

Abstract
Descending noradrenergic inhibition is an important endogenous pain-relief mechanism which can be activated by local glutamate signaling. In the present study, we examined the effect of glutamate transporter activation by riluzole in the regulation of activity of locus coeruleus (LC) neurons, which provide the major inhibitory descending noradrenergic projection to the spinal cord. Local injection of riluzole into the LC dose-dependently reduced hypersensitivity in rats after L5-L6 spinal nerve ligation (SNL). This anti-hypersensitivity effect of LC-injected riluzole was blocked by intrathecal administration of the α2-adrenoceptor antagonist idazoxan and intra-LC co-injection of the AMPA antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and the gap-junction blockers, carbenoxolone (CBX) and meclofenamic acid (MEC). In brainstem slices from normal rats, riluzole increased phosphorylated cAMP response element binding protein (pCREB) expressing nuclei in dopamine-β-hydroxylase (DβH) containing cells in the LC. This riluzole-induced pCREB activation in LC neurons was also blocked by CNQX and CBX. In the primary astrocyte culture, riluzole enhanced glutamate-induced glutamate release. Contrary to expectations, these results suggest that activation of glutamate transporters in the LC results in increase of extracellular glutamate signaling, possibly via facilitation of glutamate release from astrocytes, and activation of LC neurons to induce descending inhibition, and that this paradoxical action of glutamate transporters in the LC requires gap-junction connections.
Keywords: Riluzole, Neuropathic pain, Locus coeruleus, Descending inhibition, Glutamate transporter
1. Introduction
As an important endogenous analgesic neurotransmitter, noradrenaline is released in the spinal cord from bulbospinal noradrenergic axons which originate from the LC and adjacent nuclei in the brainstem, and suppresses the neurotransmission of pain in the spinal cord via activation of α2 adrenoceptors (Aston-Jones et al., 1991). Although various neurochemical inputs modulate neuronal activity of noradrenergic neurons in the LC, glutamate is considered as a primary excitatory regulator (Singewald and Philippu, 1998). A major glutamate input to the LC arises from the nucleus paragigantocellularis and activates noradrenergic neurons via AMPA receptors (Singewald and Philippu, 1998). We have recently demonstrated that one of the commonly used treatments for chronic pain, gabapentin, activates descending inhibition via glutamatergic signaling in the LC and induces spinal noradrenaline release in rats and human(Hayashida et al., 2007; Hayashida et al., 2008). Glutamate is, therefore, an important substance not only for physiological but also for pharmacological regulation of the LC activity. However, local regulation of glutamate signaling in the LC is poorly understood. In the central nervous system, glial cells, primarily astrocytes, regulate extracellular glutamate concentrations. Astrocytes express two types of glutamate transporters, GLT-1 and GLAST (Anderson and Swanson, 2000). Under physiological conditions, glutamate transporters take up glutamate from the extracellular space and terminate excitation from synaptically released glutamate. However, in pathological conditions such as ischemia, astrocytes release large amounts of glutamate via reverse transport into the extracellular space, which participates in neurotoxicity (Malarkey and Parpura, 2008).
Riluzole, a neuroprotective drug approved for amyotrophic lateral sclerosis (Brooks, 2009), has been recognized to possess analgesic properties in a wide range of animal pain models when systemically or intrathecally administered (Coderre et al., 2007; Irifune et al., 2007; Munro et al., 2007; Sung et al., 2003). Recent studies demonstrated that riluzole activates GLT-1 and GLAST to enhance glutamate uptake (Frizzo et al., 2004; Fumagalli et al., 2008), and reduces extracellular glutamate level in the spinal cord (Coderre et al., 2007). Based on these studies, one would hypothesize that local injection of riluzole into the LC would reduce extracellular glutamate concentrations, thereby reducing descending inhibition and potentially exacerbating hypersensitivity after peripheral nerve injury. Surprisingly, we demonstrate here that LC-injected riluzole reduced rather than exacerbated hypersensitivity in rats after L5-L6 SNL. We therefore tested whether LC-injected riluzole activates descending noradrenergic inhibition in SNL rats, and whether riluzole activates noradrenergic neurons in the LC in brainstem slices, with a focus on glutamatergic signaling.
Glutamate induces Ca2+ -dependent glutamate release from astrocytes via activation of Ca2+ permeable ionotropic AMPA receptors and group 1 metabotropic glutamate receptors (mGluRs), which release Ca2+ from internal stores through 1,4,5-inositol-trisphosphate (InsP3) signaling (Hansson et al., 2000; Verkhratsky and Kirchhoff, 2007). In some astrocytes, co-transport of sodium ions and glutamate by glutamate transporters results in Ca2+ influx via the reverse mode of Na/Ca exchange to thereby increase intracellular Ca2+ concentration (Rojas et al., 2007), which in turn lead to glutamate release from astrocytes via Ca2+ -dependent exocytosis (Malarkey and Parpura, 2008). We therefore hypothesized that glutamate-induced glutamate release in astrocytes may be enhanced by activation of glutamate transporters. To examine this hypothesis, we test effect of riluzole on glutamate-induced glutamate release from primary astrocyte culture.
Normal activity of cells in the LC has been hypothesized to reflect in part inter-cellular communication via gap junctions. Gap junctions are formed by connexons (hemichannels), which are composed of connexin, which create a pore that allows molecules such as inositol 1,4,5-trisphosphate to diffuse between cells without entering the extracellular space (Kielian, 2008). Electrical coupling of noradrenergic neurons in the LC demonstrate synchronized spontaneous oscillations in membrane potential that underlie spontaneous activity, which are disrupted by pharmacological blockade of gap junctions (Ballantyne et al., 2004). Interestingly, such oscillations also occur in adjacent astrocytes, and this oscillatory activity in both astrocytes and noradrenergic neurons requires communication via gap junctions (Alvarez-Maubecin et al., 2000). We therefore hypothesized that gap junction couplings in the LC serve as an important setting for paradoxical activation of descending inhibition by riluzole. We therefore tested effects of gap-junction blockers on riluzole-induced anti-hypersensitivity and neuronal activation in the LC to examine whether coupling or communication through gap junctions were involved in riluzole's actions in the LC.
