

Abstract
The idiosyncratic nature, severity and poor diagnosis of drug-induced liver injury (DILI) make these reactions a major safety issue during drug development, as well as the most common cause for the withdrawal of drugs from the pharmaceutical market. Elucidation of the underlying mechanism(s) is necessary for identifying predisposing factors and developing strategies in the treatment and prevention of DILI. Acetaminophen (APAP) is a widely used over the counter therapeutic that is known to be effective and safe at therapeutic doses. However, in overdose situations fatal and non-fatal hepatic necrosis can result. Evidence suggests that the chemically reactive metabolite of the drug initiates hepatocyte damage and that inflammatory innate immune responses also occur within the liver, leading to the exacerbation and progression of tissue injury. Here we investigate whether following APAP-induced liver injury (AILI) damaged hepatocytes release “danger” signals or damage associated molecular pattern molecules (DAMP), which induce pro-inflammatory activation of hepatic macrophages, further contributing to the progression of liver injury. Our study demonstrated a clear activation of Kupffer cells following early exposure to APAP (1hr.). Activation of a murine macrophage cell line, RAW cells, was also observed following treatment with liver perfusate from APAP-treated mice, or with culture supernatant of APAP-challenged hepatocytes. Moreover, in these media, DAMP molecules, heat shock protein-70 (HSP-70) and high mobility group box-1 (HMGB1) were detected. Overall, these findings reveal that DAMP molecules released from damaged and necrotic hepatocytes may serve as a crucial link between the initial hepatocyte damage and the activation of innate immune cells following APAP-exposure, and that DAMPs may represent a potential therapeutic target for AILI.
Keywords: liver, acetaminophen, damage associated molecular pattern molecules, macrophages, high mobility group box-1, heat-shock proteins
Introduction
Drug-induced liver injury (DILI) is the leading cause of acute liver failure in the United States, contributing to approximately half of all cases (Gunawan et al., 2007;Ostapowicz et al., 2002). In addition, DILI represents the primary reason for drug termination during clinical development as well as withdrawal of FDA-approved drugs from the market, with both major medical and economical consequences (Kaplowitz, 2005;Lee et al., 2005). The current detection of DILI remains difficult during the early stages of drug development due to the lack of screening methods, the relatively low incidence of these reactions and the limited knowledge regarding the underlying mechanisms (Lee, 2003). Therefore, a better understanding of the molecular and cellular mechanisms of DILI is imperative in order to identify susceptibility factors and develop successful strategies in the treatment and prevention of DILI.
Acetaminophen (APAP) is a widely used analgesic and antipyretic known to be effective and safe when consumed at therapeutic doses (1 – 4 g/day) (Kaplowitz, 2001;Rumack, 2004). However, severe liver injury resulting in liver failure can occur in some cases following an acute or cumulative overdose (10 – 15 grams) (Kaplowitz, 2001). APAP overdose accounts for more than 56,000 emergency room visits, 2,600 hospitalizations and an estimated 458 deaths due to acute liver failure each year within the United States (Lee, 2004). Overdose of APAP is also known to cause liver injury in laboratory animals with similar characteristics as those found in patients. The murine model of APAP-induced hepatotoxicity represents the most widely used model to study the pathogenesis of DILI. The initiation of APAP-induced liver injury (AILI) results from the metabolism of APAP into a reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI) (Nelson, 1990;Raucy et al., 1989). Ample evidence supports that depletion of hepatic glutathione (GSH) and the covalent binding of NAPQI to cellular macromolecules contributes to protein modification and mitochondrial dysfunction with ATP depletion, culminating in massive centrilobular necrosis (Kaplowitz, 2004;Lee et al., 1996;Pumford et al., 1997).
In addition to direct hepatic cellular dysfunction and death, the pathogenesis of APAP-induced hepatotoxicity also involves the release of a variety of inflammatory mediators that may influence individual susceptibility. It has been demonstrated that macrophage migration inhibitory factor (MIF) (Bourdi et al., 2002b), interferon (IFN)-γ (Ishida et al., 2002), and tumor necrosis factor (TNF)-α (Blazka et al., 1996) contribute to the severity of AILI, while interlukin (IL)-6 (Masubuchi et al., 2003), cyclooxygenase (COX)-2 (Reilly et al., 2001) and IL-10 (Bourdi et al., 2002a) have been shown to promote resolution and regeneration following the initial hepatic damage. Recent studies have also reported the activation and involvement of various innate immune cells, including natural killer (NK) cells and NK cells with T cell receptor (NKT) (Liu et al., 2004;Masson et al., 2008), hepatic macrophages (Ju et al., 2002;Laskin et al., 2001;Michael et al., 1999), and neutrophils (Cover et al., 2006;Ishida et al., 2006;Liu et al., 2006) during APAP-induced hepatotoxicity. However, the exact role of these cells in the pathogenesis of injury remains controversial. Importantly, it remains unknown as to the specific factors that trigger the recruitment and activation of these cells within the liver.
It is possible that damaged hepatocytes themselves can trigger inflammation through the release of various mediators. One hypothesis proposes the critical role of damage associated molecular pattern (DAMP) molecules in this process. Numerous studies have demonstrated that apoptotic and necrotic cells release DAMP molecules such as high mobility group box-1 (HMGB1), S-100 proteins, heat-shock proteins (HSPs), hyaluronan, surfactant protein, interferon-alpha, uric acid, fibronectin, beta defensin, and cardiolipin, which act as endogenous stimulators of immune responses following tissue damage (Asea et al., 2002;Biragyn et al., 2002;Lotze et al., 2005;Okamura et al., 2001;Peitsch et al., 1988;Seong et al., 2004;Shi et al., 2003;Termeer et al., 2002;Wallin et al., 2002;Wang et al., 1999). The function of HMGB1 and HSP-70 as pro-inflammatory mediators has been extensively studied in recent years. HMGB1 is a nuclear binding protein that can be passively released from necrotic cells or secreted by activated macrophages (Shi et al., 2003). HMGB1 has been demonstrated to function as both a chemokine and cytokine, thereby playing a critical role in the initiation of inflammation (Lotze and Tracey, 2005;Wang et al., 1999). HSP-70 is an inducible stress-response protein that can undergo translocation to the cell surface or release into the extra-cellular milieu during periods of cellular stress or necrotic death (Wallin et al., 2002). It has been revealed that extra-cellular HSP-70 can stimulate dendritic cells and macrophages to promote immune responses and inflammation (Asea et al., 2002;Hickman-Miller et al., 2004;Vabulas et al., 2002). However, the release of HMGB1 and HSP-70 by damaged hepatocytes and their specific role in the pathogenesis of DILI have not been investigated.
The objectives of the present study were to examine: i) whether the DAMP molecules HMGB1 and HSP-70 are released from damaged hepatocytes following APAP-challenge in vivo and in vitro, and ii) whether these molecules can trigger the activation of hepatic macrophages, Kupffer cells (KC). Our results provide evidence that APAP-challenged hepatocytes do release HMGB1 and HSP-70. In addition, rHSP-70 can activate isolated primary liver non-parenchymal cells (NPC), as well as purified KC, to produce proinflammatory mediators.
