (Fig. 2B).
In contrast to their straightforward depression-like phenotype, we showed that FBGRKO mice have a complex anxiety-like phenotype primarily characterized by altered stress reactivity (Boyle et al., 2006). These anxiety-associated symptoms were not normalized with antidepressant treatment showing a dissociation of GR action on depression-like and anxiety-like symptoms.
One major limitation of the FBGRKO system was the relatively widespread disruption of GR expression. The “forebrain” contains a number of anatomically and functionally unique structures. It is possible that some of the complexity seen in the FBGRKO phenotype may arise from the fact that we disrupted GR in areas that may have opposing effects on endocrine or behavioral output. To more specifically address the role of GR in stress adaptation, we recently characterized a lentivirus-based conditional deletion approach.
Central Nucleus of the Amygdala Knockout Mice (CeAGRKO)
One area in the forebrain to which a variety of emotional and endocrine related functions has been attributed is the amygdala. We were interested in understanding the role of GR in the amygdala in modulating both “innate” and “learned” anxiety as well as HPA axis function under a variety of situations. To circumvent the lack of any known amygdala specific promoter (Zirlinger et al., 2001), we proposed using technology developed in the gene therapy field to knockout GR in the amygdala. Specifically, in order to address the hypothesis that amygdalar GR is a primary effector in stress-related anxiety and memory changes, we packaged Cre-recombinase into a lentiviral vector and then stereotaxically injected the lentivirus into bilateral CeAs of our loxP-flanked GR mice (Boyle et al., 2005; Brewer et al., 2003; Kolber et al., 2008a) (Fig. 1). We reasoned that our viral-mediated deletion approach might have a number of advantages over GR antagonists that have been used previously to define the role of CeA GR in stress adaptation. First, our lentivirus approach provided long-term disruption of GR in contrast to the shorter-term disruption with GR antagonists. This allowed us to look at the effect of deleting GR on both basal changes and chronic changes in the same animals without having to do multiple injections. Second, we are able to quantitatively confirm that our CeAGRKO model specifically disrupted GR in the CeA while leaving nearby GR populations in the BLA intact.
Using this lentivirus-based system, we were able to reproducibly target the CeA (Figure 3) and disrupt GR expression in ~65% of the normally GR expressing neurons in the CeA while leaving the nearby BLA GR population intact (Kolber et al., 2008a). After validation of the system, our first analysis of CeAGRKO mice was in HPA axis function. We were interested in CeA driven HPA axis drive because of our observations from FBGRKO mice. To assess the role of CeA GR in modulating circadian HPA axis drive, we measured circadian nadir and peak corticosterone in CeARKO mice. We found equivalent corticosterone in CeAGRKO mice under nadir (Kolber et al., 2008a) and peak conditions (unpublished observations, Kolber BJ & LJ Muglia) (Fig. 2D).
Fig. 3.
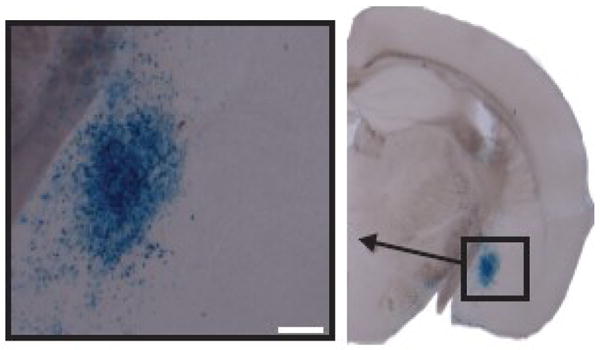
CeA is targeted with lentivirus-Cre in ROSA-26 LacZ reporter mice. LacZ expression (blue cells; evidence of Cre) seen in CeA of animal injected with lentivirus-Cre only. Scale bar = 200μm.
To evaluate the behavioral significance of CeA GR signaling, we tested mice in acute anxiety tests (e.g. open field) and Pavlovian fear conditioning. In open field (Kolber et al., 2008a), we found no differences in locomotor or anxiety-like responses comparing the CeAGRKO and control mice. However, when tested in both contextual and cued Pavlovian fear conditioning, CeAGRKO mice exhibited a marked deficit in freezing behavior under testing conditions with no alterations in training (Kolber et al., 2008a) (Fig. 2E). Furthermore, this behavioral deficit was shown to be associated with changes in extrahypothalamic CRH expression and was rescued with intracerebroventricular injection of CRH before fear conditioning training (Kolber et al., 2008a). The lack of acute anxiety changes coupled with a deficit in fear conditioning testing suggests that the CeA may play a distinct role in mediating learned anxiety, as seen in fear conditioning, versus innate anxiety, as seen in open field behavior. Targeting additional areas, including those thought to be involved in innate anxiety will reveal if this is truly a functional distinction of the CeA.