2. Results
2.1. Behavioral effect of intra-LC injection of riluzole in hypersensitivity after nerve injury
SNL strongly decreased the withdrawal threshold to pressure on the hindpaw ipsilateral to SNL from 138 ± 9 g to 72 ± 12 g (mean ± SD, P<0.0001, n=45). Intra-LC injection of riluzole dose-dependently increased withdrawal threshold in the paw ipsilateral to SNL, with significant anti-hypersensitivity efficacy from 1 to 5 μg compared to vehicle (Fig 1). The peak effect of riluzole was observed 15 min after injection. In these blinded experiments, neither vehicle nor riluzole were associated with signs of sedation.
Fig 1.
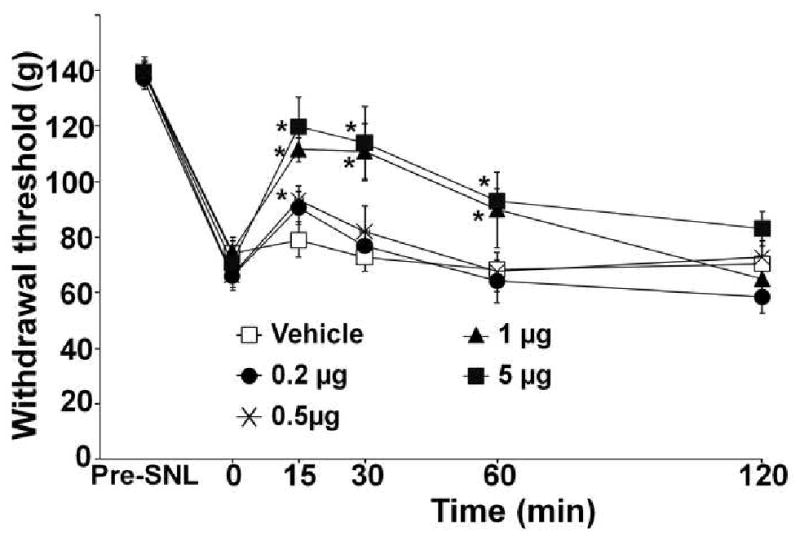
Intra-LC injected riluzole reduces mechanical hypersensitivity after SNL surgery. SNL surgery reduced withdrawal threshold to pressure applied to the paw ipsilateral to injury. After development of hypersensitivity, rats received intra-LC injection of vehicle (n=7) or riluzole (0.2-5 μg, n=7-8). *p<0.05 vs. time 0 by one-way ANOVA. Riluzole 1 μg and 5 μg groups significantly differ from the vehicle group by two-way repeated measures ANOVA (p<0.05).
Intrathecal administration of the α2-adrenoceptor antagonist idazoxan (30 μg), which itself did not affect withdrawal threshold, completely blocked the anti-hypersensitivity effect of intra-LC riluzole (1 μg, Fig. 2). Intra-LC injection of the AMPA antagonist, CNQX (0.3 μg), also blocked riluzole's effect (Fig. 2). The dose of CNQX was determined from our previous study showing that intra-LC injection of 0.3 μg CNQX itself did not affect withdrawal threshold in SNL rats (Hayashida et al., 2008).
Fig 2.
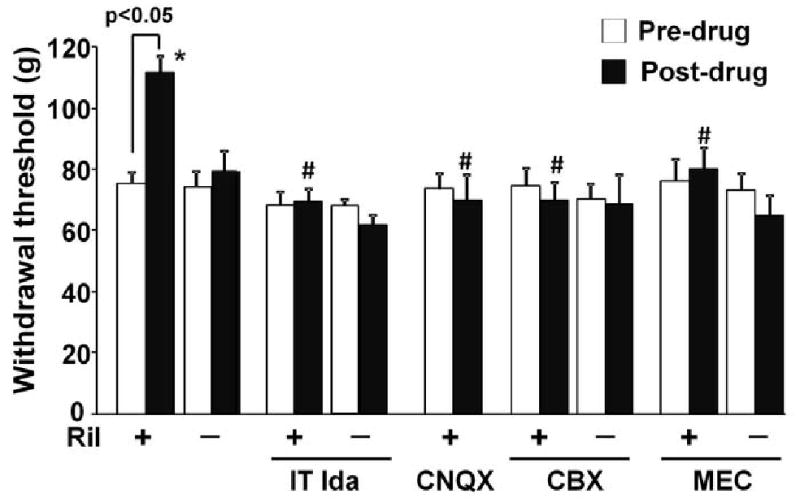
Effects of spinally and intra-LC injected antagonists on the anti-hypersensitive effect of LC-injected riluzole. Vehicle, riluzole (Ril, 1 μg), CNQX (0.3 μg), CBX (1 μg), MEC (1 μg), or their mixture were injected in the LC 15 min prior to the post-drug measurement in SNL rats (n=6-9). Intrathecal idazoxan (IT Ida, 30 μg) was administered 15 min prior to intra-LC injection of vehicle (n=7) or Ril (n=7), and the measurement was obtained 15 min later. * p<0.05 vs. vehicle. # p<0.05 vs riluzole alone.