Experimental Procedures
Cell lines and culture conditions
RAW 264.7, a murine macrophage cell line [obtained from American Type Culture Collection (Manassas, VA)] was maintained in RPMI 1640 media supplemented with 10 % fetal bovine serum, 10 mM HEPES and 1× penicillin/streptomycin. TAMH, a hepatocyte cell line [provided by Christopher C. Franklin, Department of Pharmaceutical Sciences, University of Colorado Denver (UCD), CO, USA] was maintained in Dulbecco's modified Eagle's medium/Hams's F12 (Gibco) supplemented with 5 μg/mL insulin, 5 μg/mL transferring, 5 ng/mL selenium (Fisher), 0.1 μmol/L dexamethasone, 10 mM nicotinamide, and 50 μg/mL gentamicin. Cultures were maintained at 37°C in a humidified 95 % air and 5 % CO2 atmosphere.
Animal treatment
Female C57Bl/6J mice (Jackson Laboratories) were maintained under pathogen-free conditions in the Center for Laboratory Animal Care at UCD. For all experiments, mice (7 – 10 weeks of age) were allowed food and water ad libitum until experimental use. Prior to treatment, food was withheld overnight (16 hr), as described previously (James et al., 2003), to deplete hepatic GSH stores. Mice were administered APAP (350 mg/kg, dissolved in warm saline) or saline (as control) via intraperitoneal (i.p.) injection, whereupon food was restored.
Immunoblot analysis of liver perfusate
Mice were anesthetized with Isoflurane (Vedco) and livers perfused in situ via the portal vein with a calcium free HBSS perfusion buffer. Perfusate was collected (10 mL/mouse) and concentrated using a Millipore Amicon Ultra centrifugal filter device (Fisher). Protein concentration was determined via the BCA assay. Forty μg of samples, diluted in Laemmli sample buffer, were resolved on 12 % polyacrylamide gels. Following transfer onto nitrocellulose membranes, the blots were probed with a rabbit anti-HMGB1 polyclonal antibody (1:3000, Abcam) or rat anti-Hsp70 monoclonal antibody (1:3000, Stressgen). A horseradish peroxidase-conjugated secondary antibody was then applied. Membranes were developed using ECL-plus reagent (Amersham Biosciences) and visualized using a STORM 860 scanner (GE Healthcare Bio-Science Corporation).
Immunoblot analysis of TAMH cell culture supernatant
TAMH cells were grown in T-75 flasks (Costar) to 70 % confluency prior to treatment with APAP (1, 10 or 30 mM) for 15 hr. Culture supernatant was collected and concentrated using a Millipore Amicon Ultra centrifugal filter device (Fisher). Residual APAP was removed via a desalting column (Pierce). Protein concentration was determined using the BCA assay. Forty μg of samples were diluted in Laemmli sample buffer and resolved on 12 % polyacrylamide gels. Following transfer onto nitrocellulose membranes, the blots were probed with a rabbit anti-HMGB1 polyclonal antibody (1:1000, Abcam) or rat anti-Hsc70 monoclonal antibody (1:1000, Stressgen). A horseradish peroxidase-conjugated secondary antibody was then applied. Membranes were developed using ECL-plus reagent (Amersham Biosciences) and visualized using a STORM 860 scanner (GE Healthcare Bio-Science Corporation).
KC isolation and treatment
Liver NPC were isolated following a previously established method (Fennekohl et al., 2002;Smedsrod et al., 1985) with slight modifications. In brief, mice were anesthetized, and liver tissues were perfused in situ via the superior vena cava with perfusion buffer (1× HBSS), followed by a digestion buffer (1× HBSS, supplemented with 0.05% collagenase (Type IV; Sigma Chemical Co., St. Louis, MO, USA), 1.23mM CaCl2, 4mM MgSO4, and 10mM HEPES). Following digestion, the liver was disrupted in anticoagulant-citrate-dextrose (Acd) and the cells passed through a 100 μm cell strainer before centrifugation at 30 × g for 3 min to pellet hepatocytes. Cells in the supernatant were centrifuged at 300 × g for 5 min. The pellets were collected, resuspended in Acd solution and fractionated by centrifugation using 30 % (w/v) Nycodenz (Axis-Shield) at 1.155 g/mL to yield liver NPC free of cell debris and erythrocytes. To further purify KC and liver sinusoidal endothelial cells (LSEC), liver NPC were stained with the following antibodies from eBioscience: anti-mouse CD45-FITC, anti-mouse F4/80-APC and anti-mouse CD11b-PE. KC (CD45+ F4/80high, CD11blow) and LSEC (CD45-) were then purified by fluorescence-activated cell sorting (FACS) using a MoFlo High Performance Cell Sorter (Cytomation).
For the experiments of rHSP-70 treatment, KC were purified from liver NPC by positive selection, using biotin anti-mouse F480 in conjunction with the biotin selection kit (EasySep Biotin Selection kit, Stem Cell Technologies). NPC or KC were plated 1×106 cells/well and allowed to adhere for 24 hr prior to treatment with media alone, or rHSP-70 (50 ug/ml, Stressgen) in the presence and absence of polymyxin B (PMB) (20 ug/mL, Fisher Scientific) for 24 hr.
RT-PCR analysis
Total RNA was isolated from cells using either RNeasy mini or micro kits (Qiagen, Valencia, CA, USA), as described by the manufacturer. RNA (1 μg) was reverse-transcribed to cDNA and amplified using JumpStart Taq DNA polymerase (Sigma) and gene-specific primers for β-actin, TNF-α, monocyte chemotactic protein-1 (MCP-1), IL-1β and IL-6 were used. All PCR products were resolved on 1.5 % agarose gels and visualized using ethidium bromide staining.
Data analysis
Mean ± SEM values were calculated for each experimental group and compared using unpaired Student's t-test. Probability levels were considered significant when, *, P < 0.05.
Results
Macrophage activation following APAP treatment
To investigate whether APAP treatment can activate hepatic NPC, these cells were isolated from the liver of mice at 1 hr post-APAP challenge. Compared with cells isolated from saline-treated control mice, liver NPC obtained from APAP-challenged mice exhibited a significant increase in the mRNA expression of pro-inflammatory cytokines, including TNF-α, IL-6 and IL-1β (Fig. 1). Hepatic NPC consist of both KC and LSEC. To identify which cell population represents the predominant source of these increased inflammatory cytokines, KC and LSEC were further purified by FACS. KC obtained from APAP-treated mice were observed to be responsible for the increase in the mRNA expression levels of TNF-α, IL-6 and IL-1β (Fig. 2). It appeared that the up-regulation of pro-inflammatory cytokines in KC in Fig. 2 was less profound than that in NPC shown in Fig.1. This may be explained by the increased background expression of TNF-α and IL-1β (Fig. 2), perhaps due to the slight activation of KC during the purification step by FACSorting. This discrepancy is also possibly due to pooling NPC isolated from 3 mice per group in order to obtain sufficient numbers of KC and LSEC in Fig. 2.
Fig. 1. In vivo activation of liver NPC at 1 hr post-APAP challenge.