A comparison of our FBGRKO and CeAGRKO models presents some interesting findings related to the delineation of the sub-regional function of GR. First, the lack of observed changes in fear conditioning in FBGRKO mice suggest the specificity of the CeA GR population in mediating normal fear conditioning in our mice (Kolber et al., 2008a) (Fig. 2C). Although, in the FBGRKO system, as deletion occurs over a longer period of time than with the lentiviral approach, there exists greater potential for compensatory changes that must also be considered. For example, in FBGRKO mice, deletion within an area (e.g. the hippocampus) occurs gradually between 2–6 months after birth. This is in contrast to the deletion in the CeA of CeAGRKO mice, which likely occurs within 48–72 hours after injection of LV-Cre. The slower removal of GR in the FBGRKO mice may promote compensatory changes in that area’s connectivity and circuitry.
Second, given the absence of innate anxiety-related behavior in CeAGRKO mice, the alterations in anxiety seen in FBGRKO mice are unlikely to be caused by changes in CeA GR signaling in those mice and may instead be related to GR populations in the BLA or other areas. However, it should be noted that deletion of GR in the CeAGRKO mice occurs in ~65% of the CeA neurons (Kolber et al., 2008a) compared to the nearly 90% GR disruption that occurs in the BLA, hippocampus and cortex of FBGRKO mice (Boyle et al., 2006). The remaining 35% of GR positive neurons in the CeAGRKO mice may be sufficient to maintain normal adaptation to innate anxiety. In the future, it will be interesting to evaluate despair-like behavior in the CeAGRKO mice to determine what role CeA GRs may play in mediating depression-like behavior.
3. Conclusions
Overall, evidence from numerous observations in humans, pharmacological studies in rodents and mutant models in mice has revealed the important role of the HPA axis in modulating behavior associated with stress adaptation. Using conditional loss-of-function studies, we have begun, along with other groups (Berger et al., 2006; Ridder et al., 2005; Rozeboom et al., 2007; Wei et al., 2004), to disentangle the sub-regional impact of HPA axis-regulated molecules on behavior and endocrine function. Future studies using our lentiviral-based technique should be useful in identifying specific roles for GR in other areas including the BLA, bed nucleus of the stria terminalis, hippocampus and PVN. Ultimately, with a thorough understanding of GR function in the nervous system, it may be possible to design therapeutic agents that optimize treatment for psychiatric disorders.
Acknowledgments
This work was supported by grants from the NIH to BJK (F31MH075250) and LJM (AG18876).
Abbreviations
- ACTH
adrenocorticotrophic hormone
- BLA
basolateral nucleus of the amygdala
- CeA
central nucleus of the amygdala
- CRH
corticotropin-releasing hormone
- DST
dexamethasone suppression test
- GR
glucocorticoid receptor
- HPA
hypothalamic-pituitary-adrenal
- MDD
major depressive disorder
- PVN
paraventricular nucleus of the hypothalamus
- TST
tail suspension test
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arato M, Banki CM, Bissette G, Nemeroff CB. Elevated CSF CRF in suicide victims. Biol Psychiatry. 1989;25:355–9. doi: 10.1016/0006-3223(89)90183-2. [DOI] [PubMed] [Google Scholar]
- Beaulieu S, Di Paolo T, Barden N. Control of ACTH secretion by the central nucleus of the amygdala: implication of the serotoninergic system and its relevance to the glucocorticoid delayed negative feedback mechanism. Neuroendocrinology. 1986;44:247–54. doi: 10.1159/000124652. [DOI] [PubMed] [Google Scholar]