To test whether functional coupling through gap-junction is required for the anti-hypersensitivity effect of riluzole in the LC, we utilized two structurally different gap-junction blockers, CBX (1 μg) and MEC (1 μg). CBX and MEC, neither of which affected withdrawal threshold alone, inhibited the anti-hypersensitivity effect of intra-LC riluzole (Fig. 2).
2.2. Effect of riluzole on pCREB expression in noradrenergic neurons in the LC
Brainstem slice experiments were performed to examine whether riluzole acts in the local circuits in the brainstem to activate LC neurons. We utilized DβH as a marker for noradrenergic neurons and pCREB as a marker for neuronal activation as previously reported (Hayashida et al.) Figure 3 depicts DβH and pCREB expression in the LC in the brainstem slices from normal animals treated with vehicle or riluzole (1 μM). The riluzole-treated brainstem slice showed clear pCREB-immunoreactive nuclei in DβH-immunoreactive cells (Fig 3 D, E, and F), while very few cells expressed pCREB in the vehicle-treated (Fig 3 A, B, and C). As shown in Figure 4, riluzole increased in a concentration-related manner the percentage of DβH-immunoreactive cells which expressed pCREB in the LC compared to the vehicle. Riluzole showed its maximum efficacy at 1 μM, however, its efficacy decreased at 10 μM, possibly due to inhibition of voltage-dependent sodium channels and AMPA-mediated current at this higher concentration (Albo et al., 2004; Lamanauskas and Nistri, 2008).
Fig 3.
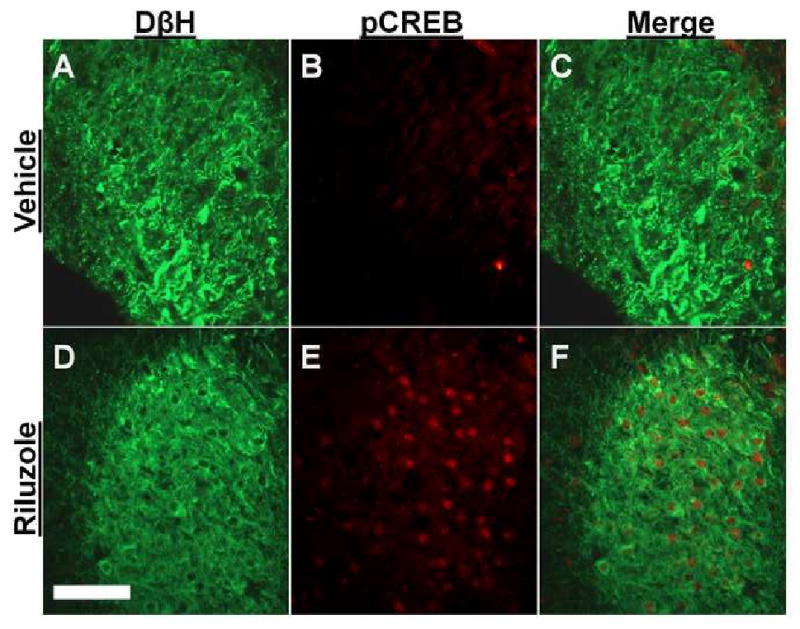
Photomicrographs of DβH- and pCREB-immunoreactive neurons in brainstem slices from normal rats. Brainstem slices treated with vehicle (A-C) or riluzole (1 μM, D-F) for 30 min were stained with antibodies for DβH (Green) and pCREB (Red). Scale bar = 100 μm.
Fig 4.
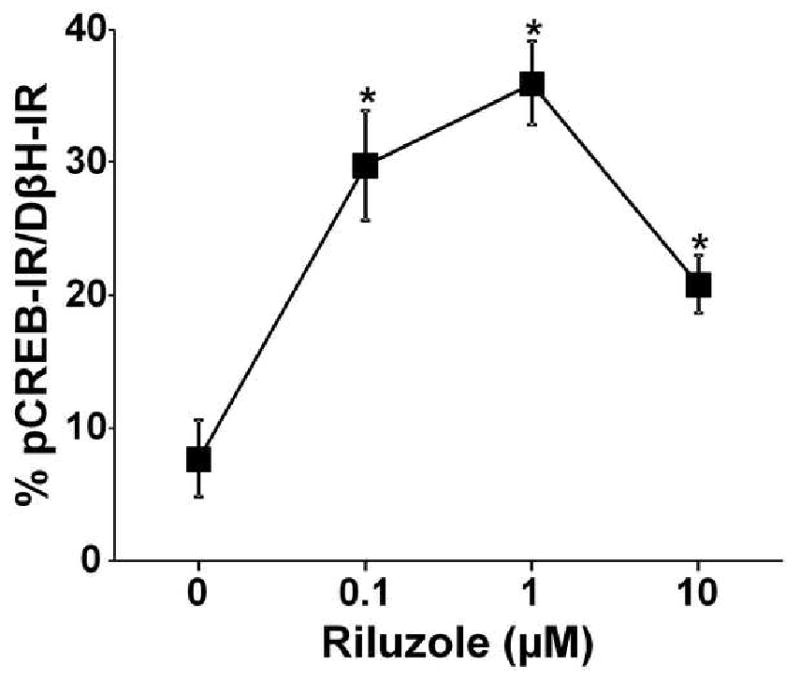
Quantification of riluzole concentration response on the number of pCREB expressing cells in brainstem slices from normal rats. Data are presented as percentage of pCREB-immunoreactivity in DβH-immunoreactive neurons. Brainstem slices were treated with vehicle (n=4) or riluzole (0.1 - 10 μM, n=4-5) for 30 min. *p<0.05 vs. vehicle.
Figure 5 A, B, and C show representative photomicrographs of the LCs treated with riluzole (1 μM) in the presence or absence of CNQX (50 μM) and CBX (50 μM). Consistent with behavioral observations in SNL rats, CNQX and CBX, neither of which affected basal pCREB expression in DβH-immunoreactive cells in the LC of normal rats, abolished riluzole-induced pCREB activation (Fig 5 D).