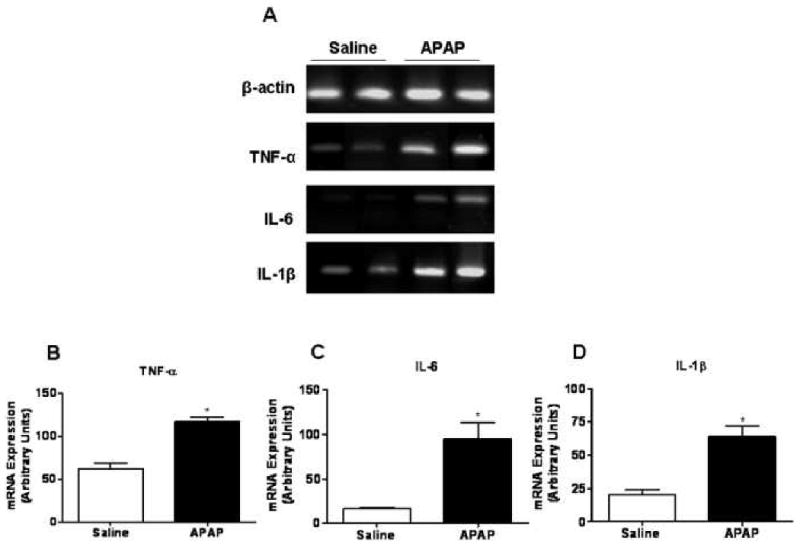
(A) NPCs were isolated from female C57Bl/6J mice treated with either saline or APAP (350 mg/kg) at 1 hr post-treatment. Total RNA was extracted and the mRNA expression of inflammatory mediators by NPC was determined by RT-PCR analysis. Densitometric quantification of (B) TNF-α, (C) IL-6 and (D) IL-1β mRNA expression by NPC. *, P < 0.05, compared with NPC isolated from saline-treated mice.
Fig. 2. In vivo activation of KC at 1 hr post-APAP challenge.
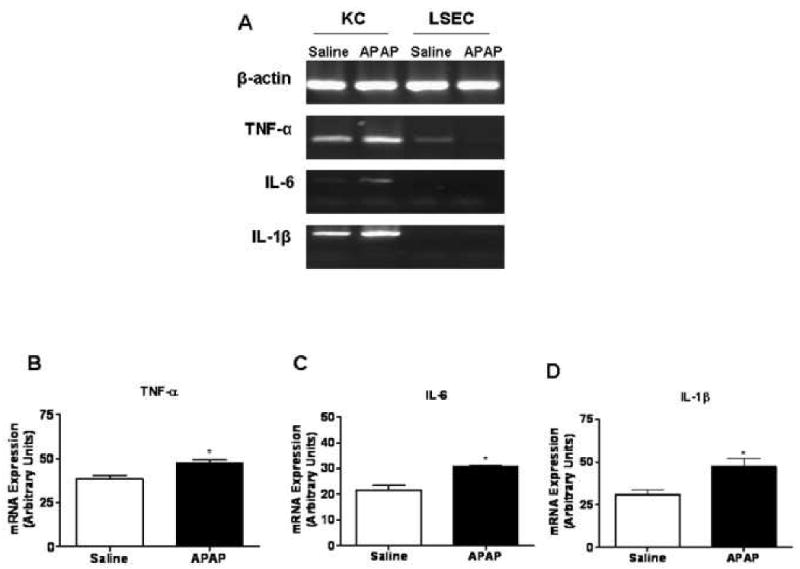
(A) KC and LSEC were purified via FACS from NPC isolated from female C57Bl/6J mice treated with either saline or APAP (350 mg/kg) at 1 hr post-treatment. Total RNA was extracted and the mRNA expression of inflammatory mediators by KC and LSEC was determined by RT-PCR analysis. Densitometric quantification of (B) TNF-α, (C) IL-6 and (D) IL-1β mRNA expression by KC. * P < 0.05, compared with KC isolated from saline-treated mice.
We hypothesized that the activation of KC in APAP-treated mice was triggered by DAMP molecules released from damaged hepatocytes. To examine this hypothesis, liver perfusate was collected from mice at 6 hr following either saline or APAP challenge.
This perfusate was used to treat RAW 264.7 cells, a macrophages cell line, for 3 hr and the induction of pro-inflammatory cytokines was determined by RT-PCR analysis. The data demonstrated that liver perfusate obtained from APAP-challenged mice induced a pro-inflammatory response in RAW cells, as evidenced by the statistically significant up-regulation of mRNA expression levels of MCP-1 and IL-1β (Fig. 3).
Fig. 3. Activation of RAW 264.7 cells by liver perfusate obtained from APAP-challenged mice.
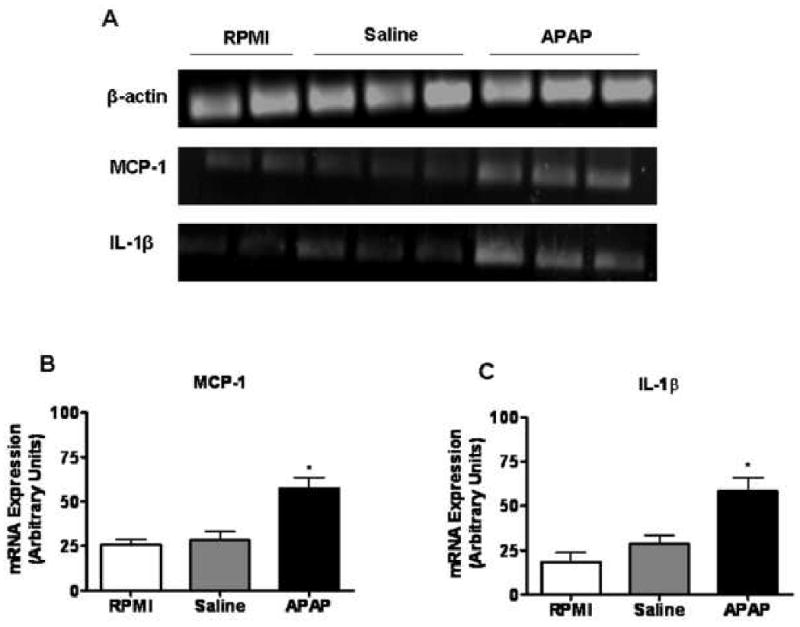
(A) RAW cells were plated in a 48-well cell culture plate and treated for 3 hr with liver perfusate obtained from mice challenged for 6 hr with either saline or APAP (350 mg/kg). Cells cultured in RPMI media alone served as control. Total RNA was extracted and the mRNA expression of inflammatory mediators by RAW cells was determined by RT-PCR analysis. Densitometric quantification of (B) MCP-1 and (C) IL-1β mRNA expression by RAW cells. *, P < 0.05, compared with cells cultured in RMPI and cells treated with liver perfusate obtained from saline-treated mice.
Release of HSP-70 from APAP-challenged hepatocytes
To determine whether HSP-70 represents a DAMP molecule released during AILI in mice, immunoblot analysis was performed using liver perfusate obtained from mice treated with either saline or APAP. The release of HSP-70 from the liver was observed as early as 1 hr following APAP challenge (data not shown) and remained detectable by 24 hr (data not shown). The level of HSP-70 released into the extracellular milieu peaked at 3-6 hr (Fig.4), at which time significant elevation of serum alanine transaminase (ALT) activities were observed 3000 and 5000 IU/L respectively (data not shown). Multiple types of cells could be responsible for the release of HSP-70 within the liver. To investigate whether APAP-induced necrotic hepatocytes can release HSP-70, a mouse hepatocyte cell line (TAMH cells) were treated in vitro with various concentrations of APAP. Fifteen hr after treatment, culture supernatants were collected to measure aspartate transaminase (AST) levels and detect the release of HSP-70. The AST levels were 328 and 556 IU/L in media collected from TAMH cells after treatment with 10 and 30mM APAP respectively (data not shown), suggesting the occurrence of cell stress and/or death. Correlating with APAP-induced cytotoxicity, the release of HSP-70 was detected in the supernatant collected from cells treated with 10 and 30mM APAP (Fig. 5). The data confirmed that damaged hepatocytes can release HSP-70 following APAP-challenge.