- Berger S, Wolfer DP, Selbach O, Alter H, Erdmann G, Reichardt HM, Chepkova AN, Welzl H, Haas HL, Lipp HP, Schutz G. Loss of the limbic mineralocorticoid receptor impairs behavioral plasticity. Proc Natl Acad Sci U S A. 2006;103:195–200. doi: 10.1073/pnas.0503878102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard RJ, McKittrick CR, Blanchard DC. Animal models of social stress: effects on behavior and brain neurochemical systems. Physiol Behav. 2001;73:261–71. doi: 10.1016/s0031-9384(01)00449-8. [DOI] [PubMed] [Google Scholar]
- Boyle MP, Brewer JA, Funatsu M, Wozniak DF, Tsien JZ, Izumi Y, Muglia LJ. Acquired deficit of forebrain glucocorticoid receptor produces depression-like changes in adrenal axis regulation and behavior. Proc Natl Acad Sci U S A. 2005;102:473–8. doi: 10.1073/pnas.0406458102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle MP, Kolber BJ, Vogt SK, Wozniak DF, Muglia LJ. Forebrain glucocorticoid receptors modulate anxiety-associated locomotor activation and adrenal responsiveness. J Neurosci. 2006;26:1971–8. doi: 10.1523/JNEUROSCI.2173-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer JA, Khor B, Vogt SK, Muglia LM, Fujiwara H, Haegele KE, Sleckman BP, Muglia LJ. T-cell glucocorticoid receptor is required to suppress COX-2-mediated lethal immune activation. Nat Med. 2003;9:1318–22. doi: 10.1038/nm895. [DOI] [PubMed] [Google Scholar]
- Brown ES, Varghese FP, McEwen BS. Association of depression with mental illness: does cortisol play a role. Biol Psychiatry. 2004;55:1–9. doi: 10.1016/s0006-3223(03)00473-6. [DOI] [PubMed] [Google Scholar]
- Carpenter WT, Bunney WE. Adrenal cortical activity in depressive illness. Am J Psychiatry. 1971;128:31–40. doi: 10.1176/ajp.128.1.31. [DOI] [PubMed] [Google Scholar]
- Carroll BJ, Greden JF, Feinberg M. Neuroendocrine disturbances and the diagnosis and aetiology of endogenous depression. Lancet. 1980;1:321–2. doi: 10.1016/s0140-6736(80)90826-0. [DOI] [PubMed] [Google Scholar]
- Claes SJ. CRH, stress, and major depression: a psychobiological interplay. Vitam Horm. 2004;69:117–50. doi: 10.1016/S0083-6729(04)69005-4. [DOI] [PubMed] [Google Scholar]
- Corcoran C, Walker E, Huot R, Mittal V, Tessner K, Kestler L, Malaspina D. The stress cascade and schizophrenia: etiology and onset. Schizophr Bull. 2003;29:671–92. doi: 10.1093/oxfordjournals.schbul.a007038. [DOI] [PubMed] [Google Scholar]
- De Kloet R, Wallach G, McEwen BS. Differences in corticosterone and dexamethasone binding to rat brain and pituitary. Endocrinology. 1975;96:598–609. doi: 10.1210/endo-96-3-598. [DOI] [PubMed] [Google Scholar]
- Feldman S, Weidenfeld J. Further evidence for the central effect of dexamethasone at the hypothalamic level in the negative feedback mechanism. Brain Res. 2002;958:291–6. doi: 10.1016/s0006-8993(02)03581-3. [DOI] [PubMed] [Google Scholar]
- Holsboer F, Liebl R, Hofschuster E. Repeated dexamethasone suppression test during depressive illness. Normalisation of test result compared with clinical improvement. J Affect Disord. 1982;4:93–101. doi: 10.1016/0165-0327(82)90039-8. [DOI] [PubMed] [Google Scholar]
- Kolber BJ, Roberts MS, Howell MP, Wozniak DF, Sands MS, Muglia LJ. Central amygdala glucocorticoid receptor action promotes fear-associated CRH activation and conditioning. Proc Natl Acad Sci U S A. 2008a;105:12004–9. doi: 10.1073/pnas.0803216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolber BJ, Wieczorek L, Muglia LJ. Hypothalamic-pituitary-adrenal axis dysregulation and behavioral analysis of mouse mutants with altered glucocorticoid or mineralocorticoid receptor function. Stress. 2008b;1 doi: 10.1080/10253890701821081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretz O, Schmid W, Berger S, Gass P. The mineralocorticoid receptor expression in the mouse CNS is conserved during development. Neuroreport. 2001;12:1133–7. doi: 10.1097/00001756-200105080-00017. [DOI] [PubMed] [Google Scholar]