Fig 5.
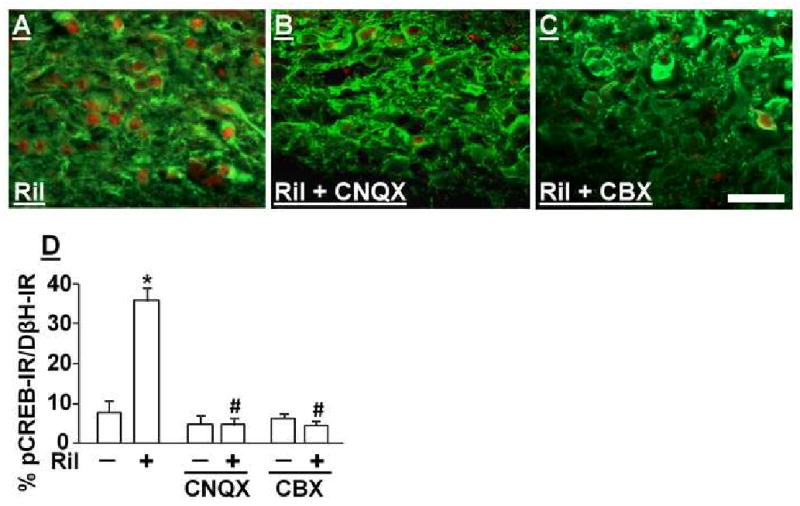
Effects of AMPA and Gap-junction antagonists on riluzole-induced pCREB activation in brainstem slices from normal rats. (A, B, and C) Photomicrographs of DβH (Green)- and pCREB (Red)-immunoreactive neurons in brainstem slices treated with riluzole (Ril, 1 μM) alone (A) or riluzole plus CNQX (50 μM, B) and CBX (50 μM, C) for 30 min. Scale bar = 50 μm. (D) Quantification of effects of CNQX and CBX on riluzole-induced pCREB activation in brainstem slices. Data are presented as percentage of pCREB-immunoreactivity in DβH-immunoreactive neurons. Brainstem slices were treated with vehicle or riluzole (Ril, 1 μM) in the presence or absence of CNQX and CBX for 30 min. *p<0.05 vs. vehicle. # p<0.05 vs. riluzole alone.
2.3. Effect of riluzole on glutamate-induced glutamate release from astrocytes
In vehicle-treated astrocytes, glutamate (1-100 μM) increased [3H]-glutamate release in a concentration-dependent manner (Fig. 6). Riluzole (1 μM) significantly enhanced glutamate-induced [3H]-glutamate release from astrocytes compared to the vehicle-treated (p<0.01 by two-way ANOVA). Maximum difference in [3H]-glutamate release between vehicle and riluzole groups was observed at 1 μM glutamate.
Fig. 6.
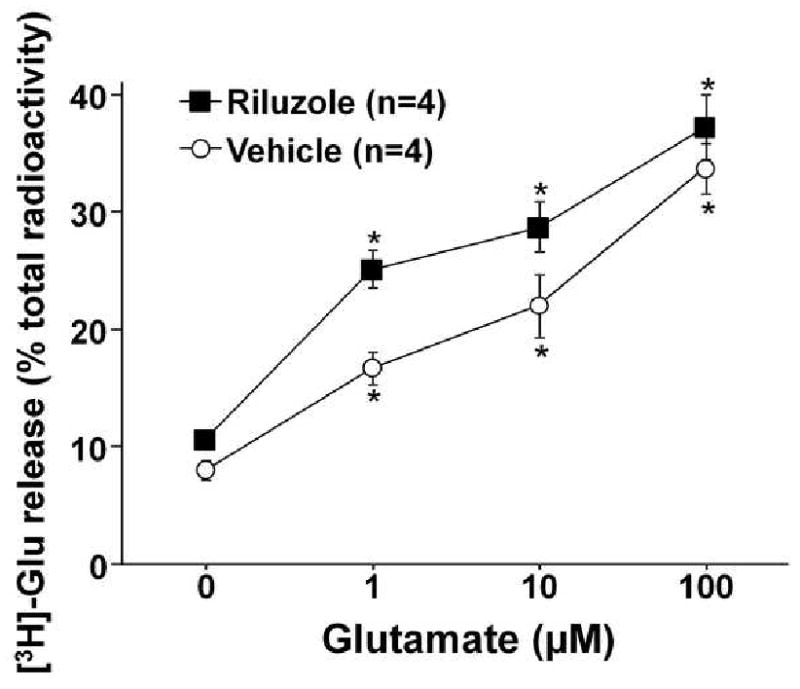
Effect of riluzole in glutamate-induced glutamate release from primary astrocyte culture. After loading with 1 μM glutamate (combination of both tritiated and unlabeled glutamate) for 1 hr, astrocytes were treated with glutamate (1-100 μM) with or without riluzole (1 μM) for 10 min. [3H]-glutamate (Glu) release is presented as a percentage of total radioactivity. [3H]-glutamate release in riluzole-treated group significantly differ from the vehicle group by two-way ANOVA (p<0.01). *p<0.05 vs. 0 μM.
3. Discussion
Glutamate is the most ubiquitous excitatory neurotransmitter in the central nervous system and its regulation by glutamate transporters has been intensely investigated in the spinal cord and brain. In the spinal cord, blockade of spinal glutamate transporters induces hypersensitivity to mechanical and thermal stimuli in normal rats (Weng et al., 2006), whereas activation or gene-transfer of GLT-1 in the spinal cord reduces hypersensitivity in rats after peripheral nerve injury and inflammation (Coderre et al., 2007; Maeda et al., 2008). These results suggest that glutamate transporters in the spinal cord play critical roles in both physiological and pathological pain states by reducing extracellular glutamate concentrations and reducing pain neurotransmission.