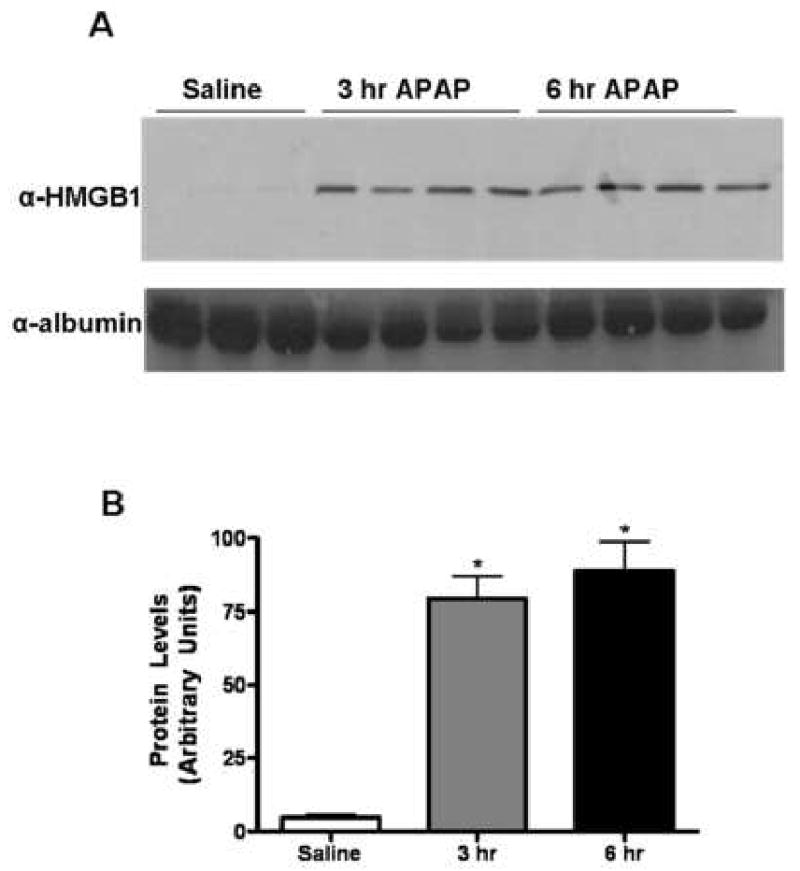
Fig. 4. Release of HSP-70 into the extra-cellular milieu of APAP-challenged mice.
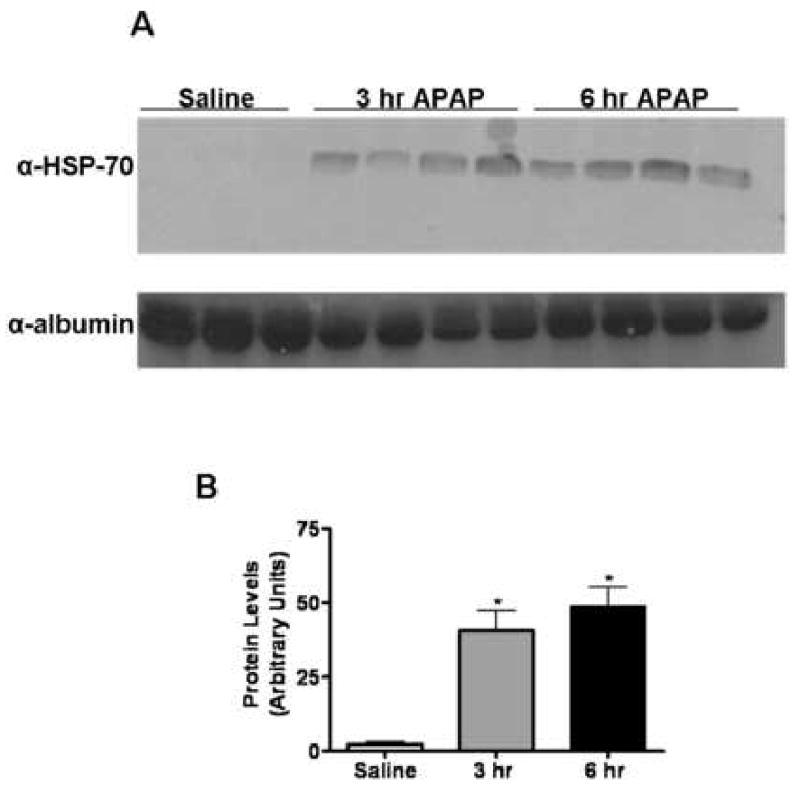
Liver perfusate was obtained from mice challenged with either saline or APAP (350 mg/kg) for 3 and 6 hr. (A) Liver perfusates were immunoblotted with the anti-HSP70 antibody (1:3000) and anti-albumin antibody (1:3000), which served as a loading control. (B) Quantification of HSP-70 release into the extra-cellular milieu by densitometry. *, P < 0.05, compared with saline group.
Fig. 5. Release of HSP-70 into the culture supernatant of APAP-treated TAMH cells.
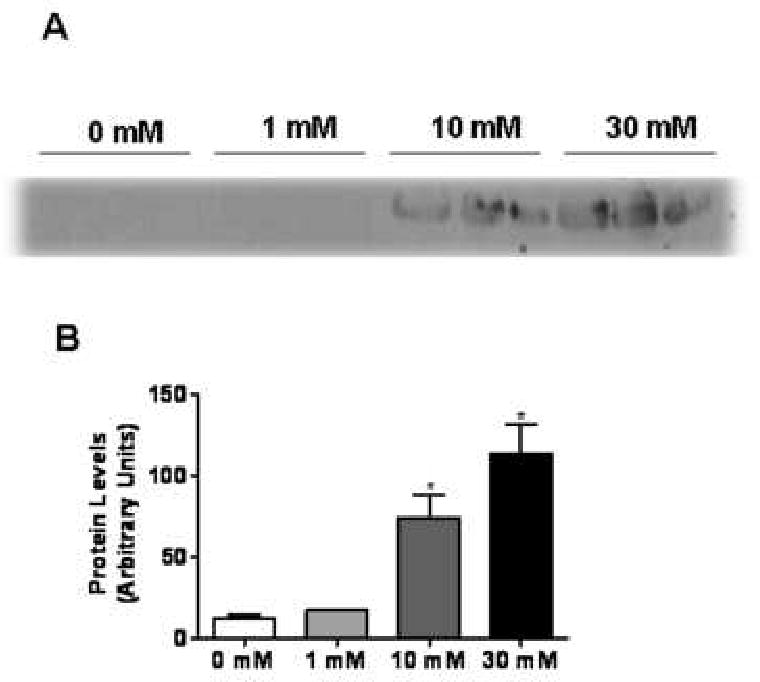
TAMH cells were treated with APAP (0, 1, 10 or 30 mM) for 15 hr. Following treatment, cellular supernatant was collected, concentrated and residual APAP was removed via a desalting column. (A) Immunoblot analysis of HSP-70 release into the cellular supernatant. (B) Quantification of HSP-70 release into the cellular supernatant by densitometry. *, P < 0.05, compared with cells treated with 0 mM APAP.
Pro-inflammatory activation of NPC and KC in vitro by rHSP-70
To determine whether DAMP molecules released from necrotic hepatocytes serve to promote further tissue inflammation during AILI, we examined the ability of recombinant HSP-70 (rHSP-70) to stimulate NPC and KC in vitro. Hepatic NPC were isolated from naïve mice and treated in vitro with low endotoxin containing rHSP-70 (50 ug/mL).