- Morimoto M, Morita N, Ozawa H, Yokoyama K, Kawata M. Distribution of glucocorticoid receptor immunoreactivity and mRNA in the rat brain: an immunohistochemical and in situ hybridization study. Neurosci Res. 1996;26:235–69. doi: 10.1016/s0168-0102(96)01105-4. [DOI] [PubMed] [Google Scholar]
- Murphy BE, Filipini D, Ghadirian AM. Possible use of glucocorticoid receptor antagonists in the treatment of major depression: preliminary results using RU 486. J Psychiatry Neurosci. 1993;18:209–13. [PMC free article] [PubMed] [Google Scholar]
- Nemeroff CB. Neurobiological consequences of childhood trauma. J Clin Psychiatry. 2004;65(Suppl 1):18–28. [PubMed] [Google Scholar]
- Nemeroff CB, Krishnan KR, Ree D, Leder R, Beam C, Dunnick NR. Adrenal gland enlargement in major depression: A computed tomographic study. Arch Gen Psychiatry. 1992;49:384–387. doi: 10.1001/archpsyc.1992.01820050048008. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry. 2001;49:391–404. doi: 10.1016/s0006-3223(00)01088-x. [DOI] [PubMed] [Google Scholar]
- Raadsheer FC, Hoogendijk WJ, Stam FC, Tilders FJ, Swaab DF. Increased numbers of corticotropin-releasing hormone expressing neurons in the hypothalamic paraventricular nucleus of depressed patients. Neuroendocrinology. 1994;60:436–44. doi: 10.1159/000126778. [DOI] [PubMed] [Google Scholar]
- Ridder S, Chourbaji S, Hellweg R, Urani A, Zacher C, Schmid W, Zink M, Hortnagl H, Flor H, Henn FA, Schutz G, Gass P. Mice with genetically altered glucocorticoid receptor expression show altered sensitivity for stress-induced depressive reactions. J Neurosci. 2005;25:6243–50. doi: 10.1523/JNEUROSCI.0736-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozeboom AM, Akil H, Seasholtz AF. Mineralocorticoid receptor overexpression in forebrain decreases anxiety-like behavior and alters the stress response in mice. Proc Natl Acad Sci U S A. 2007;104:4688–93. doi: 10.1073/pnas.0606067104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard JD, Barron KW, Myers DA. Stereotaxic localization of corticosterone to the amygdala enhances hypothalamo-pituitary-adrenal responses to behavioral stress. Brain Res. 2003;963:203–13. doi: 10.1016/s0006-8993(02)03978-1. [DOI] [PubMed] [Google Scholar]
- Sonino N, Fava GA. Psychiatric disorders associated with Cushing’s syndrome. Epidemiology, pathophysiology and treatment. CNS Drugs. 2001;15:361–73. doi: 10.2165/00023210-200115050-00003. [DOI] [PubMed] [Google Scholar]
- Steckler T, Holsboer F. Corticotropin-releasing hormone receptor subtypes and emotion. Biol Psychiatry. 1999;46:1480–508. doi: 10.1016/s0006-3223(99)00170-5. [DOI] [PubMed] [Google Scholar]
- Stokes PE. The potential role of excessive cortisol induced by HPA hyperfunction in the pathogenesis of depression. Eur Neuropsychopharmacol. 1995;5(Suppl):77–82. doi: 10.1016/0924-977x(95)00039-r. [DOI] [PubMed] [Google Scholar]
- Wei Q, Lu XY, Liu L, Schafer G, Shieh KR, Burke S, Robinson TE, Watson SJ, Seasholtz AF, Akil H. Glucocorticoid receptor overexpression in forebrain: a mouse model of increased emotional lability. Proc Natl Acad Sci U S A. 2004;101:11851–6. doi: 10.1073/pnas.0402208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AH, Gallagher P, Watson S, Del-Estal D, Owen BM, Ferrier IN. Improvements in neurocognitive function and mood following adjunctive treatment with mifepristone (RU-486) in bipolar disorder. Neuropsychopharmacology. 2004;29:1538–45. doi: 10.1038/sj.npp.1300471. [DOI] [PubMed] [Google Scholar]
- Zirlinger M, Kreiman G, Anderson DJ. Amygdala-enriched genes identified by microarray technology are restricted to specific amygdaloid subnuclei. Proc Natl Acad Sci U S A. 2001;98:5270–5. doi: 10.1073/pnas.091094698. [DOI] [PMC free article] [PubMed] [Google Scholar]