Activation of the LC results in noradrenaline release in the spinal cord, which produces antinociception and anti-hypersensitivity activation of α2 adrenoceptors. Although descending inhibitory mechanisms have been investigated for several decades (Fields and Basbaum, 1978), the regulation of descending modulation of pain transmission is poorly understood. Glutamate is a primary excitatory neurotransmitter in the LC (Singewald and Philippu, 1998), and one would anticipate that activation of glutamate transporters in the LC would reduce glutamatergic signaling, LC activity, and descending inhibition. In contrast, we observed in the present study that intra-LC injection of glutamate transporter activator riluzole in nerve-injured rats reduced hypersensitivity and that this likely reflected paradoxically increased neuronal activity, since the anti-hypersensitivity effect which was reversed by a spinally administered α2 adrenoceptor antagonist. The effect of riluzole clearly involves glutamate availability and signaling, since both the anti-hypersensitivity and LC neuronal activation, as evidenced by pCREB expression, from riluzole were blocked by an AMPA receptor antagonist.
Riluzole is the only clinically approved drug for amyotrophic lateral sclerosis with neuroprotective effects as a glutamate modulator (Brooks, 2009). In addition to activation of glutamate transporters (Frizzo et al., 2004; Fumagalli et al., 2008), high concentrations of riluzole (>10 μM) exert actions on other targets, including inhibition of voltage-dependent sodium channels (Lamanauskas and Nistri, 2008; Urbani and Belluzzi, 2000; Zona et al., 1998), high-voltage activated calcium and potassium channels (Huang et al., 1997; Zona et al., 1998), and glutamate receptors (Albo et al., 2004; De Sarro et al., 2000). All of those effects of riluzole would induce neuronal inhibition rather than activation. However, we did not observe inhibition by riluzole (0.1-1 μM) in the LC in the current study, but paradoxically observed activation of noradrenergic neurons in this nucleus via actions on AMPA receptors. Since riluzole inhibits post-synaptic currents mediated by AMPA receptors in spinal motor neurons (Albo et al., 2004), it is unlikely that riluzole directly activates AMPA receptors or amplifies their signaling in noradrenergic neurons. We therefore consider that riluzole increases extracellular glutamate concentrations in the LC to activate descending inhibition. Although cultured astrocytes differ from the in vivo conditions, the present study supports this idea that activation of glutamate transporters by riluzole enhances glutamate-induced glutamate release from astrocytes.
GLT-1 and GLAST expressed in astrocytes take up glutamate from the extracellular space under physiological conditions, but during pathological states such as ischemia, perturbed ionic conditions (e.g. increased extracellular K+ levels) can increase extracellular glutamate concentrations through reverse transport (Malarkey and Parpura, 2008). However, since riluzole activated LC neurons in brainstem slices from normal animals in the present study, it is unlikely that riluzole induces glutamate release via reverse transport. In astrocytes, AMPA receptors and group 1 mGluRs are functionally important for increased intracellular Ca2+ concentration following exposure to glutamate and the coincident activation of those receptors produces a positive enhancement for Ca2+ -dependent glutamate release (Hansson et al., 2000; Verkhratsky and Kirchhoff, 2007). Previous study in cerebellar astrocytes demonstrated that co-transport of sodium ions and glutamate by glutamate transporters results in the reverse mode of Na/Ca exchange to increase intracellular Ca2+ concentration (Rojas et al., 2007), suggesting activation of glutamate transporters could enhance Ca2+ -dependent glutamate release by glutamate. We therefore consider that riluzole may utilize this mechanism to enhance glutamate-induced glutamate release by activation of glutamate transporters.
In the LC, electrical coupling of neurons promotes synchronized spontaneous activity, which is blocked by CBX (Ballantyne et al., 2004). This indicates that gap-junction between LC neurons may be required for the maintenance of endogenous rhythmic activity in the LC. Several lines of evidence also support an astrocyte-neuronal gap junction communication in the LC. As such, pharmacologic blockade of gap junctions disrupted oscillations in both glial and neuronal membrane potential in this structure, selective inhibition of LC neuronal activity by a μ-opioid agonist coincidentally reduced astrocyte membrane potential oscillations, and selective depolarization of astrocytes with a glutamate transporter substrate (L-α-aminoadipic acid) increased the neuronal firing rate in the LC(Alvarez-Maubecin et al., 2000). These observations indicate that activation of LC neurons and astrocytes by glutamate can reciprocally increase their activities via gap-junction connections and may result in glutamate release from astrocytes. Since most gap-junction blockers have different non-selective effects (Pan et al., 2007), we used two structurally different gap-junction blockers, CBX and MEC, in the present study. Both gap junction blockers reversed anti-hypersensitivity effect of intra-LC riluzole and CBX inhibited riluzole-induced neuronal activation in the LC, suggesting riluzole's efficacy relies on neuro-neuro and/or neuro-glia gap junction connections. Of course, the current study does not identify the location of the gap junctions in the LC relevant to riluzole's action. Imaging or patch-clamp study in astrocytes and LC neurons in a brainstem slice preparation or in vivo would more directly address effects of riluzole on neuronal activity and glutamate release from astrocytes. Future studies using such methods will be required.
In summary, the present study has demonstrated that riluzole activates LC neurons via action on AMPA receptors and induces descending inhibition to reduce hypersensitivity in nerve-injured rats, and that this paradoxical action of riluzole may be due to facilitation of glutamate-induced glutamate release from astrocytes and also requires neuro-neuro and/or neuroglia gap-junction connections in the LC. These results suggest that riluzole is a useful probe to understand the local regulation of glutamate in the LC to activate descending noradrenergic inhibition and should be considered for clinical investigation in patients with chronic pain.