Twenty-four hr after treatment, NPC demonstrated a significant increase in the expression levels of TNF-α and IL-1β (Fig. 6). To further investigate whether KC were the source of increased expression of pro-inflammatory cytokines, KC were further purified from NPC by F480+ biotin bead selection. KC were treated with rHSP-70 (50 ug/ml) in the presence or absence of PMB for 24 hr. PMB, a known neutralizer of lipopolysaccharide (LPS), was used to deplete residue LPS contamination within rHSP-70, to ensure that the stimulatory effect was due to rHSP-70. KC demonstrated a significant increase in the expression levels of TNF-α, MCP-1 and IL-1β (Fig. 6) following treatment with rHSP-70. Additionally, PMB did not appear to decrease this stimulatory effect, confirming that LPS did not contribute to the induction of proinflammatory cytokine expression by KC.
Fig. 6. Activation of isolated NPC and KC following treatment with rHSP-70.
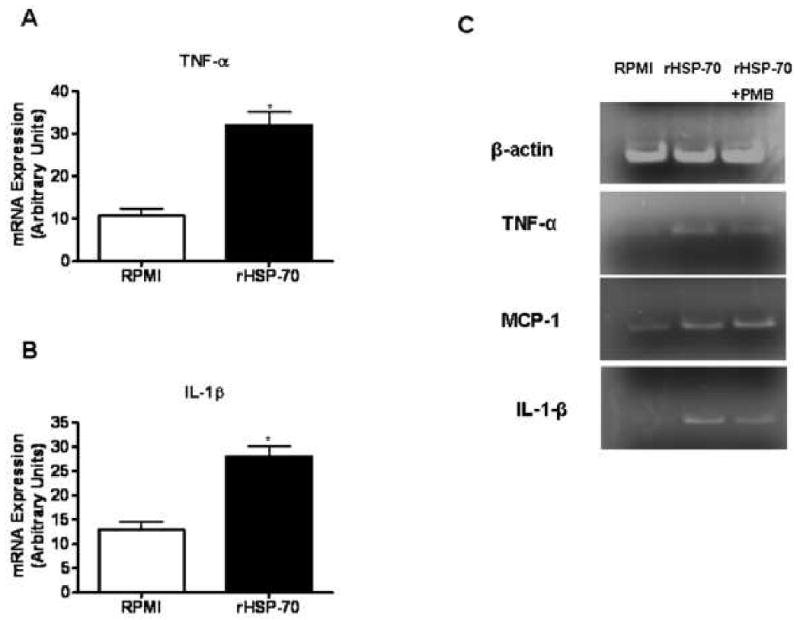
NPCs were isolated from naïve female C57Bl/6J mice, cultured in a 96 well plate, and treated with rHSP-70 for 24 hr. Cells cultured in RPMI media alone served as control. Total RNA was extracted and the mRNA expression of inflammatory mediators by NPC was determined by RT-PCR analysis. Densitometric quantification of (A) TNF-α and (B) IL-1β mRNA expression by NPC. *, P < 0.05, compared with cells cultured in media alone (C) KC were purified via F480+ bead selection from NPC isolated from naïve female C57Bl/6J mice, cultured in a 96 well plate, and treated with rHSP-70 in the presence or absence of PMB. mRNA expression of TNF-α, MCP-1 and IL-1β by KC was determined by RT-PCR analysis.
Release of HMGB1 from APAP-challenged Hepatocytes
Aside from HSP-70, there may exist additional DAMP molecules released during APAP-induced hepatotoxicity. HMGB1 represents one such molecule that is known to possess adjuvant-like effects when released into the extra-cellular milieu. To investigate the involvement of HMGB1 in the pro-inflammatory response following AILI, immunoblot analysis was performed using liver perfusate obtained from mice treated with either saline or APAP. Similar to HSP-70, HMGB1 release from the liver was detected as early as 1 hr following APAP challenge (data not shown), peaked at 3 – 6 hr (Fig. 7) and remained present up to 24 hr (data not shown). To further examine the release of HMGB1 by APAP-induced necrotic hepatocytes, TAMH cells were treated in vitro with various concentrations of APAP and the culture supernatant was collected for immunoblot analysis, as described above. HMGB1 was detected in media collected from TAMH cells treated with 10 and 30mM APAP (Fig. 8), which induced cell damage and the release of AST. The data revealed that damaged hepatocytes can release HMGB1 upon APAP challenge.
Fig. 7. Release of HMGB1 into the extra-cellular milieu of APAP-challenged mice.
Liver perfusate was obtained from mice challenged with either saline or APAP (350 mg/kg) for 3 and 6 hr. (A) Liver perfusates were immunoblotted with the anti-HMGB1 antibody (1:3000) and anti-albumin antibody (1:3000), which served as a loading control. (B) Quantification of HMGB1 release into the extra-cellular milieu by densitometry. *, P < 0.05, compared with saline group.
Fig. 8. Release of HMGB1 into the cellular supernatant of APAP-treated TAMH cells.
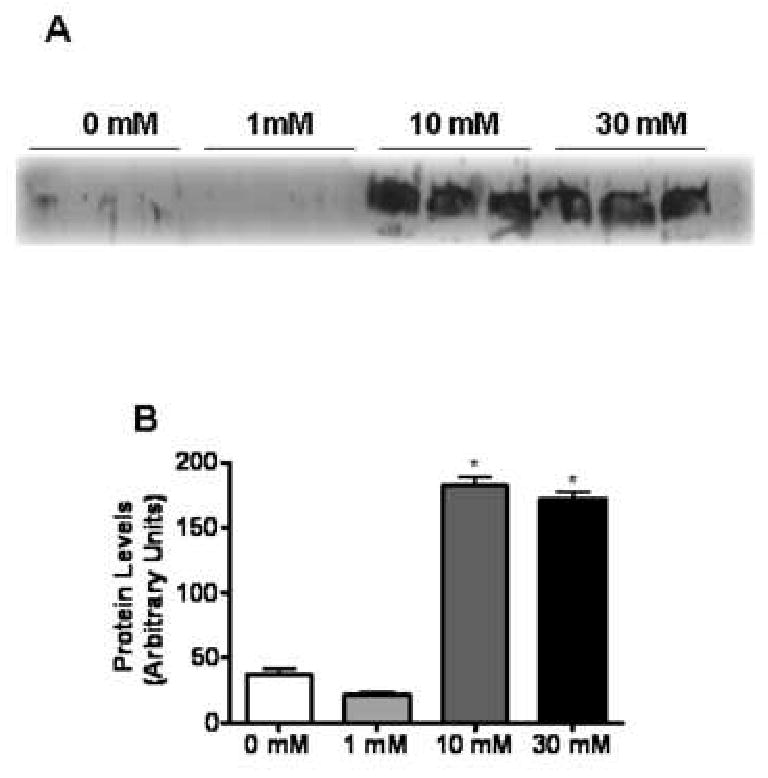
TAMH cells were treated with APAP (0, 1, 10 or 30 mM) for 15 hr. Following treatment, cellular supernatant was collected, concentrated and residual APAP was removed via a desalting column. (A) Immunoblot analysis of HMGB1 release into the cellular supernatant. (B) Quantification of HMGB1 release into the cellular supernatant by densitometry. *, P < 0.05, compared with cells treated with 0 mM APAP.
Discussion
APAP overdose represents a major cause of acute liver failure in the United States each year.8 While hepatocyte damage is initiated by the formation of NAPQI, studies using the murine model of AILI have suggested a role of inflammatory responses in the progression of hepatic injury (Blazka et al., 1995;Blazka et al., 1996;Bourdi et al., 2002b;Bourdi et al., 2002a;Ishida et al., 2002;Laskin and Laskin, 2001;Liu et al., 2004;Liu et al., 2006;Masubuchi et al., 2003;Michael et al., 1999 However, the mechanism by which the innate immune cells are activated is not clear. A recent report showed that DNA isolated from UV irradiation-induced apoptotic hepatocytes could stimulate LSEC production of IL-1β and IL-18 through activating Toll-like receptor (TLR)9 (Imaeda et al., 2009). Nonetheless, it has not been investigated whether APAP-challenged hepatocytes could release immunogenic DNA or other DAMP molecules. The present study provides evidence to support the hypothesis that the initial hepatocyte damage following APAP overdose causes the subsequent activation of innate immune cells via the release of DAMP molecules, such as HSP-70 and HMGB1.
Our data demonstrated that hepatic KC were activated to express TNF-α, IL-6 and IL-1β as early as 1 hr after APAP challenge to mice (Fig. 2). Although it cannot be ruled out that KC can metabolize APAP, resulting in the direct activation of these cells, it is widely believed that APAP is metabolized by hepatocytes, and that the generation of NAPQI leads to cellular dysfunction. Damaged hepatocytes could release DAMP molecules that act as immuno-stimulatory danger signals (Matzinger, 1994) and elicit activation of innate immune cells within the liver. In support of this hypothesis, our data revealed the induction of IL-1β and MCP-1 expression in RAW cells following treatment with the liver perfusate obtained from APAP-challenged mice (Fig. 3). This result suggests that certain mediators in the liver perfusate can stimulate macrophage activation. Our data further demonstrated that DAMP molecules, HMGB1 and HSP-70, were released into the extracellular milieu in the liver of mice treated with APAP (Figs. 4 and 7), as well as in the culture supernatant of APAP-challenged hepatocytes (Figs. 5 and 8). Hepatocyte damage upon APAP challenge in vitro and in vivo was assessed by the release of AST and ALT, respectively. The increase of such enzymes correlated with the observed release of DAMP molecules. In addition, we found that rHSP-70 could activate primary hepatic KC and up-regulate their expression of pro-inflammatory mediators (Fig. 6). Collectively, these results suggest a role for DAMP molecules in the activation of KC during APAP-induced hepatotoxicity. Since APAP is predominantly metabolized within the liver, the release of DAMP molecules is expected to reach a much higher level within the liver, thereby exerting much greater effects on nearby KC. However, it is plausible that DAMP molecules could be released systemically and off target reaction may occur.
In the present study, we examined KC expression of TNF-α, MCP-1, and IL-1β as indicators for the activation of these cells. It is possible that they also express anti-inflammatory cytokines, as it has been shown that KC represent a major source of both pro- and anti-inflammatory mediators, and play both pro-toxicant and protective roles during AILI (Ju et al., 2002;Laskin and Laskin, 2001;Michael et al., 1999). While our finding provides insight into the mechanism by which KC are activated, it does not indicate the precise role of KC in the pathogenesis of AILI. Aside from macrophages, other innate immune cells, particularly NK and NKT cells, reside in the liver in large numbers. Although their pathological role in AILI remains controversial (Liu et al., 2004;Masson et al., 2008), NKT cells have been shown to become activated by known DAMPs, such as catecholamines (Minagawa et al., 2000) and NAD+ (Kawamura et al., 2006). While our study presents hepatic KC as a major target of DAMP molecules, it is possible that other innate immune cells within the liver can also be activated as a result of DAMP molecule release following the initial tissue injury.
Several DAMP molecules have been demonstrated to act as endogenous stimulators of immune responses following tissue damage (Asea et al., 2002;Biragyn et al., 2002;Lotze and Tracey, 2005;Okamura et al., 2001;Peitsch et al., 1988;Seong and Matzinger, 2004;Shi et al., 2003;Termeer et al., 2002;Wallin et al., 2002;Wang et al., 1999). HSPs represent one class of DAMP molecules that have been extensively studied over the recent years. HSPs were first considered possible DAMP molecules due to their up-regulation during periods of cellular stress throughout a variety of disorders (Mambula et al., 2006;Sapozhnikov et al., 1999). HSP-60, -70 and -90 are presented on the cell surface (Martin et al., 2003;Theriault et al., 2005;Vabulas et al., 2001;Vabulas et al., 2002) and have been shown to activate antigen presenting cells (APC), specifically macrophages and dendritic cells, via engagement of Toll-like receptors (TLR) (Asea et al., 2000;Chen et al., 1999;Vabulas et al., 2001;Vabulas et al., 2002). A marked increase of HSP-70 within whole liver homogenate has been demonstrated following APAP-induced liver injury (Welch et al., 2006). However, the release of HSP-70 into the extra-cellular milieu during APAP-induced hepatotoxicity has not been assessed. Our detection of HSP-70 within the liver perfusate of APAP-challenged mice (Fig. 4) is the first to demonstrate its release during this pathological condition. The detection of HSP-70 in the supernatant of TAMH cells treated with APAP (Fig. 5) further confirmed that APAP-challenged hepatocytes represent a major source of HSP-70. Moreover, rHSP-70 was able to stimulate pro-inflammatory responses in vitro of purified NPC and KC (Fig. 6), which is consistent with previous studies on the ability of HSPs to activate APC (Chen et al., 1999;Martin et al., 2003). To address the concern that the immuno-stimulatory ability of HSP-70 might be due to LPS contaminants, we chose to use a rHSP-70 protein containing an extremely low level of endotoxin (< 50 EU/mg protein). This yielded the total amount of endotoxin present during cell treatment to be less than 2.5 EU. Further, PMB was included in some cultures to remove any residual LPS contamination. The addition of PMB did not inhibit rHSP-70-induced activation of hepatic KC (Fig. 6), confirming that the pro-inflammatory response of KC was due to the stimulatory ability of HSP-70 and not due to LPS contamination. In order to further confirm the effects of HSP70 on macrophages, we performed the experiment of treating RAW cells with APAP-liver perfusate in the presence of a commercially available anti-HSP70 neutralizing antibody. While the HSP70 neutralizing antibody was able to attenuate the pro-inflammatory effect of the APAP-liver perfusate, decreases were also observed in samples treated with the control IgG, thus lending our data inconclusive (data not shown). The data suggest that HSP70 within the liver perfusate does not play a predominant role in the induction of pro-inflammatory cytokines. It is likely that both HSP70 and HMGB1 together contribute to the activation of RAW cells, and that depletion of HSP70 alone is not sufficient to significantly attenuate the stimulatory effect of the liver perfusate.
In addition to HSP-70, HMBG1 represents another DAMP molecule that would otherwise be retained within the cell, except during periods of pathological damage. HMGB1 is normally tightly bound to chromatin, thereby facilitating the coiling of DNA. HMGB1 was first identified as a DAMP molecule due to its release from necrotic cells and its ability to further stimulate a pro-inflammatory state in monocytes and dendritic cells (Scaffidi et al., 2002;Tsung et al., 2005;Yang et al., 2007). Our data showed that HMGB1 could be detected in the liver perfusate of APAP -challenged mice (Fig. 7), as well as in culture supernatant of APAP-treated TAMH cells (Fig. 8), suggesting that hepatocytes represent a critical source of HMGB1 release. While our study is the first to demonstrate the release of HMGB1 into extracellular milieu, the role of HMGB1 in propagating AILI has been implicated in two published studies. Antibodies directed against HMGB1 have been able to reduce tissue inflammation, as indicated by a decrease in the myeloperoxidase (MPO)/alanine transaminase (ALT) ratio, following APAP-induced hepatotoxicity (Scaffidi et al., 2002). A more recent publication provides insight into the mechanism of HMGB1-induced inflammation (Chen et al., 2009). HMGB1 was proposed to from a tri-molecular complex with CD24 and Siglec-G, which act as negative regulators of the pro-inflammatory function of HMGB-1, as both CD24-/- and Siglec-G-/- mice were more susceptible to AILI than their WT counterparts. Administration of an anti-HMGB1 neutralizing antibody was able to attenuate AILI in WT, as well as in CD24-/- and Siglec-G-/- mice.