4. Experimental procedure
4.1. Animals
Male Sprague-Dawley rats (weighing 180-250 g) from Harlan Industries (Indianapolis, IN, USA) were used. All experiments were approved by Animal Care and Use Committee at Wake Forest University (Winston Salem, NC). Animals were housed under a 12-h light-dark cycle, with food and water ad libitum.
4.2. Surgical preparations
SNL was performed as previously described (Kim and Chung, 1992). Animals were anesthetized with 2% isoflurane, the right L5 and L6 spinal nerves were tightly ligated using 5-0 silk suture. Two weeks after SNL, intra-LC and intrathecal catheterization were performed as previously described (Hayashida et al., 2008; Yaksh and Rudy, 1976). Under anesthesia with intraperitoneal sodium pentobarbital (50 mg/kg), animals were placed securely in a stereotaxic frame (KOPF, Tujunga, CA). A sterile stainless steel guide cannula (26-gauge needle shaft; Plastics One, Roanoke, VA) was implanted into the right LC. The coordinates for placement of the tip of the guide cannula were 9.8 mm posterior and 1.4 mm lateral to the bregma, and 7.0 mm ventral from the surface of the dura mater, according to the rat brain atlas (Paxinos and Watson, 2005). For intrathecal catheterization, a polyethylene catheter (ReCathCO LLC, Allison Park, PA), 7.5 cm, was inserted to reach the lumbar enlargement of the spinal cord. Animals were allowed at least 5 days to recover from the surgery. After the experiment, rats received a methylene blue injection (0.5 μL) into the LC and killed by intraperitoneal pentobarbital (150 mg/kg) injection. The brainstem was removed, post-fixed with 8% buffered paraformaldehyde, sectioned, and the placement of the cannula was verified microscopically. In total 47 rats, 2 rats were excluded from the study due to weight loss after surgery and LC cannula misplacement.
4.3. Behavioral test
The person performing the behavioral test was blinded to drug and dose. Withdrawal threshold to pressure applied to the hind paw was measured using an analgesimeter (Ugo Basile, Comerio, Italy) as previously described (Randall and Selitto, 1957). The device applies increasing pressure to the hind paw. When the animal withdrew the paw or vocalized, the pressure was immediately released and the withdrawal threshold, expressed in grams, was obtained. Training of animals for this test was performed for 2-3 days before the drug treatment. A cut-off of 250 g was used to avoid potential tissue injury. We used these animals two or three times on different days and experiments in the same animals were separated by at least 4 days. Drugs and the doses were randomly assigned. Riluzole (Sigma Chemical CO., St. Louis, MO), carbenoxolone disodium (CBX, Sigma), and meclofenamic acid sodium (MEC, Sigma) were dissolved in saline and 6-cyano-7-nitroquinoxaline-2,3-dione disodium salt (CNQX-2Na, Sigma) was dissolved in dimethyl sulfoxide, and diluted with artificial cerebrospinal fluid (in mM: NaCl 126.0, KCl 2.5, MgSO4 1.3, CaCl2 2.5, Glucose 12, NaH2PO4 1.2, and NaHCO3 25). A volume of 0.5 μl solution was injected into the LC at the speed of 0.5 μl per minute using a syringe pump (Model 200, KD Scientific, Holliston, MA) 15 min prior to the behavioral measurement. For intrathecal administration, idazoxan hydrochloride (Sigma Chemical CO.) was dissolved in saline and injected in a volume of 10 μl followed by 10 μl of saline 30 min prior to the measurement.
4.4. Brainstem slice study
Brainstem slice experiments were performed as previously reported (Hayashida et al., 2008). Under deep anesthesia with 5 % isoflurane, animals were killed by decapitation to remove brainstem. Transverse brainstem slices containing the LC (600 μm thickness) were obtained from normal rats using a vibratome (VT1000S, Leica Mircosystems, Nussloch, Germany) and perfused in the chamber with artificial cerebrospinal fluid (2 mL/min, 37 ± 0.5 °C, 95% O2 / 5% CO2 saturated). After 3 hrs of baseline perfusion, brainstem slices were transferred to the drug challenge chamber and incubated with drugs for 30 min and then fixed with 4% paraformaldehyde for 1 hr. The fixed tissues were then cryoprotected with 30% sucrose for 48 hrs and sectioned (16 μm). Because of potential damage of tissue, the first and last 7 sections from the tissue surface were discarded and the rest were used for the immunocytochemistry. For DβH and pCREB staining, 4 brainstem sections were randomly selected from each animal. Sections were pre-treated with 1.5% normal donkey serum (NDS, Vector, Burlingame, CA) and then incubated for 24 hours at 4°C with a mouse monoclonal anti-DβH antibody (1:1,000, MAB308, Chemicon International Inc., Temecula, CA, USA) and a rabbit anti-pCREB antibody (1:1,000, #06-519, UPSTATE, Lake Placid, NY) diluted in phosphate buffered saline containing 0.3% Triton-X 100 and 1.5% NDS. Subsequently, the sections were incubated for 1 hr with a donkey anti-mouse fluorscein (1:100, Chemicon International Inc.) and then incubated with a donkey anti-rabbit rhodamine (1:100, Chemicon International Inc.). Finally DβH and pCREB immunostaining were visualized under an inverted microscope (200 × magnifications) with standard FITC and TRITC filters. Images of both sides of LCs were captured using a digital CCD camera with a consistent setting. Cells with DβH and pCREB immunostaining were counted in the entire LCs in each section. In each animal, 83-204 DβH immunoreactive LC cells with visible nuclei were counted. The person performing immunohistochemistry and counting cells was blinded to drug and treatment.
4.5. Glutamate release study
Primary astrocyte cultures were prepared from brains from neonatal rats between postnatal days 1 and 2 as previously described (Holtje et al., 2008) with minor modifications. After removal of meninges, brains were mechanically dissociated in ice-cold Hank's buffered salt solution (HBSS, pH=7.2) by fire-polished glass pipettes and centrifuged at 240×G for 5 min. Tissues were redissociated in ice-cold HBSS, and the procedure were repeated two times using smaller pipette tip diameters. Cells were first seeded onto T-50 flasks and incubated in Dulbecco's modified Eagle's medium, supplemented with 10% fetal bovine serum, 100 units/ml penicillin/streptomycin, 2 mM L-glutamine, and 10 ng/ml epidermal growth factor (EGF, Fisher Scientific, Pittsburgh, PA, USA), included to maintain GLT-1 protein expression in astrocytes (Zelenaia et al., 2000), at 37 °C and 5 % CO2. Microglial cells were detached from the astrocyte monolayer by shaking and decanting. After 5 days in culture with two changes of medium, astrocytes were harvested and reseeded in 24-well plates at a density of 4×104 cells/well and cultured for 48 hrs. Glutamate release experiments were performed as previously described (Holtje et al., 2008) with minor modifications. Astrocytes were incubated with Krebs-Hepes buffer (in mM: NaCl 140, KCl 4.7, CaCl2 2.5, MgSO4 1.2, and Hepes 15, pH=7.4) containing 1 μM glutamate (combination of both tritiated and unlabeled glutamate) for 1 hr at 37 °C, and then washed twice with Krebs-Hepes buffer. Astrocytes were incubated with vehicle (saline) or riluzole (1 μM) in the presence of unlabeled glutamate (0-100 μM) for 10 min at 37 °C. Supernatants were collected and the amount of radioactivity was measured by scintillation spectrometry. Astrocytes were lysed with 0.4% Triton X-100 for 10 min at 42 °C and the amount of radioactivity in lysates were measured by scintillation spectrometry.[3H]-glutamate release was presented as a percentage of total radioactivity (supernatant and lysates) in each well.
4.6. Statistical analyses
Unless otherwise stated, data were normally distributed and are presented as mean ± SE. Differences among groups were determined using one or two-way ANOVA as appropriate. P< 0.05 was considered significant.
Acknowledgments
This work was supported by grants DA024826 to KH and NS59574 to JE from the National Institute of Health, Bethesda, Maryland.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albo F, Pieri M, Zona C. Modulation of AMPA receptors in spinal motor neurons by the neuroprotective agent riluzole. J Neurosci Res. 2004;78:200–7. doi: 10.1002/jnr.20244. [DOI] [PubMed] [Google Scholar]
- Alvarez-Maubecin V, Garcia-Hernandez F, Williams JT, Van Bockstaele EJ. Functional coupling between neurons and glia. J Neurosci. 2000;20:4091–8. doi: 10.1523/JNEUROSCI.20-11-04091.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- Aston-Jones G, Shipley MT, Chouvet G, Ennis M, van Bockstaele E, Pieribone V, Shiekhattar R, Akaoka H, Drolet G, Astier B, et al. Afferent regulation of locus coeruleus neurons: anatomy, physiology and pharmacology. Prog Brain Res. 1991;88:47–75. doi: 10.1016/s0079-6123(08)63799-1. [DOI] [PubMed] [Google Scholar]
- Ballantyne D, Andrzejewski M, Muckenhoff K, Scheid P. Rhythms, synchrony and electrical coupling in the Locus coeruleus. Respir Physiol Neurobiol. 2004;143:199–214. doi: 10.1016/j.resp.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Brooks BR. Managing amyotrophic lateral sclerosis: slowing disease progression and improving patient quality of life. Ann Neurol. 2009;65 1:S17–23. doi: 10.1002/ana.21544. [DOI] [PubMed] [Google Scholar]
- Coderre TJ, Kumar N, Lefebvre CD, Yu JS. A comparison of the glutamate release inhibition and anti-allodynic effects of gabapentin, lamotrigine, and riluzole in a model of neuropathic pain. J Neurochem. 2007;100:1289–99. doi: 10.1111/j.1471-4159.2006.04304.x. [DOI] [PubMed] [Google Scholar]
- De Sarro G, Siniscalchi A, Ferreri G, Gallelli L, De Sarro A. NMDA and AMPA/kainate receptors are involved in the anticonvulsant activity of riluzole in DBA/2 mice. Eur J Pharmacol. 2000;408:25–34. doi: 10.1016/s0014-2999(00)00709-3. [DOI] [PubMed] [Google Scholar]
- Fields HL, Basbaum AI. Brainstem control of spinal pain-transmission neurons. Annu Rev Physiol. 1978;40:217–48. doi: 10.1146/annurev.ph.40.030178.001245. [DOI] [PubMed] [Google Scholar]
- Frizzo ME, Dall'Onder LP, Dalcin KB, Souza DO. Riluzole enhances glutamate uptake in rat astrocyte cultures. Cell Mol Neurobiol. 2004;24:123–8. doi: 10.1023/B:CEMN.0000012717.37839.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli E, Funicello M, Rauen T, Gobbi M, Mennini T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol. 2008;578:171–6. doi: 10.1016/j.ejphar.2007.10.023. [DOI] [PubMed] [Google Scholar]
- Hansson E, Muyderman H, Leonova J, Allansson L, Sinclair J, Blomstrand F, Thorlin T, Nilsson M, Ronnback L. Astroglia and glutamate in physiology and pathology: aspects on glutamate transport, glutamate-induced cell swelling and gap-junction communication. Neurochem Int. 2000;37:317–29. doi: 10.1016/s0197-0186(00)00033-4. [DOI] [PubMed] [Google Scholar]