In summary, the present study is the first to demonstrate the release of DAMP molecules, HSP-70 and HMGB1, from APAP-challenged hepatocytes in vitro and in vivo. The data also provide evidence for a link between APAP-induced hepatocyte death and activation of hepatic macrophages. Further studies to establish whether one or multiple DAMP molecules are responsible for the activation of hepatic innate immune cells are warranted.
Acknowledgments
This work was supported by U.S. National Institutes of Health grant Ruth L. Kirschstein National Service Award F31DK082269 (to Brittany Martin-Murphy) and R01ES012914 (to Cynthia Ju).
Nonstandard Abbreviations used
- DILI
drug-induced liver injury
- APAP
acetaminophen
- AILI
acetaminophen-induced liver injury
- NAPQI
N-acetyl-p-benzoquinone imine
- GSH
glutathione
- MIF
migration inhibitory factor
- IFN
interferon
- TNF
tumor necrosis factor
- IL
interleukin
- COX
cyclooxygenase
- NK
natural killer
- DAMP
damage associated molecular pattern
- HMGB1
high mobility group box-1
- HSP
heat shock protein
- KC
Kupffer cells
- NPC
non-parenchymal cells
- LSEC
liver sinusoidal endothelial cells
- FACS
fluorescence-activated cell sorting
- MCP-1
monocyte chemotactic protein-1
- rHSP
recombinant heat shock protein
- PMB
polymyxin B
- LPS
lipopolysaccharide
- APC
antigen presenting cells
- TLR
toll-like receptor
- MPO
myeloperoxidase
- ALT
alanine transaminase
Footnotes
Conflict of Interest Statement: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6:435–442. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, Shirakawa AK, Farber JM, Segal DM, Oppenheim JJ, Kwak LW. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. 2002;298:1025–1029. doi: 10.1126/science.1075565. [DOI] [PubMed] [Google Scholar]
- Blazka ME, Elwell MR, Holladay SD, Wilson RE, Luster MI. Histopathology of acetaminophen-induced liver changes: role of interleukin 1 alpha and tumor necrosis factor alpha. Toxicol Pathol. 1996;24:181–189. doi: 10.1177/019262339602400206. [DOI] [PubMed] [Google Scholar]
- Blazka ME, Wilmer JL, Holladay SD, Wilson RE, Luster MI. Role of proinflammatory cytokines in acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 1995;133:43–52. doi: 10.1006/taap.1995.1125. [DOI] [PubMed] [Google Scholar]
- Bourdi M, Masubuchi Y, Reilly TP, Amouzadeh HR, Martin JL, George JW, Shah AG, Pohl LR. Protection against acetaminophen-induced liver injury and lethality by interleukin 10: role of inducible nitric oxide synthase. Hepatology. 2002a;35:289–298. doi: 10.1053/jhep.2002.30956. [DOI] [PubMed] [Google Scholar]
- Bourdi M, Reilly TP, Elkahloun AG, George JW, Pohl LR. Macrophage migration inhibitory factor in drug-induced liver injury: a role in susceptibility and stress responsiveness. Biochem Biophys Res Commun. 2002b;294:225–230. doi: 10.1016/S0006-291X(02)00466-7. [DOI] [PubMed] [Google Scholar]
- Chen GY, Tang J, Zheng P, Liu Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science. 2009;323:1722–1725. doi: 10.1126/science.1168988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Syldath U, Bellmann K, Burkart V, Kolb H. Human 60-kDa heat-shock protein: a danger signal to the innate immune system. J Immunol. 1999;162:3212–3219. [PubMed] [Google Scholar]
- Cover C, Liu J, Farhood A, Malle E, Waalkes MP, Bajt ML, Jaeschke H. Pathophysiological role of the acute inflammatory response during acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 2006;216:98–107. doi: 10.1016/j.taap.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Fennekohl A, Sugimoto Y, Segi E, Maruyama T, Ichikawa A, Puschel GP. Contribution of the two Gs-coupled PGE2-receptors EP2-receptor and EP4-receptor to the inhibition by PGE2 of the LPS-induced TNFalpha-formation in Kupffer cells from EP2-or EP4-receptor-deficient mice. Pivotal role for the EP4-receptor in wild type Kupffer cells. J Hepatol. 2002;36:328–334. doi: 10.1016/s0168-8278(01)00277-x. [DOI] [PubMed] [Google Scholar]
- Gunawan BK, Kaplowitz N. Mechanisms of drug-induced liver disease. Clin Liver Dis. 2007;11:459–75. doi: 10.1016/j.cld.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Hickman-Miller HD, Hildebrand WH. The immune response under stress: the role of HSP-derived peptides. Trends Immunol. 2004;25:427–433. doi: 10.1016/j.it.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, Mehal WZ. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest. 2009;119:305–314. doi: 10.1172/JCI35958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y, Kondo T, Kimura A, Tsuneyama K, Takayasu T, Mukaida N. Opposite roles of neutrophils and macrophages in the pathogenesis of acetaminophen-induced acute liver injury. Eur J Immunol. 2006;36:1028–1038. doi: 10.1002/eji.200535261. [DOI] [PubMed] [Google Scholar]
- Ishida Y, Kondo T, Ohshima T, Fujiwara H, Iwakura Y, Mukaida N. A pivotal involvement of IFN-gamma in the pathogenesis of acetaminophen-induced acute liver injury. FASEB J. 2002;16:1227–1236. doi: 10.1096/fj.02-0046com. [DOI] [PubMed] [Google Scholar]
- James LP, McCullough SS, Lamps LW, Hinson JA. Effect of N-acetylcysteine on acetaminophen toxicity in mice: relationship to reactive nitrogen and cytokine formation. Toxicol Sci. 2003;75:458–467. doi: 10.1093/toxsci/kfg181. [DOI] [PubMed] [Google Scholar]
- Ju C, Reilly TP, Bourdi M, Radonovich MF, Brady JN, George JW, Pohl LR. Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem Res Toxicol. 2002;15:1504–1513. doi: 10.1021/tx0255976. [DOI] [PubMed] [Google Scholar]
- Kaplowitz N. Drug-induced liver disorders: implications for drug development and regulation. Drug Saf. 2001;24:483–490. doi: 10.2165/00002018-200124070-00001. [DOI] [PubMed] [Google Scholar]
- Kaplowitz N. Acetaminophen hepatoxicity: what do we know, what don't we know, and what do we do next? Hepatology. 2004;40:23–26. doi: 10.1002/hep.20312. [DOI] [PubMed] [Google Scholar]
- Kaplowitz N. Idiosyncratic drug hepatotoxicity. Nat Rev Drug Discov. 2005;4:489–499. doi: 10.1038/nrd1750. [DOI] [PubMed] [Google Scholar]
- Kawamura H, Aswad F, Minagawa M, Govindarajan S, Dennert G. P2X7 receptors regulate NKT cells in autoimmune hepatitis. J Immunol. 2006;176:2152–2160. doi: 10.4049/jimmunol.176.4.2152. [DOI] [PubMed] [Google Scholar]
- Laskin DL, Laskin JD. Role of macrophages and inflammatory mediators in chemically induced toxicity. Toxicology. 2001;160:111–118. doi: 10.1016/s0300-483x(00)00437-6. [DOI] [PubMed] [Google Scholar]
- Lee SS, Buters JT, Pineau T, Fernandez-Salguero P, Gonzalez FJ. Role of CYP2E1 in the hepatotoxicity of acetaminophen. J Biol Chem. 1996;271:12063–12067. doi: 10.1074/jbc.271.20.12063. [DOI] [PubMed] [Google Scholar]
- Lee WM. Drug-induced hepatotoxicity. N Engl J Med. 2003;349:474–485. doi: 10.1056/NEJMra021844. [DOI] [PubMed] [Google Scholar]
- Lee WM. Acetaminophen and the U.S. Acute Liver Failure Study Group: lowering the risks of hepatic failure. Hepatology. 2004;40:6–9. doi: 10.1002/hep.20293. [DOI] [PubMed] [Google Scholar]
- Lee WM, Senior JR. Recognizing drug-induced liver injury: current problems, possible solutions. Toxicol Pathol. 2005;33:155–164. doi: 10.1080/01926230590522356. [DOI] [PubMed] [Google Scholar]
- Liu ZX, Govindarajan S, Kaplowitz N. Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology. 2004;127:1760–1774. doi: 10.1053/j.gastro.2004.08.053. [DOI] [PubMed] [Google Scholar]
- Liu ZX, Han D, Gunawan B, Kaplowitz N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology. 2006;43:1220–1230. doi: 10.1002/hep.21175. [DOI] [PubMed] [Google Scholar]
- Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- Mambula SS, Calderwood SK. Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J Immunol. 2006;177:7849–7857. doi: 10.4049/jimmunol.177.11.7849. [DOI] [PubMed] [Google Scholar]
- Martin CA, Carsons SE, Kowalewski R, Bernstein D, Valentino M, Santiago-Schwarz F. Aberrant extracellular and dendritic cell (DC) surface expression of heat shock protein (hsp)70 in the rheumatoid joint: possible mechanisms of hsp/DC-mediated cross-priming. J Immunol. 2003;171:5736–5742. doi: 10.4049/jimmunol.171.11.5736. [DOI] [PubMed] [Google Scholar]
- Masson MJ, Carpenter LD, Graf ML, Pohl LR. Pathogenic role of natural killer T and natural killer cells in acetaminophen-induced liver injury in mice is dependent on the presence of dimethyl sulfoxide. Hepatology. 2008;48:889–897. doi: 10.1002/hep.22400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masubuchi Y, Bourdi M, Reilly TP, Graf ML, George JW, Pohl LR. Role of interleukin-6 in hepatic heat shock protein expression and protection against acetaminophen-induced liver disease. Biochem Biophys Res Commun. 2003;304:207–212. doi: 10.1016/s0006-291x(03)00572-2. [DOI] [PubMed] [Google Scholar]
- Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- Michael SL, Pumford NR, Mayeux PR, Niesman MR, Hinson JA. Pretreatment of mice with macrophage inactivators decreases acetaminophen hepatotoxicity and the formation of reactive oxygen and nitrogen species. Hepatology. 1999;30:186–195. doi: 10.1002/hep.510300104. [DOI] [PubMed] [Google Scholar]
- Minagawa M, Oya H, Yamamoto S, Shimizu T, Bannai M, Kawamura H, Hatakeyama K, Abo T. Intensive expansion of natural killer T cells in the early phase of hepatocyte regeneration after partial hepatectomy in mice and its association with sympathetic nerve activation. Hepatology. 2000;31:907–915. doi: 10.1053/he.2000.5850. [DOI] [PubMed] [Google Scholar]
- Nelson SD. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin Liver Dis. 1990;10:267–278. doi: 10.1055/s-2008-1040482. [DOI] [PubMed] [Google Scholar]
- Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF., III The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001;276:10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- Ostapowicz G, Fontana RJ, Schiodt FV, Larson A, Davern TJ, Han SH, McCashland TM, Shakil AO, Hay JE, Hynan L, Crippin JS, Blei AT, Samuel G, Reisch J, Lee WM. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137:947–954. doi: 10.7326/0003-4819-137-12-200212170-00007. [DOI] [PubMed] [Google Scholar]
- Peitsch MC, Tschopp J, Kress A, Isliker H. Antibody-independent activation of the complement system by mitochondria is mediated by cardiolipin. Biochem J. 1988;249:495–500. doi: 10.1042/bj2490495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pumford NR, Halmes NC, Hinson JA. Covalent binding of xenobiotics to specific proteins in the liver. Drug Metab Rev. 1997;29:39–57. doi: 10.3109/03602539709037572. [DOI] [PubMed] [Google Scholar]
- Raucy JL, Lasker JM, Lieber CS, Black M. Acetaminophen activation by human liver cytochromes P450IIE1 and P450IA2. Arch Biochem Biophys. 1989;271:270–283. doi: 10.1016/0003-9861(89)90278-6. [DOI] [PubMed] [Google Scholar]
- Reilly TP, Brady JN, Marchick MR, Bourdi M, George JW, Radonovich MF, Pise-Masison CA, Pohl LR. A protective role for cyclooxygenase-2 in drug-induced liver injury in mice. Chem Res Toxicol. 2001;14:1620–1628. doi: 10.1021/tx0155505. [DOI] [PubMed] [Google Scholar]
- Rumack BH. Acetaminophen misconceptions. Hepatology. 2004;40:10–15. doi: 10.1002/hep.20300. [DOI] [PubMed] [Google Scholar]
- Sapozhnikov AM, Ponomarev ED, Tarasenko TN, Telford WG. Spontaneous apoptosis and expression of cell surface heat-shock proteins in cultured EL-4 lymphoma cells. Cell Prolif. 1999;32:363–378. doi: 10.1111/j.1365-2184.1999.tb01354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–521. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- Smedsrod B, Pertoft H. Preparation of pure hepatocytes and reticuloendothelial cells in high yield from a single rat liver by means of Percoll centrifugation and selective adherence. J Leukoc Biol. 1985;38:213–230. doi: 10.1002/jlb.38.2.213. [DOI] [PubMed] [Google Scholar]
- Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriault JR, Mambula SS, Sawamura T, Stevenson MA, Calderwood SK. Extracellular HSP70 binding to surface receptors present on antigen presenting cells and endothelial/epithelial cells. FEBS Lett. 2005;579:1951–1960. doi: 10.1016/j.febslet.2005.02.046. [DOI] [PubMed] [Google Scholar]
- Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201:1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Hacker H, Wagner H. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem. 2001;276:31332–31339. doi: 10.1074/jbc.M103217200. [DOI] [PubMed] [Google Scholar]
- Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002;277:15107–15112. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- Wallin RP, Lundqvist A, More SH, von Bonin A, Kiessling R, Ljunggren HG. Heat-shock proteins as activators of the innate immune system. Trends Immunol. 2002;23:130–135. doi: 10.1016/s1471-4906(01)02168-8. [DOI] [PubMed] [Google Scholar]
- Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- Welch KD, Reilly TP, Bourdi M, Hays T, Pise-Masison CA, Radonovich MF, Brady JN, Dix DJ, Pohl LR. Genomic identification of potential risk factors during acetaminophen-induced liver disease in susceptible and resistant strains of mice. Chem Res Toxicol. 2006;19:223–233. doi: 10.1021/tx050285z. [DOI] [PubMed] [Google Scholar]
- Yang D, Chen Q, Yang H, Tracey KJ, Bustin M, Oppenheim JJ. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J Leukoc Biol. 2007;81:59–66. doi: 10.1189/jlb.0306180. [DOI] [PubMed] [Google Scholar]