- Hayashida K, DeGoes S, Curry R, Eisenach JC. Gabapentin activates spinal noradrenergic activity in rats and humans and reduces hypersensitivity after surgery. Anesthesiology. 2007;106:557–62. doi: 10.1097/00000542-200703000-00021. [DOI] [PubMed] [Google Scholar]
- Hayashida K, Obata H, Nakajima K, Eisenach JC. Gabapentin acts within the locus coeruleus to alleviate neuropathic pain. Anesthesiology. 2008;109:1077–84. doi: 10.1097/ALN.0b013e31818dac9c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtje M, Hofmann F, Lux R, Veh RW, Just I, Ahnert-Hilger G. Glutamate uptake and release by astrocytes are enhanced by Clostridium botulinum C3 protein. J Biol Chem. 2008;283:9289–99. doi: 10.1074/jbc.M706499200. [DOI] [PubMed] [Google Scholar]
- Huang CS, Song JH, Nagata K, Yeh JZ, Narahashi T. Effects of the neuroprotective agent riluzole on the high voltage-activated calcium channels of rat dorsal root ganglion neurons. J Pharmacol Exp Ther. 1997;282:1280–90. [PubMed] [Google Scholar]
- Irifune M, Kikuchi N, Saida T, Takarada T, Shimizu Y, Endo C, Morita K, Dohi T, Sato T, Kawahara M. Riluzole, a glutamate release inhibitor, induces loss of righting reflex, antinociception, and immobility in response to noxious stimulation in mice. Anesth Analg. 2007;104:1415–21. doi: 10.1213/01.ane.0000263267.04198.36. table of contents. [DOI] [PubMed] [Google Scholar]
- Kielian T. Glial connexins and gap junctions in CNS inflammation and disease. J Neurochem. 2008;106:1000–16. doi: 10.1111/j.1471-4159.2008.05405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–63. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Lamanauskas N, Nistri A. Riluzole blocks persistent Na+ and Ca2+ currents and modulates release of glutamate via presynaptic NMDA receptors on neonatal rat hypoglossal motoneurons in vitro. Eur J Neurosci. 2008;27:2501–14. doi: 10.1111/j.1460-9568.2008.06211.x. [DOI] [PubMed] [Google Scholar]
- Maeda S, Kawamoto A, Yatani Y, Shirakawa H, Nakagawa T, Kaneko S. Gene transfer of GLT-1, a glial glutamate transporter, into the spinal cord by recombinant adenovirus attenuates inflammatory and neuropathic pain in rats. Mol Pain. 2008;4:65. doi: 10.1186/1744-8069-4-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malarkey EB, Parpura V. Mechanisms of glutamate release from astrocytes. Neurochem Int. 2008;52:142–54. doi: 10.1016/j.neuint.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro G, Erichsen HK, Mirza NR. Pharmacological comparison of anticonvulsant drugs in animal models of persistent pain and anxiety. Neuropharmacology. 2007;53:609–18. doi: 10.1016/j.neuropharm.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Pan F, Mills SL, Massey SC. Screening of gap junction antagonists on dye coupling in the rabbit retina. Vis Neurosci. 2007;24:609–18. doi: 10.1017/S0952523807070472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxix coordinates. Academc Press; San Diego, CA: 2005. [Google Scholar]
- Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther. 1957;111:409–19. [PubMed] [Google Scholar]
- Rojas H, Colina C, Ramos M, Benaim G, Jaffe EH, Caputo C, DiPolo R. Na+ entry via glutamate transporter activates the reverse Na+/Ca2+ exchange and triggers Ca(i)2+-induced Ca2+ release in rat cerebellar Type-1 astrocytes. J Neurochem. 2007;100:1188–202. doi: 10.1111/j.1471-4159.2006.04303.x. [DOI] [PubMed] [Google Scholar]
- Singewald N, Philippu A. Release of neurotransmitters in the locus coeruleus. Prog Neurobiol. 1998;56:237–67. doi: 10.1016/s0301-0082(98)00039-2. [DOI] [PubMed] [Google Scholar]
- Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci. 2003;23:2899–910. doi: 10.1523/JNEUROSCI.23-07-02899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbani A, Belluzzi O. Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur J Neurosci. 2000;12:3567–74. doi: 10.1046/j.1460-9568.2000.00242.x. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Kirchhoff F. Glutamate-mediated neuronal-glial transmission. J Anat. 2007;210:651–60. doi: 10.1111/j.1469-7580.2007.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng HR, Chen JH, Cata JP. Inhibition of glutamate uptake in the spinal cord induces hyperalgesia and increased responses of spinal dorsal horn neurons to peripheral afferent stimulation. Neuroscience. 2006;138:1351–60. doi: 10.1016/j.neuroscience.2005.11.061. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17:1031–6. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- Zelenaia O, Schlag BD, Gochenauer GE, Ganel R, Song W, Beesley JS, Grinspan JB, Rothstein JD, Robinson MB. Epidermal growth factor receptor agonists increase expression of glutamate transporter GLT-1 in astrocytes through pathways dependent on phosphatidylinositol 3-kinase and transcription factor NF-kappaB. Mol Pharmacol. 2000;57:667–78. doi: 10.1124/mol.57.4.667. [DOI] [PubMed] [Google Scholar]
- Zona C, Siniscalchi A, Mercuri NB, Bernardi G. Riluzole interacts with voltage-activated sodium and potassium currents in cultured rat cortical neurons. Neuroscience. 1998;85:931–8. doi: 10.1016/s0306-4522(97)00604-0. [DOI] [PubMed] [Google Scholar]